Self-association of the antitumour agent novatrone (mitoxantrone) and its hetero-association with caffeine
David B. Davies*a, Dennis A. Veselkova, Maxim P. Evstigneevb and Alexei N. Veselkovb
aSchool of Biological and Chemical Sciences, Birkbeck College, University of London, Gordon House, 29 Gordon Square, London, UK WC1H 0PP
bDepartment of Physics and Chemistry, Sevastopol State Technical University, Sevastopol, 99053, Crimea, Ukraine
First published on 5th December 2000
Abstract
The self-association of the antitumour drug, novatrone, NOV (mitoxantrone) and its hetero-association with caffeine (CAF![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) ) have been investigated by 1D and 2D 500 MHz 1H NMR spectroscopy. Two-dimensional homonuclear correlation NMR spectroscopy (2D TOCSY and 2D ROESY) has been used for complete assignment of proton signals and for a qualitative analysis of the mutual arrangements of the aromatic drug molecules in the aggregates. The structural and thermodynamical parameters of molecular self- and hetero-association of the aromatic compounds have been determined from measurements of the NMR chemical shifts of the drug protons as a function of concentration and temperature. The self-association of NOV has been analysed using both the indefinite cooperative and non-cooperative models, and the hetero-association of NOV and CAF has been analysed in terms of a statistical-thermodynamical model, in which molecules form indefinite aggregates for both self- and hetero-association. The magnitudes of parameters (equilibrium reaction constants, enthalpy (ΔH) and entropy (ΔS)) have been calculated for self-association of NOV and its complexation with CAF; at 318 K the equilibrium constant for self-association of NOV is 12400 (±4000) l mol−1 and for hetero-association with CAF is 256 (±30) l mol−1. The most favourable structures of the NOV dimer and the 1∶1 NOV–CAF hetero-association complexes have been determined from the calculated limiting values of the induced chemical shifts of the drug protons.
) have been investigated by 1D and 2D 500 MHz 1H NMR spectroscopy. Two-dimensional homonuclear correlation NMR spectroscopy (2D TOCSY and 2D ROESY) has been used for complete assignment of proton signals and for a qualitative analysis of the mutual arrangements of the aromatic drug molecules in the aggregates. The structural and thermodynamical parameters of molecular self- and hetero-association of the aromatic compounds have been determined from measurements of the NMR chemical shifts of the drug protons as a function of concentration and temperature. The self-association of NOV has been analysed using both the indefinite cooperative and non-cooperative models, and the hetero-association of NOV and CAF has been analysed in terms of a statistical-thermodynamical model, in which molecules form indefinite aggregates for both self- and hetero-association. The magnitudes of parameters (equilibrium reaction constants, enthalpy (ΔH) and entropy (ΔS)) have been calculated for self-association of NOV and its complexation with CAF; at 318 K the equilibrium constant for self-association of NOV is 12400 (±4000) l mol−1 and for hetero-association with CAF is 256 (±30) l mol−1. The most favourable structures of the NOV dimer and the 1∶1 NOV–CAF hetero-association complexes have been determined from the calculated limiting values of the induced chemical shifts of the drug protons.
Introduction
The synthetic antitumour drug novatrone, NOV (mitoxantrone) has proven clinical antineoplastic activity in patients with breast cancer, acute leukemia and non-Hodgkin’s lymphoma.1 NOV is one of the promising drugs that have been developed to overcome the cardiotoxicity associated with anthracyclines.2 The molecular mechanisms and intracellular targets associated with the antitumour effects of NOV are still unclear,3 and the possibilities suggested involve formation of NOV–DNA complexes,4–7 complexes of NOV with cytoskeleton proteins8 and/or accumulation of cytotoxic metabolites.9 However, most of the evidence indicates that nucleic acids are the main targets of NOV in living cells and both DNA transcription and RNA processing are affected.1,6,7 The antibiotic NOV, unlike many other intercalating drugs, does not show pronounced sequence specificity in complexation with DNA,5–7,10 although it has been suggested that hydroxy groups in the chromophore of NOV play a significant role in the recognition of preferred nucleotide sequences.10 Prior to determination of the structural and thermodynamical characteristics of the intercalative binding of aromatic drugs to defined DNA sequences by NMR spectroscopy,11–13 it is necessary to determine the self-association of the drugs in solution. There are a number of investigations of the self-association of NOV which give a large range of values of equilibrium self-association constants in aqueous solution5,14,15 and so the equilibrium constant and thermodynamic parameters of self-association of NOV are measured under the same solution conditions (0.1 M phosphate buffer, pD 7.1) as used for our drug–DNA studies.12,13Caffeine (1,3,7-trimethylxanthine, CAF) represents a class of aromatic molecules that constitute the most widely distributed naturally occurring methylxanthines. It is generally accepted that some of the biological activity of CAF results from its interactions with biopolymers such as enzymes and nucleic acids.16 It has also been shown that CAF is capable of reducing the toxicity of a typical DNA intercalator, ethidium bromide,17 and the efficacy of a number of anti-cancer aromatic drugs, such as novatrone, doxorubicin and its analogues, ellipticine and others.18,19 It was suggested19–21 that CAF forms hetero-association complexes with aromatic molecules, which effectively lowers the concentration of free ligand and thereby reduces the pharmacological activity of the drugs, i.e. it was concluded21 that CAF acts as an “interceptor” of biologically active aromatic molecules, which bind to DNA by intercalation. Although novatrone was included in this general “interceptor” mechanism, there have been no experimental determinations of the hetero-association of novatrone with caffeine and so, in this work, their complexation is investigated in order to provide further understanding of the basis of the reduction of the efficacy of the drug.
