Comparative spectroscopic and theoretical studies on the conformation of some α-diethoxyphosphoryl carbonyl compounds and their α-ethylsulfonyl analogues![[hair space]](https://www.rsc.org/images/entities/h2_char_200a.gif) 1
1
Paulo R.
Olivato
*a,
Rubens Ruiz
Filho
a,
Julio
Zukerman-Schpector
b,
Maurizio
Dal Colle
c and
Giuseppe
Distefano
*c
aInstituto de Química, Universidade de São Paulo, C.P. 26077 - CEP 05513-970, São Paulo, SP Brazil. Fax: +55 11 3815 5579; E-mail: prolivat@iq.usp.br
bDepartamento de Química, Universidade Federal de São Carlos, Brazil
cDipartimento di Chimica, Università di Ferrara, Via Borsari, 46, 44100, Ferrara, Italy
First published on 11th December 2000
Abstract
Comparative νCO IR analysis of β-carbonylphosphonates [XC(O)CH2P(O)(OR)2: X = Me 1, Ph 2, OEt 3, NEt24 and SEt 5; R = Et] (series I) and β-carbonylsulfones [XC(O)CH2SO2R: X = Me 6, Ph 7, OEt 8, NEt29 and SEt 10; R = Et] (series II) along with ab initio 6-31G** calculations on 1a and 6a (R = Me) suggest the existence of only a single gauche conformer for series I. The negative carbonyl frequency shifts for both series follow approximately the electron-affinities of the π*CO orbital of the parent compounds MeC(O)X 11–15. The less positive asymmetric sulfonyl frequency shifts (ΔνSO2) for II in relation to the phosphoryl frequency shifts (ΔνPO) for I and the larger negative carbonyl frequency shifts for II with respect to the corresponding values for I are in line with the upfield 13C NMR chemical shifts of the carbonyl carbon for II compared to I. These trends agree with the shorter O(SO2)![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) ⋯C(CO) contact in comparison with the O(PO)⋯C(CO) one and are discussed in terms of Olp→π*CO charge transfer and electrostatic interactions, which are stronger for series II than for I, indicating that the sulfonyl oxygen atom is a better electron donor than the phosphoryl oxygen atom. Intrinsic geometrical parameters of O
⋯C(CO) contact in comparison with the O(PO)⋯C(CO) one and are discussed in terms of Olp→π*CO charge transfer and electrostatic interactions, which are stronger for series II than for I, indicating that the sulfonyl oxygen atom is a better electron donor than the phosphoryl oxygen atom. Intrinsic geometrical parameters of O![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) S–CH2 and OP–CH2 moieties seem to be responsible for this behaviour as indicated by X-ray study and ab initio calculations of dialkyl (methylsulfonyl)methylphosphonate MeSO2CH2P(O)(OR)2 (R = Et 18, Me 18a).
S–CH2 and OP–CH2 moieties seem to be responsible for this behaviour as indicated by X-ray study and ab initio calculations of dialkyl (methylsulfonyl)methylphosphonate MeSO2CH2P(O)(OR)2 (R = Et 18, Me 18a).
Our previous spectroscopic (IR, 13C NMR, UV and UPS), X-ray diffraction and theoretical studies of some β-carbonyl-sulfides and -sulfones XC(O)CH2SOnR
2–13 (X = Me, Ar, NR2, OR and SR; R = Me, Ar; n = 0 and 2) indicated that these compounds in the gas phase, in solution and in the solid state (for the β-carbonylsulfones) prefer a gauche conformation between the CO and CH2–S bonds. For the corresponding β-carbonylsulfoxides XC(O)CH2S(O)R2,5,9,13–15 the gauche rotamer is also the more stable, but the cis one prevails for some X or R substituents due to electronic16,17 or steric17 factors.
In general, the stability of the gauche rotamers of β-carbonyl-sulfides, -sulfoxides and -sulfones has been ascribed to π*CO/σC–S and πCO/σ*C–S orbital interactions. However, in the case of β-carbonyl-sulfones10,11 and -sulfoxides,14,15 additional stabilisation of the gauche (or cis) rotamer derives from crossed electrostatic and charge transfer interactions between oppositely charged atoms i.e. O(SO2)→C(CO) and (or) O(CO)→S(SOn) (n = 1 and 2).