The hetero-association of CAF with different aromatic molecules has been investigated using different mathematical models and analytical procedures to interpret the experimental results.20–22 However most of the proposed models of molecular hetero-association have some limitations in either their use or theoretical approach as discussed in detail elsewhere.23,24 A statistical-thermodynamical model of hetero-association, in which molecules form indefinite aggregates for both self-association and hetero-association, has recently been developed in our laboratory to analyse the NMR parameters of component molecules in aqueous solution,23,24 and this model will be used to analyse the hetero-association of CAF with NOV in this work.
We report the NMR analysis of the self-association of the antibiotic NOV and its complexation with CAF (structures of both molecules are presented in Fig. 1) in buffered aqueous solution. Two-dimensional homonuclear correlation 500 MHz 1H NMR spectroscopy (2D TOCSY and 2D ROESY) has been used for complete assignment of proton signals and for a qualitative analysis of the mutual arrangements of the aromatic drug molecules in the aggregates. 1D 1H NMR measurements have been made as a function of both concentration and temperature, from which the structural and thermodynamical parameters of NOV self-association and its hetero-association with CAF have been determined.
 | ||
| Fig. 1 Structures of caffeine (CAF) and novatrone (NOV).
| ||
Results
1 Self-association of novatrone in aqueous solution
The structure of the aggregate and thermodynamics of self-association of NOV have been determined in this work in the same way as done previously25 for other aromatic drugs under the same experimental conditions. 1H NMR signal assignments of NOV have been made using homonuclear 2D TOCSY and 2D ROESY experiments and are in good agreement with published 1D NMR data.14 The NOV molecule has a plane of symmetry and so chemical equivalence is found for resonances of H2/H3, H6/H7 and the protons of the two aminoalkyl side chains. The 2D ROESY spectrum of NOV solution (Fig. 2) obtained at initial drug concentration (x0 = 1.01 mM) exhibits an intermolecular ROE contact between the H2/H3 and H11 protons of the antibiotic (shown by arrow), which provides unambiguous evidence of the inverse (“head to tail”) orientation of the antibiotic chromophores in the aggregate. A similar conclusion about the mutual arrangement of the NOV chromophores in the dimer aggregate was made previously14 on the basis of 1D NOE experiments.![2D ROESY (500 MHz, τm = 240 ms) of NOV solution ([x0] = 1.01 mmol l−1) in 0.1 M phosphate buffer, pD = 7.1 at T = 318 K. The intermolecular cross-peak (H2/3–H11) is shown by the arrow.](/image/article/2001/P2/b007042o/b007042o-f2.gif) | ||
| Fig. 2 2D ROESY (500 MHz, τm = 240 ms) of NOV solution ([x0] = 1.01 mmol l−1) in 0.1 M phosphate buffer, pD = 7.1 at T = 318 K. The intermolecular cross-peak (H2/3–H11) is shown by the arrow. | ||
The changes with concentration and temperature of the chemical shifts of the non-exchangeable aromatic protons and the H11 protons of the side chains of the NOV chromophore are presented in Fig. 3. The concentration dependences of the proton chemical shifts of NOV show shifts to lower frequencies at higher concentrations, which result from intermolecular stacked complexes in solution in common with other aromatic drug molecules.25,26 The self-association properties of the drug have been analysed using the indefinite non-cooperative model, in which the equilibrium constants Kj for the equilibria (eqn. (1)) are assumed to be equal for j = (1; ∞), as done previously25 for other aromatic drugs. In this model the dependence of the observed chemical shift δ on concentration is given by eqn. (2),25,26 where δi is the proton chemical shift for the drug molecule in the where δi is the proton chemical shift for the drug molecule in the complex, δm is the proton chemical shift of the monomer, i.e. at infinite dilution. It was taken into account that (δi − δm) = 2(δd − δm), where δd is the proton chemical shift of the molecule at the end of the aggregate and also in the dimer.
 | (1) |
 | (2) |
 | ||
| Fig. 3 Experimental proton chemical shifts for self-association of NOV (empty circles) and in the mixed solution of NOV with CAF (NOV, filled circles; CAF, filled triangles): (a) dependence on NOV concentration at T = 318 K (CCAF = p0 = 2.0 mM); (b) dependence on temperature (self-association: CNOV = x0 = 0.47 mM, empty circles; hetero-association: CNOV = a0 = 0.9 mM (filled circles), CCAF = p0 = 2.0 mM (filled triangles)). | ||
The parameters δm, δi and K in eqn. (2) were calculated from the experimental concentration dependences of chemical shifts for different drug protons (Fig. 3(a)) using the variational method of data analysis by minimization of a quadratic discrepancy function as described previously,25,27 and the calculated values of the self-association parameters of NOV are presented in Table 1.