The relevant electronic properties of closely related molecules differing only in the nature of their third row element, such as P or S, are quite similar. In fact, the ionisation energies of the outermost MO (oxygen lone pair) for dimethyl sulfone Me2SO2 (10.65 eV)18–20 and dimethyl methylphosphonate (MeO)2P(O)Me (10.71 eV)21 are almost identical. Similarly, the field-inductive parameters for the ethylsulfonyl EtSO2– and diethoxyphosphoryl (EtO)2P(O)– groups are equal (F ≡ 0.55).22 In addition, the attachment energy (i.e. the negative of the electron affinity) values for electron capture into the σ*C–S and σ*C–P orbitals of Me2S (3.25 eV)8 and Me3P (3.10)23,24 are similar, and the σC–S and σC–P ionisation energies (12.78 and 11.325 eV, respectively) are not very different. Therefore, it was interesting to study the α-diethoxyphosphoryl carbonyl compounds (EtO)2P(O)CH2C(O)X (X = Me 1, Ph 2, OEt 3, NEt24 and SEt 5) by means of IR and 13C NMR spectroscopies and ab initio calculations in order to compare these data with those previously reported for the corresponding α-ethylsulfonyl carbonyl compounds2,9,11 EtSO2CH2C(O)X (X = Me 6, Ph 7, OEt 8, NEt29 and SEt 10). This paper also reports the X-ray diffraction data and the results of ab initio calculations on dialkyl (methylsulfonyl)methylphosphonates (EtO)2P(O)CH2SO2R (R = Et 18, Me 18a), necessary to obtain the experimental geometric parameters of the (EtO)2P(O)CH2– group which cannot be easily obtained from the liquid compounds 1–5. Moreover, compound 18 allows a comparison of the relative electron-donor abilities of the sulfonyl and phosphoryl oxygen atoms.
Experimental
Materials
All solvents for IR measurements were spectrograde and were used without further purification. The already known α-diethoxyphosphoryl carbonyl compounds (EtO)2P(O)CH2C(O)X (X = Me 1,26 Ph 2,27 OEt 3,28 NEt2428,29 and SEt 530) were prepared by an adaptation of the method described for 5, i.e., by reacting equimolar quantities of triethyl phosphite with the corresponding α-bromocarbonyl compound in benzene at room temperature, followed by reflux for 8 hours. The α-diethoxyphosphoryl carbonyl compound was chromatographically separated from isomeric diethyl 1-substituted-vinyl phosphate H2CC[X][OP(O)(OEt)2] through a silica gel column using hexane–acetone as eluent. After solvent evaporation, the liquid α-diethoxyphosphoryl carbonyl compounds 1–5 were obtained in a pure form by distillation under reduced pressure. Diethyl (methylsulfonyl)methylphosphonate 18 (mp 92–93 °C, colourless crystals from chloroform) was synthesised following a literature procedure.31
IR measurements
The IR spectra were obtained using an FT-IR Nicolet Magna 550 Spectrometer with 1.0 cm−1 resolution. For the carbonyl phosphonates 1–5 the carbonyl stretching region (1800–1600 cm−1) was recorded in 2.0 × 10−2 mol dm−3 carbon tetrachloride, chloroform and acetonitrile solution, using a 0.519 mm sodium chloride cell. The phosphoryl (PO) stretching region (1300–1100 cm−1) was analysed in carbon tetrachloride solution. The existence of a single symmetric carbonyl stretching band for the series 1–5, in all solvents, was confirmed by Fourier Self Deconvolution (FSD) and second derivative analysis performed on each carbonyl band using the OMNIC Version 1.0 FT-IR software of the Nicolet instrument.