| Non-cooperative model | Cooperative model | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Protons | δm (ppm) | δd (ppm) | 103K/l mol−1 | δm (ppm) | δd (ppm) | 103K/l mol−1 | σ | ΔH°/kJ mol−1 | ΔS°/J mol−1 K−1 |
| H6/H7 | 7.68 | 7.18 | 7.67 | 7.18 | |||||
| H2/H3 | 7.30 | 6.91 | 12.4 (±4.0) | 7.31 | 6.91 | 12.2 (±3.9) | 0.98 (±0.04) | −31.1 (±2.4) | −17.9 (±5.2) |
| H11 | 3.96 | 3.69 | 3.96 | 3.69 | |||||
In order to estimate the probability of formation of complexes of higher order than dimers, the experimental results have also been analysed using the indefinite cooperative model25 of molecular self-association, where the reaction constants for equilibrium (1) are assumed to be equal for all j ≥ 2 (K2 = K3 = . . . = Kj = K) and K1 = σK. This model gives the following dependence of the observed chemical shift δ on concentration (eqn. (3)),25 where x0 and x1 are the initial and monomer concentrations of the drug in solution, respectively. The cooperative model contains four unknown parameters (δm, δi, σ and K) which have been determined from the experimental concentration dependences of drug proton chemical shifts (Fig. 3(a)), as in previous work.25 The mean values of the calculated parameters using the cooperative model are also presented in Table 1.
 | (3) |
25 in the form of eqn. (4), where fm(T) and fi(T) are the equilibrium molar fractions of the drug at temperature T in the monomer (m) and in the aggregate (i), respectively. It was shown previously25 for different aromatic drugs that the values of δmj and δij in eqn. (4) do not depend on temperature in the temperature range studied and the same considerations apply to NOV in this work. Hence, the influence of temperature on the observed values of δ summarized in eqn. (4) results from changes in the mole fractions, fm and fi, which are simply related to the equilibrium association constant K at a given temperature. Analysis of the temperature dependence of the equilibrium constant enables the thermodynamic parameters ΔH and ΔS of drug self-association to be estimated. A detailed description of the method of calculation of thermodynamical parameters of drug self-association has been given previously.25 The calculated values of ΔH and ΔS for self-association of NOV in aqueous solution are presented in Table 1.| δj(T) = fm(T)·δmj + fi(T)·δij | (4) |
2 Hetero-association of novantrone with caffeine in aqueous solution
The structures and thermodynamics of complexation between NOV and CAF have been investigated by analysis of the proton chemical shifts of both drugs in mixed solutions as a function of concentration and temperature (Fig. 3). No intermolecular cross-peaks between the NOV and CAF protons were detected in 2D ROESY (or NOESY) experiments in the mixed solutions even at the highest concentrations studied (CNOV = a0 = 1 mM, CCAF = p0 = 2 mM). The absence of the cross-peaks may be due to formation of various hetero-association complexes between NOV and CAF molecules, in which there is a relatively small content of any particular complex in solution. In the titration experiments NMR measurements were made by keeping the CAF concentration constant (p0 = 2 mM) and changing the concentration of NOV in solution; this experimental procedure was adopted because the equilibrium self-association constant of NOV (Table 1) is over a thousand times greater than that of CAF (KCAF = KP = 6.9 (±0.2) mol l−1 at T = 318 K)28 at the same temperature and therefore changes of concentration of NOV affect the equilibrium distribution of the aggregates more than keeping it constant and varying the concentration of CAF in solution.Comparison of the concentration dependences of the proton chemical shifts of NOV with and without CAF in solutions (Fig. 3(a)) shows that the presence of CAF in solution shifts the proton resonances of NOV to high frequencies (low field) due to hetero-association between NOV and CAF molecules. This can be explained by the smaller shielding effect of CAF molecules on the protons of NOV in the hetero-complexes compared with that of molecules of NOV in the self-association aggregates. At relatively low concentration of NOV in solution the contribution of CAF molecules to the shielding of the antibiotic protons becomes substantial, inducing the shift to higher frequencies for the NOV protons. The statistical thermodynamical model of hetero-association of aromatic molecules23,24 has been used to analyse the experimental data. The dynamic equilibrium considered in the model includes indefinite self-association as well as indefinite hetero-association reactions of different types, as shown in eqn. (5), where A1 and P1 correspond to the monomers of NOV and CAF, and Ai, Ak, Pj, Pl are the aggregates containing i, k monomers of NOV and j, l monomers of CAF, respectively. The equilibrium constants for the self-association reactions of NOV (KA) and CAF (KP) and for the hetero-association of drug molecules (Khet) are assumed to be independent of the number of molecules in the aggregates and complexes. As the self-association constant for CAF, KP, is substantially smaller than KA for NOV (Table 1), estimates have shown23 that the hetero-complexes, AkPjAi, where Pj aggregates of CAF are flanked by the aggregates of NOV (Ak and Ai), are unlikely to form in solution, and consequently reaction 5(e) can be neglected in the present case.
 | (5) |
Taking into consideration the mass conservation law for the reactions in eqn. (5), the additive model for the proton chemical shifts and the assumption that only neighbouring molecules contribute to the chemical shift changes, the dependence on concentration of the observed proton chemical shifts of NOV in the mixed solution can be written in the form of eqn. (6)23 and the corresponding expression for CAF is given by eqn. (7).
 | (6) |
 | (7) |
The values of δmA, δdA, δcA and δmP, δdP, δcP are the proton chemical shifts of NOV/CAF in the monomer, dimer and hetero-complex, respectively. The equilibrium self-association constants KA and KP as well as δmA, δdA and δmP, δdP have been determined independently (Table 1) for the same experimental conditions. It follows that the observed concentration dependence of the proton chemical shifts of NOV and CAF in mixed solutions (Fig. 3(a)) is a function of two unknown quantities, δc and Khet, which have been determined using the computational procedure described previously.23 The magnitudes of the calculated parameters Khet and δc at T = 318 K are summarised in Table 2.