NMR measurements
1H and 13C NMR spectra of 0.5 mol dm−3 solutions in chloroform with TMS as an internal standard were recorded at 200 and 50 MHz, respectively, using a Bruker AC-200 spectrometer in the FT mode. The conditions for recording 1H and 13C NMR spectra have been described elsewhere.12Calculations
The most stable conformation, the geometric parameters and the electron charge distribution at the various atoms of α-dimethoxyphosphorylacetone 1a, dimethyl (methylsulfonylmethyl)phosphonate 18a, α-methylsulfonylacetone 6a, dimethyl sulfone 19 and dimethyl methylphosphonate 20 were computed at the HF/6-31G** level using the Gaussian 98 series of programs.32 To save computer time, the ethyl groups were replaced by methyl groups. C2v and Cs symmetry was used for 19 and 20, respectively, while several (gauche and cis) starting geometries were used for the other compounds.
Crystal data
C6H15O5PS, M = 230.21, monoclinic, space group P21/a, a = 10.074(1), b = 8.3570(9), c = 13.991(2) Å, β = 108.46(1)°, V = 1117.3(2) Å3, Z = 4, Dx = 1.369 Mg m−3, λ(Mo-Kα) = 0.71073 Å, μ = 0.423 mm−1, R = 0.0453.†Data collection and processing
X-Ray diffraction data were collected on a CAD4 Mach3 diffractometer with the θ/2θ scan technique at 293 K; solution by direct methods (SIR92),33 full-matrix least-squares refinement on F2. 2040 measured reflections (2θmax = 50°) yielded 1958 unique and 1463 with Fo2 ![[greater than or equal, slant]](https://www.rsc.org/images/entities/char_2a7e.gif) 4σFo2. Anisotropic displacement parameters for all non-H atoms were applied. H atoms were located on stereochemical grounds and refined with fixed geometry, each riding on a carrier atom, with an isotropic displacement parameter amounting to 1.5 (for methyl H atoms) or 1.2 (for the other H atoms) times the value of the equivalent isotropic displacement parameter of the atom to which they are attached. 121 parameters were refined and the final conventional R was 0.0453. Structure refinement, final geometrical calculations were carried out with SHELXL97,34 PARST-9535 and WinGX.36Fig. 1 was produced using ZORTEP.37
4σFo2. Anisotropic displacement parameters for all non-H atoms were applied. H atoms were located on stereochemical grounds and refined with fixed geometry, each riding on a carrier atom, with an isotropic displacement parameter amounting to 1.5 (for methyl H atoms) or 1.2 (for the other H atoms) times the value of the equivalent isotropic displacement parameter of the atom to which they are attached. 121 parameters were refined and the final conventional R was 0.0453. Structure refinement, final geometrical calculations were carried out with SHELXL97,34 PARST-9535 and WinGX.36Fig. 1 was produced using ZORTEP.37
 | ||
| Fig. 1 ZORTEP view of compound 18 showing the thermal ellipsoid at 50% probability and the heavy atom labelling. | ||
Results and discussion
Table 1 collects the carbonyl stretching frequencies for the α-diethoxyphosphoryl carbonyl compounds 1–5, in carbon tetrachloride, chloroform and acetonitrile. This table also includes the carbonyl stretching frequencies of the predominant gauche conformer of the α-ethylsulfonyl carbonyl compounds2,5,96–10 and the corresponding data for the parent carbonyl compounds911–15. Only a symmetrical carbonyl band is observed for the whole series 1–5 in all solvents. The existence of a good linear correlation between the carbonyl frequencies of 6–10 and 1–5 in carbon tetrachloride [r = 0.990; s = 5.8 cm−1] suggests that the latter compounds also exist in solution in the gauche conformation.38
a in the IR spectra of α-diethoxyphosphoryl carbonyl compounds (EtO)2P(O)CH2C(O)X 1–5, for the gauche conformerb of the α-ethylsulfonyl carbonyl compounds EtSO2CH2C(O)–X 6–10 and of the parent carbonyl compoundsc CH3C(O)–X 11–15
| ν/cm−1 | ||||
|---|---|---|---|---|
| Compound | X | CCl4 | CHCl3 | CH3CN |
| a Each carbonyl frequency corresponds to the maximum of a single symmetrical band (see Experimental section). b The gauche conformer is the more abundant one (conc. >80%). c From ref. 9. d From refs. 2,5 and 9, respectively. e From refs. 2,5 and 9, respectively. f From refs. 2,5 and 9, respectively. | ||||
| 1 | Me | 1719.3 | 1714.7 | 1716.1 |
| 6d |
1720.5 | 1720.0 | 1723.0 | |
| 11 | 1718.5 | 1711.5 | 1714.5 | |
| 2 | Ph | 1685.0 | 1681.9 | 1683.4 |
| 7e |
1680.0 | 1679.0 | 1682.0 | |
| 12 | 1691.0 | 1683.0 | 1693.0 | |
| 3 | OEt | 1740.8 | 1735.0 | 1736.6 |
| 8f |
1738.0 | 1739.0 | 1743.0 | |
| 13 | 1742.0 | 1732.5 | 1736.6 | |
| 4 | NEt2 | 1646.5 | 1636.8 | 1637.4 |
| 9f |
1650.0 | 1644.0 | 1645.0 | |
| 14 | 1650.0 | 1640.0 | 1644.0 | |
| 5 | SEt | 1687.2 | 1680.6 | 1683.7 |
|
10f |
1678.0 | 1677.5 | 1681.0 | |
| 15 | 1695.0 | 1687.0 | 1690.0 | |
Table 2 lists the carbonyl frequency shifts (ΔνCO/cm−1) for 1–5 and 6–10 in relation to the parent compounds 11–15 together with the attachment energy value for the latter compounds. The ΔνCO values for both series are negative, or slightly positive for the methyl derivatives 1 and 6. The two series are reasonably well correlated (r = 0.912) and follow approximately the electron affinity trend of the parent carbonyl compounds8,39 (except in the case of the methyl derivatives 1 and 6). These data suggest that the geometry of the α-phosphoryl carbonyl compounds (structure I) is similar to that of the gauche conformer of the α-sulfonyl carbonyl compounds (structure II), whose geometry was previously determined by theoretical calculations and X-ray diffraction analysis. The trends of Table 2 are in line with previous propositions2,9,12,13 on β-keto sulfones and suggest that the O(PO)→π*CO and O(SO2)→π*CO charge transfer and π*CO/σC–Het hyperconjugative2,40 interactions are the main factors which stabilise the gauche conformation (structures I and II).
 a for the α-diethoxyphosphoryl (EtO)2P(O)CH2C(O)–X 1–5 and for the α-ethylsulfonyl EtSO2CH2C(O)–X 6–10 carbonyl compounds, in CCl4, and the attachment energy values (EA)b for the π*C(O)X orbital of the parent carbonyl compounds CH3C(O)–X 11–15
a for the α-diethoxyphosphoryl (EtO)2P(O)CH2C(O)–X 1–5 and for the α-ethylsulfonyl EtSO2CH2C(O)–X 6–10 carbonyl compounds, in CCl4, and the attachment energy values (EA)b for the π*C(O)X orbital of the parent carbonyl compounds CH3C(O)–X 11–15
| Compound | X | ΔνCO/cm−1 | Compound | ΔνCO/cm−1 | E A/eV |
|---|---|---|---|---|---|
| a ΔνCO refers to the difference: νsubstituted carbonyl compound − νparent compound. b From ref. 8. c The value for acetophenone is not detectable by ETS, from ref. 39. | |||||
| 1–11 | Me | +0.8 | 6–11 | +2.0 | 1.26 |
| 2–12 | Ph | −6.0 | 7–12 | −11.0 | <0c |
| 3–13 | OEt | −1.2 | 8–13 | −4.0 | 2.09 |
| 4–14 | NEt2 | −3.5 | 9–14 | 0.0 | 2.26 |
| 5–15 | SEt | −7.8 | 10–15 | −17.0 | 0.95 |
The frequencies of the phosphoryl (νPO) and asymmetric sulfonyl (νSO2) stretching bands of 1–5 and 6–10 in carbon tetrachloride, and the corresponding frequency shifts with respect to their respective parent compounds 16 and 17 are collected in Table 3. All the frequency shifts are positive and the ΔνSO2 values are ca. 1.7 times smaller than the corresponding ΔνPO ones. This behaviour is in line with the absolute carbonyl gauche shifts for 6–10 being larger than the corresponding values for 1–5 (Table 2) and strongly suggests that the Olp→π*CO charge transfer interaction in the gauche rotamer of β-carbonyl sulfones is stronger than the corresponding interaction for β-carbonyl phosphonates. In fact, a stronger O(SO2)→π*CO charge transfer than the O(PO)→π*CO one should lead to a large decrease in the bond order of both CO and OSO oscillators in compounds 6–10 and, therefore, in their frequencies, compared to the CO and PO oscillators for compounds 1–5.