a
| NOV Protons | δc(NOV) (ppm) | δm(NOV) (ppm) | CAF Protons | δc(CAF) (ppm) | δm(CAF) (ppm) | Khet/l mol−1 | ΔH°het/kJ mol−1 | ΔS°het/J mol−1 K−1 |
|---|---|---|---|---|---|---|---|---|
| a The self-association parameters of NOV used for the calculations are presented in Table 1. Those for CAF were taken from ref. 28 (ΔH°CAF = −21.0 ± 0.4 kJ mol−1, ΔS°CAF = −50 ± 1 J K−1 mol−1; K = 6.9 ± 0.2 l mol−1 at T = 318 K). | ||||||||
| H6/H7 | 7.61 | 7.68 | H8 | 6.90 | 7.89 | |||
| H2/H3 | 7.12 | 7.30 | 7-CH3 | 3.18 | 3.95 | |||
| H11 | 3.95 | 3.96 | 3-CH3 | 2.60 | 3.54 | 256 (±30) | −9.3 (±0.8) | 15.3 (±4.0) |
| 1-CH3 | 2.46 | 3.35 | ||||||
The thermodynamical parameters ΔH°het and ΔS°het of the hetero-association of NOV with CAF were determined from measurements of the proton chemical shifts of the molecules in the mixed solution as a function of temperature (Fig. 3(b)) using the additive model for the experimental proton chemical shifts, as in previous work.23 The derived values of enthalpy and entropy of hetero-association between NOV and CAF are also presented in Table 2.
Discussion
Structural and thermodynamical properties of the self-association of novatrone in aqueous solution
The self-association constant of NOV (Table 1) is substantially higher than K values determined previously23,25,28 for other aromatic drug molecules under similar experimental conditions. It is likely that the high K value for self-association of NOV not only is connected with stacking interactions of the chromophores of antibiotics similar to other molecules containing three aromatic rings in the chromophores,23,25 but also might be due to some additional stabilization of the aggregates. Inspection of the structure of NOV (Fig. 1) indicates that intermolecular hydrogen bonds may be able to form in the molecular complexes. The equilibrium constant K for NOV self-association (Table 1) is similar in magnitude to the dimerization constant Kd of this drug determined previously by optical studies.5,15 However, the absolute values of dimerization constant for NOV5,15 and Kd determined in this work differ by about a factor of two at the same temperature under similar experimental conditions, i.e.Kd = K/2 = 16500 (±5200) l mol−1 at T = 298 K as recalculated using the thermodynamical parameters of NOV self-association (Table 1). This difference may be due, in part, to the assumption in the previous work that the dimer model is appropriate to analyse the aggregation of NOV (Kd = 30070 ± 360 l mol−1) in the μM concentration range.5 Using the expression of Delbarre et al.29 for the indefinite non-cooperative model of molecular association, estimates show that, even at 10 μM concentration of the drug, the contribution of higher order aggregates (higher than dimers) is ≈20% due to the high self-association constant of NOV, thus leading to relatively large errors in the determination of self-association constants using the dimer model. The same conclusion can be drawn for the K-values derived by use of a dimer + trimer model for emission studies in the 10−4–10−5 M concentration range i.e.K2 = 27000 l mol−1 and K3 = 70000 l mol−1, in which the authors quoted a 20–30% error limit.15 Limitation of the number of molecules in the stack leads to magnitudes of association constants being greater than for the indefinite association model because, if some reactions are not taken into account, the effective fraction of interacting molecules used in the concentration equations is lower, giving rise to a different value of the self-association constant, e.g. the estimated amount of stacks higher than dimers/trimers for NOV self-association is ca. 90% at a concentration of 10−4 M.Limiting the self-association of NOV to dimers and trimers15 also leads to the surprising result that K3 is much greater than K2. The validity of this approximation was checked by calculation of the self-association of NOV using the cooperative model of indefinite association (Table 1). The cooperativity parameter, σ, close to 1, indicates that the self-association of NOV is non-cooperative, i.e. the dimerisation constant equals the equilibrium constants of formation of higher order associates (K3, K4, . . ., Kj). A similar situation was observed for the phenanthridine drugs, ethidium bromide (EB) and propidium iodide (PI), whose chromophores do not contain bulky side groups or chains,23,25 whereas self-association is evidently non-cooperative (σ > 1)25,28 for the antibiotics daunomycin, nogalamycin and actinomycin D, which have large substituents attached to the chromophores. The value of σca. 0.98 (±0.04) shows that the previous approximation15 of only considering dimers and trimers is incorrect and the reason that K3 was found to be greater than K2 is that the contribution of higher order aggregates was neglected. It is also worth noting that NOV was found to exhibit a high tendency to self-aggregation within intracellular media,3 which can be explained by the large value of the self-association constant and consequently high aggregation affinity of this drug.
The dimer structure of NOV in aqueous solution has been determined using the calculated values of the induced proton chemical shifts of the antibiotic (Δδ = δm − δd, Table 1) and the results of 2D ROESY experiments (Fig. 2) similar to previous work for other aromatic drugs.23,25 The mutual orientation of the chromophores of NOV in the dimer was calculated by comparison of Δδ and their theoretical values from quantum-mechanical calculations of iso-shielding curves for aromatic molecules.30 To be consistent with the 2D ROESY data (Fig. 2) only the anti-parallel orientation of the planes of the NOV chromophores was considered. The structure of the monomer form (chromophore and side chains) of the NOV molecule was determined theoretically by energy minimization of the interactions between structural groups of the antibiotic using the MM2 force field31 and additional parametrization introduced by Dudek and Pounder.32 In the calculated most favourable structure of the NOV dimer (Fig. 4) the planes of the chromophores of NOV are parallel to each other and situated 0.34 nm apart; there is significant overlap of the aromatic rings of the chromophore and the amino alkyl side chains tend to be well separated. The structure shows that H-bonds may be formed between the side chain secondary amino groups (NH5/NH8) and hydroxy groups OH1/OH4 of the antibiotic chromophore which are situated sufficiently close to each other in space (Fig. 4). The intermolecular hydrogen bonding also looks as though it could be stabilized by a hydrogen-bond network between OH1, 9-CO and 8-NH of one molecule and the same groups on the adjacent molecule in the stack; by symmetry consideration the same situation would also occur for the OH4, 10-CO and 5-NH groups. Indirect confirmation of the assumption of intermolecular hydrogen bonding is shown by optical investigations of the self-association of the pharmacologically less active derivative of NOV, ametantrone.5 The self-association of ametantrone, whose structure differs by the substitution of H-atoms in positions 1 and 4 instead of hydroxy groups in NOV, is characterized by an approximately five times smaller dimerization constant compared to NOV under the same experimental conditions.5 It should be noted that in the calculations of the dimer structure of NOV, only the values of induced chemical shifts Δδ for the three non-exchangeable protons can be used, thus allowing some freedom in the shift of the planes of the molecules perpendicular to the longitudinal axis of the chromophore of the antibiotic. However, such displacements of the chromophores of the molecules would lead to a decrease in the overlap of the aromatic ring systems of NOV and a decrease in the stacking interactions and, consequently, to a less stable dimer complex.
 | ||
| Fig. 4 The calculated NMR structure of the self-associated dimer complex of NOV: (a) side view of the dimer complex; (b) view, looking perpendicular to the planes of the chromophores of the antibiotic molecules. | ||
A geometric arrangement of the NOV dimer in solution has been also proposed using the exciton model.15 Although “head to tail” orientation of the NOV molecules in the dimer15 resembles the calculated structure in Fig. 4, the separation between the planes of the chromophores in the proposed dimer structure is much higher (≈0.62 nm) than that obtained in this work. Our experimental NMR results show that the dimer structure presented by Lee and Dutta15 is unlikely to be correct, because at such a large separation between the chromophores, the magnetic shielding of the drug protons in the aggregates, as well as changes of proton chemical shifts with concentration and temperature, should be negligible.30 The ring current magnetic field which gives the main contribution to the nuclear shielding in aromatic molecules30 decreases rapidly with distance (≈1/r3) and becomes insignificant at r ![[greater than or equal, slant]](https://www.rsc.org/images/entities/char_2a7e.gif) 0.5 nm, whereas the experimental dependences of proton chemical shifts of NOV as a function of both concentration and temperature (Fig. 3) show pronounced changes of chemical shifts due to stacking interactions of the aromatic molecules of antibiotic in aqueous solution.
0.5 nm, whereas the experimental dependences of proton chemical shifts of NOV as a function of both concentration and temperature (Fig. 3) show pronounced changes of chemical shifts due to stacking interactions of the aromatic molecules of antibiotic in aqueous solution.
The change in enthalpy of aggregation of NOV exceeds, in absolute value, ΔH for self-association of the phenanthridine drugs EB and PI23,25 and is comparable with those obtained for the anthracycline antibiotics, DAU and NOG.33 It is likely that the relatively high negative value of enthalpy of self-association of NOV is determined by dispersive interactions between aromatic chromophores and the possible formation of intermolecular H-bonds in the antibiotic aggregate. Dispersive interactions are characterized by both negative enthalpy and negative entropy,34 and the enthalpy of hydrogen-bond formation in aqueous solution is estimated to be between −8 and −13 kJ mol−1.34,35
Hetero-association of novatrone with caffeine in aqueous solution
It is seen from Table 2 that the magnitude of the hetero-association constant for complexation of NOV with CAF is intermediate between the values of self-association constants of the interacting aromatic drug molecules, as found for all the systems studied previously,28i.e. for hetero-association of CAF with different aromatic dyes and drugs. At the same time the equilibrium constant of hetero-association between NOV and CAF is somewhat higher than Khet for other drug–CAF systems.28 The largest value of hetero-association constant among all the systems studied previously28 was observed for the complexation between acridine orange and caffeine, Khet = 158 l mol−1 at T = 318 K, which was attributed, in the main, to hydrophobic interactions between methyl groups of the dye chromophores and caffeine molecules. For hetero-association of NOV and CAF it is possible that stabilization of the hetero-complex is also caused by hydrophobic interactions between the methyl groups of caffeine and the alkyl side chains of the antibiotic. It is feasible that some contribution to the stabilizing effect of the 1∶1 NOV–CAF complex may also be due to hydrogen-bond formation between the molecules. The thermodynamical parameters determined for the NOV–CAF molecular system (Table 2) are consistent with these conclusions. The absolute value of ΔH°het (Table 2) was found to be substantially smaller than the enthalpies of self-association reactions for both NOV and CAF under the same experimental conditions (Tables 1, 2). At the same time, the entropy of hetero-association of the molecules has a small positive value. Positive entropy contributions are mainly determined by hydrophobic interactions.36 Thus, it is likely that the hydrophobic effect plays a significant role in the hetero-association between NOV and CAF molecules, giving a positive contribution to ΔS°het and probably resulting from interactions of methyl groups of CAF and alkyl side chains of the anthraquinone chromophore of NOV.The relative content of each molecular complex in solution has been calculated as a function of r (= a0/p0, the ratio of the initial concentrations of NOV and CAF in the mixed solution) and temperature using the calculated values of reaction constants (Tables 1, 2) determined in this work. It can be seen in Fig. 5(a) that, with increasing r, there is an increase in content of all associated forms of NOV and a concomitant decrease of the monomer and self-associated forms of CAF. Over 65% of the CAF (P) molecules exist in solution in the monomer form, whilst the self-association (Ai) and hetero-association (AiPj) complexes of NOV are predominant in the molecular equilibrium compared with any other associated forms of the molecules at NOV concentrations higher than a0 = 0.12 mM; this behaviour is due to the relatively small self-association constant of CAF compared to the relatively high self-association constant of NOV (Table 1). The temperature dependence of the relative content of different complexes (Fig. 5(b)) shows that dissociation of both the self- and hetero-association complexes in the mixed solution occurs at higher temperatures, giving a concomitant increase in the monomer concentrations of NOV (A1) and CAF (P1).