a in relation to the parent compounds 16 and 17
| Compound | X | ν PO/cm−1 | ΔνPO/cm−1 | Compound |
ν
SO2(as)/cm−1b |
ΔνSO2(as)/cm−1 |
|---|---|---|---|---|---|---|
| a Refers to the difference: να-phosphoryl or νsulfonyl compound − νparent compound. b From ref. 9. c Refers to the parent compounds (EtO)2P(O)Me and Et2SO2, respectively. d Refers to the parent compounds (EtO)2P(O)Me and Et2SO2, respectively. | ||||||
| 1 | Me | 1261 | +15 | 6 | 1331 | +10 |
| 2 | Ph | 1267 | +21 | 7 | 1332 | +11 |
| 3 | OEt | 1270 | +24 | 8 | 1335 | +14 |
| 4 | NEt2 | 1253 | +7 | 9 | 1325 | +4 |
| 5 | SEt | 1265 | +19 | 10 | 1335 | +14 |
|
16c |
— | 1246 | — |
17d |
1321 | — |
Table 4 shows the carbonyl 13C chemical shifts in deuterochloroform for 1–5 and 6–10 together with the differences (Δδ) between the chemical shift of each α-substituted carbonyl compound and the chemical shift of the corresponding parent compound 11–15. The smaller carbonyl upfield shifts (Δδ) of ca. 2.0 ppm for the α-phosphoryl derivatives compared to the α-sulfonyl derivatives, in spite of the quasi equal field-inductive effect22 for the diethylphosphoryl and the ethylsulfonyl groups, indicate that the O(PO)→π*CO CT interaction in the gauche rotamers of series 1–5 is weaker than the O(SO2)→π*CO CT in the corresponding rotamers of series 6–10, supporting the IR frequency shift analysis.

| X | Compound | δ CO | Compound | δ CO | ΔδCOa |
Compound | δ CO | ΔδCOa |
|---|---|---|---|---|---|---|---|---|
| a ΔδCO refers to the difference: δsubstituted carbonyl compound − δreference compound. | ||||||||
| Me | 11 | 203.7 | 1 | 200.0 | −3.7 | 6 | 198.0 | −5.7 |
| Ph | 12 | 196.7 | 2 | 192.0 | −4.7 | 7 | 189.1 | −7.6 |
| OEt | 13 | 169.8 | 3 | 165.8 | −4.0 | 8 | 163.1 | −6.7 |
| NEt2 | 14 | 164.8 | 4 | 162.5 | −2.3 | 9 | 161.4 | −3.4 |
| SEt | 15 | 193.6 | 5 | 190.4 | −3.2 | 10 | 188.2 | −5.4 |
In order to confirm the gauche conformer assignment of the single carbonyl band of the diethoxyphosphoryl carbonyl compounds 1–5, and to have precise geometries for the gauche rotamer of these compounds, ab initio calculations on α-dimethoxyphosphorylacetone 1a (chosen as a representative compound for the whole series) were carried out. The relevant data are presented in Table 5 along with the corresponding data for the gauche rotamer of α-methylsulfonylacetone 6a. The two most stable minima of 1a have the gauche conformation (Structures III and IV). It seems reasonable, therefore, to decide that the single carbonyl band observed in solution for the whole α-diethoxyphosphoryl carbonyl series 1–5 should correspond to the more abundant g1 rotamer of 1a in the gas phase.