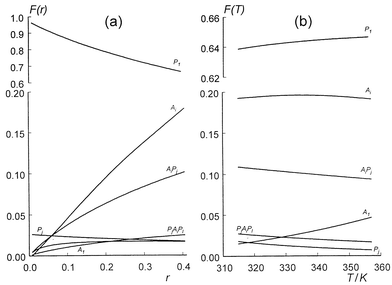 | ||
| Fig. 5 Calculated relative content F (the ratio of the concentration of a given type of molecular component to the total concentration of all types of aggregates in solution) of self- and hetero-associated complexes of NOV (A) and CAF (P) as a function of: a) r = a0/p0, the ratio of concentrations of NOV and CAF; b) temperature at a0 = 0.9 mM, p0 = 2.0 mM.
| ||
Structure of the 1∶1 NOV–CAF hetero-complex in aqueous solution
The values of the limiting proton chemical shifts, δcA for NOV and δcP for CAF protons in Table 2, have been used to calculate the most favourable structures of the 1∶1 hetero-association complex of NOV + CAF in aqueous solution in the same way as used for the dimer of NOV, i.e. by comparison of induced proton chemical shifts Δδ with those values calculated from quantum-mechanical iso-shielding curves for aromatic molecules.30 One of the most favourable calculated structures of the 1∶1 complex of NOV with CAF in aqueous solution is presented in different spatial projections in Fig. 6. As the NOV molecule has a plane of symmetry there is an equivalent structure with CAF rotated 180° in the plane. In both structures the planes of the chromophores of NOV and CAF molecules in the 1∶1 hetero-complex are parallel to each other at a distance of about 0.34 nm. Intensive overlap of the aromatic parts of the chromophores is found for the CAF–NOV hetero-complex, which indicates substantial stacking interactions of the drug molecules. The calculated structure (Fig. 6) shows that two of the methyl groups of CAF (in positions 1 and 3) are situated close in space to the alkyl side chains of NOV, thus indicating possible hydrophobic contacts between them. Moreover, the calculated structure could also be stabilized by hydrogen bonds between CAF oxygens (in positions 2 and 6) and the NH groups of the side chains of NOV. It should be noted that, due to the symmetry of the iso-shielding curves for the NOV molecule and the approximate symmetrical shape of the iso-shielding curves for CAF, there may be two possible orientations of the CAF chromophore in the 1∶1 hetero-complex, i.e. when the plane of the CAF chromophore is rotated by 180° with respect to the transverse axis. This structure of the NOV–CAF complex is consistent with that obtained using molecular modelling calculations for the same system,21 from which it was also concluded that “rotation of the caffeine by 180° relative to the DNA intercalator ring system does not significantly alter the relative binding energy”.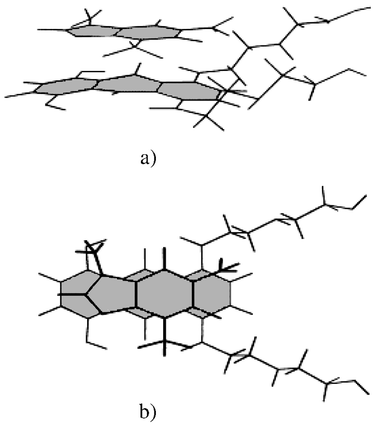 | ||
| Fig. 6 A structure of the 1∶1 CAF–NOV hetero-association complex calculated from NMR limiting chemical shifts: (a) side view of the hetero-complex; (b) view looking perpendicular to the planes of the chromophores of aromatic molecules. An equally probable structure is found by 180° in-plane rotation of the CAF molecule. | ||
Taken together, the NMR results of the structural and thermodynamical analysis of the complexation of caffeine with the DNA-intercalating drug, novatrone, have shown that CAF forms stacked complexes with the antibiotic in aqueous solution. Stabilization of the 1∶1 NOV–CAF hetero-complex in aqueous solution results from van der Waals dispersive forces, giving a substantial overlap of the aromatic ring systems of the chromophores, hydrophobic interactions and the possibility of intermolecular hydrogen bonding.
Experimental
Materials
Novatrone (mitoxantrone) and caffeine were purchased from the Sigma Chemical Company and used without further purification. The samples were lyophilized from D2O and re-dissolved in 0.1 M phosphate buffer in 99.95% D2O, pD = 7.1, containing 10−4 M EDTA. The concentrations of the stock solutions of the aromatic molecules were measured spectrophotometrically on appropriate dilution using the following molar absorption coefficients: ε = 8360 l mol−1 cm−1 (λ = 682 nm) for NOV4,5 and ε = 9740 l mol−1 cm−1 (λ = 273 nm) for CAF.37NMR experiments
500 MHz 1H-NMR spectra were recorded on a Bruker DRX spectrometer with the residual water peak saturated during relaxation. 2D ROESY experiments were measured at 318 K, with a mixing time of τm = 240 ms, using standard pulse sequences described previously.38 NOV showed limited solubility in aqueous buffered solution and, therefore, chemical shift measurements of the non-exchangeable protons of the aromatic molecules were made as a function of concentration of NOV in the appropriate concentration range (from 1.01 to 0.026 mM) and at a relatively high temperature T = 318 K. The measurements as a function of temperature were made at constant drug concentrations in the temperature range 312–357 K. The sample temperature was regulated using a Bruker BVT-3000 unit. Chemical shifts were measured relative to an internal reference TMA (tetramethylammonium bromide) and recalculated with respect to DSS (sodium 2,2-dimethyl-2-silapentane-5-sulfonate), i.e.δDSS = δTMA + 3.178 (ppm). All NMR measurements were made in the fast-exchange condition on the NMR time-scale.Calculation of association parameters
The calculated fits of the models of the equilibrium reactions to the experimental chemical shift data were assessed by the magnitude of the discrepancy function Δ, where n is the number of experimental points; δei and δi are the experimental drug proton chemical shifts for the i-th concentration or temperature and the calculated values using the theoretical models.27 Minimization of Δ was performed using initial approximations produced by the law of accidental numbers over a wide range of variations of the parameters. A large statistical set of data enabled the whole field of possible values of minimization parameters to be investigated over a wide range of their variations, in order to determine the global, and not a local, minimum. The values of Δ obtained in the calculations are in the range 10−5–10−6 for at least 14 experimental points in each data set, which corresponds to an average deviation between observed and modelled chemical shifts in the range 0.002–0.0003 ppm, assuming an error of 0.001 ppm in the measurements of the chemical shifts; these results indicate a very good fit to the experimental data. Magnitudes of the equilibrium constants were calculated for each proton independently and the average value and mean deviation determined.
Determination of the molecular structures
The structures of the NOV dimer and 1∶1 NOV–CAF hetero-association complexes have been determined by analysis of the calculated values of induced proton chemical shifts in the dimer, Δδ = (δm − δd), or in the hetero-complex, Δδ = (δm − δc), where δm, δd and δc are chemical shifts of the drug in the monomer, dimer and 1∶1 hetero-complex forms, respectively. The mutual orientations of the molecules in the dimers or 1∶1 complexes are determined by comparison of the induced proton chemical shifts and their theoretical values, as described in previous work.25 A detailed description of calculations of iso-shielding curves for aromatic molecules is given by Giessner-Prettre and Pullman.30 The discrepancy between experimental and theoretical values of induced proton chemical shifts of the molecules in the complexes did not exceed 5%. The spatial representation of structures was obtained with the help of the “Mathematica 2.2” software (Wolfram Res. Inc.).Acknowledgements
We thank the CVCP for an ORS Award (D. A. V.) and the University of London Intercollegiate Research Service for access to the 500 MHz Biomedical NMR facility (at Birkbeck College) and the Optical Spectroscopy Service (at King’s College). This work was supported, in part, by INTAS (Grant No. INTAS-97 31753).References
- F. E. Cotter, Br. J. Clin. Pract., 1988, 42, 207 Search PubMed; G. Ehninger, U. Schuler, B. Proksch, K.-P. Zeller and J. Blanz, Clin. Pharmacokinet., 1990, 18, 365 Search PubMed; Z. Arlin, D. C. Case, J. Moore, P. Wiernik, E. Feldman, S. Saletan, P. Desai, L. Sia and K. Cartwright, Leukemia, 1990, 4, 177 CAS; D. Faulds, J. A. Balfour, P. Chrisp and H. D. Langtry, Drugs, 1991, 41, 400 Search PubMed.
- B. Drewinko, L.-Y. Yang, B. Barlogie and J. M. Trujillo, Cancer Res., 1983, 43, 2648 Search PubMed; I. E. Smith, Cancer Treat. Rev., 1983, 10, 103 CrossRef CAS.
- A. Feofanov, S. Sharonov, I. Kudelina, F. Fleury and I. Nabiev, Biophys. J., 1997, 73, 3317 Search PubMed; A. Feofanov, S. Sharonov, F. Fleury, I. Kudelina and I. Nabiev, Biophys. J., 1997, 73, 3328 Search PubMed.
- J. Kapuscinsky, Z. Darzynkiewicz, F. Traganos and M. R. Melamed, Biochem. Pharmacol., 1981, 30, 231 CrossRef.
- J. Kapuscinsky and Z. Darzynkiewicz, Biochem. Pharmacol., 1985, 34, 4203 CrossRef.
- J. Kapuscinsky and Z. Darzynkiewicz, Proc. Natl. Acad. Sci. USA, 1986, 83, 6302.
- P. J. Smith, S. A. Morgan, M. E. Fox and J. V. Watson, Biochem. Pharmacol., 1990, 40, 2069 CrossRef CAS.
- R. A. Roberts, A. E. Cress and W. S. Dalton, Biochem. Pharmacol., 1989, 38, 4283 CrossRef CAS; C. K. Ho, S. L. Law, H. Chiang, M. L. Hsu, C. C. Wang and S. Y. Wang, Biochem. Biophys. Res. Commun., 1991, 180, 118 CAS.
- K. Mewes, J. Blanz, G. Ehninger, R. Gebhard and K.-P. Zeller, Cancer Res., 1993, 53, 5135 Search PubMed.
- C. Bailly, S. Routier, J.-L. Bernier and M. J. Waring, FEBS Lett., 1996, 379, 269 CrossRef CAS.
- D. B. Davies and A. N. Veselkov, J. Chem. Soc., Faraday Trans., 1996, 92, 3545 RSC.
- D. B. Davies, L. Karawajew and A. N. Veselkov, Biopolymers, 1996, 38, 745 CrossRef CAS.
- D. B. Davies, V. I. Pahomov and A. N. Veselkov, Nucleic Acids Res., 1997, 25, 4523 CrossRef CAS.
- N. Birliakis and B. Perly, Carbohydr. Res., 1992, 235, C1 CrossRef.
- B. S. Lee and P. K. Dutta, J. Phys. Chem., 1989, 93, 5665 CrossRef CAS.
- L.-S. Kan, P. N. Borer, D. M. Cheng and P. O. P. Ts’o, Biopolymers, 1980, 19, 1641 CrossRef CAS; C. P. Selby and A. Sancar, Proc. Natl. Acad. Sci. USA, 1990, 87, 3522 CAS.
- H. Kimura and T. Aoyama, J. Pharmacobio-Dyn., 1989, 12, 589 Search PubMed.
- W. E. Ross, L. A. Zwelling and K. W. Kohn, Int. J. Radiat. Oncol. Biol. Phys., 1979, 5, 1221 CAS; R. Ganapathi, D. Grabowski, H. Schmidt, A. Yen and G. Iliakis, Cancer Res., 1979, 46, 5553 Search PubMed; G. M. Iliakis, M. Nusse, R. Ganapathi, J. Egner and A. Yen, Int. J. Radiat. Oncol. Biol. Phys., 1986, 12, 1987 CAS.
- F. Traganos, J. Kapuscinsky and Z. Darzynkiewicz, Cancer Res., 1991, 51, 3682 Search PubMed; F. Traganos, B. Kaminska-Eddy and Z. Darzynkiewicz, Cell Proliferation, 1991, 24, 305 Search PubMed.
- J. Kapuscinsky and M. Kimmel, Biophys. Chem., 1993, 46, 153 Search PubMed.
- R. W. Larsen, R. Jasuja, R. Hetzler, P. T. Muraoka, V. G. Andrada and D. M. Jameson, Biophys. J., 1996, 70, 443 Search PubMed.
- K. Weller, H. Shutz and I. Petri, Biophys. Chem., 1984, 19, 289 Search PubMed; F. Aradi and A. Foldesi, Magn. Reson. Chem., 1985, 23, 375 CAS; F. Aradi and A. Foldesi, Magn. Reson. Chem., 1989, 27, 249 CAS; J.-S. Chen and J.-Ch. Shiao, J. Chem. Soc., Faraday Trans., 1994, 90, 429 RSC; N. J. Baxter, M. P. Williamson, T. H. Lilley and E. Haslam, J. Chem. Soc., Faraday Trans., 1996, 92, 231 RSC; M. Zdunek, J. Piosik and J. Kapuscinski, Biophys. Chem., 2000, 84, 77 Search PubMed.
- D. B. Davies, D. A. Veselkov and A. N. Veselkov, Mol. Phys., 1999, 97, 439 CrossRef CAS.
- D. B. Davies, D. A. Veselkov, V. K. Kodintsev, M. P. Evstigneev and A. N. Veselkov, Mol. Phys., 2000, 98, 1961 CrossRef CAS.
- D. B. Davies, L. N. Djimant and A. N. Veselkov, J. Chem. Soc., Faraday Trans., 1996, 92, 383 RSC.
- R. B. Martin, Chem. Rev., 1996, 96, 3043 CrossRef CAS.
- A. N. Veselkov, L. N. Djimant, L. Karawajew and E. L. Kulikov, Stud. Biophys., 1985, 106, 171 Search PubMed.
- D. B. Davies, D. A. Veselkov, L. N. Djimant and A. N. Veselkov, Eur. Biophys. J., submitted. Search PubMed.
- A. Delbarre, B. P. Roques, J. B. LePecq, J. Y. Lallemand and Nquyen-Dat-Xuong, Biophys. Chem., 1976, 4, 275 Search PubMed.
- C. Giessner-Prettre and B. Pullman, Q. Rev. Biophys., 1987, 20, 113 CAS.
- U. Burkert and N. Allinger, Molecular mechanics, ACS, Washington, DC, 1982, p. 339. Search PubMed.
- M. J. Dudek and J. W. Pounder, J. Comput. Chem., 1995, 16, 791 CrossRef CAS.
- D. B. Davies, R. J. Eaton and A. N. Veselkov, to be submitted. .
- P. D. Ross and S. Subramanian, Biochemistry, 1981, 20, 3096 CrossRef.
- M. T. Record Jr., C. F. Andersen and T. M. Lohman, Q. Rev. Biophys., 1978, 11, 103 CrossRef CAS.
- H. P. Hopkins, J. Fumero and W. D. Wilson, Biopolymers, 1990, 29, 445 CrossRef.
- T. H. Lilley, H. Linsdell and A. Maestre, J. Chem. Soc., Faraday Trans., 1992, 88, 2865 RSC.
- A. Bax and D. G. Davis, J. Magn. Reson., 1985, 63, 207 CAS.
| This journal is © The Royal Society of Chemistry 2001 |