| Dihedral anglese/° |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| μ/D | α | β | γ | γ′ | O(6)⋯C(2)/Åf |
O(1)⋯S(4)/Åg |
||||||
| a Refers to the gauche conformation. b Relative energy. c Molar fraction of each rotamer as a percentage. d α = O(1)–C(2)–C(3)–P(4); β = C(2)–C(3)–P(4)–O(5); γ = C(2)–C(3)–P(4)–O(7); γ′ = C(2)–C(3)–P(4)–O(6). e α = O(1)–C(2)–C(3)–S(4); β = C(2)–C(3)–S(4)–C(5); γ = C(2)–C(3)–S(4)–O(6); γ′ = C(2)–C(3)–S(4)–O(7). f Sum of van der Waals radii = 3.22 Å. g Sum of van der Waals radii = 3.32 Å. h The second minimum energy conformation corresponds to another gauche rotamer whose concentration is less than 1%. | ||||||||||||
|
6ah |
g | 0.0 | >99 | 3.03 | 78.8 | −70.3 | 44.9 | 174.5 | 2.973 | 3.298 | ||
The higher stability of the g1 with respect to the g2 rotamer is in line with a propitious geometry (structure III) giving an intramolecular distance (3.128 Å) between the negatively charged phosphoryl oxygen (qO = −0.740 e) and the positively charged carbonyl carbon (qC = +0.498 e), which is shorter than the sum of the van der Waals radii (3.22 Å) (see Table 5). This close contact produces significant Oδ−PO→Cδ+CO Coulombic and charge transfer interactions. Further stabilisation derives from the distance (3.331 Å) between the carbonyl oxygen (qO = −0.523 e) and the phosphoryl phosphorus (qP = +1.577 e) which is very close to the sum of the van der Waals radii (3.32 Å).
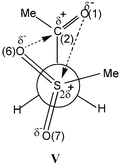
The geometry of the gauche conformer of 6a (see Table 5 and structure V) is very close to that of the g1 conformer of 1a (structure III). However, the O(6)⋯C(2) and O(1)⋯S(4) contacts between pairs of oppositely charged atoms are shorter than the corresponding distances for 1a (g1) (see Tables 5 and 6). Thus, the HF/6-31G** calculations for 1a and 6a corroborate the IR and 13C NMR data for 1–5 and 6–10, indicating that both series of compounds exist, in the gas phase and in solution, in the gauche conformation and that the Oδ−SO2→Cδ+CO charge transfer and Coulombic interactions in β-carbonyl sulfones are stronger than the Oδ−PO→Cδ+CO CT and Coulombic interactions in β-carbonyl phosphonates.
| e/C | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound | Conf. | O(5)[PO] | O(6)[OR] | O(7)[OR] | P(4)[PO] | C(2)[CO] | O(1)[CO] | O(6)[SO2] | O(7)[SO2] | S(4)[SO2] |
| 1a | g 1 | −0.740 | −0.718 | −0.700 | +1.577 | +0.498 | −0.523 | |||
| g 2 | −0.714 | −0.716 | −0.717 | +1.574 | +0.512 | −0.520 | ||||
| 6a | g | +0.513 | −0.514 | −0.690 | −0.672 | +1.457 | ||||
In the less stable g2 conformer of 1a the alkoxy oxygens O(6) and O(7) are the donor atoms (structure IV). Their interatomic distances from the oppositely charged carbonyl carbon atom C(2) are close to and shorter than, respectively, the sum of the van der Waals radii and shorter than the corresponding distances in the g1 conformer (see Tables 5 and 6). The lower stability of the g2 with respect to the g1 rotamer is probably related to the oxygen lone pair IE values which are higher for the methoxy than for the phosphoryl oxygen (12.0 and 10.71 eV, respectively24 for dimethyl methylphosphonate taken as a reference compound).
The existence of only the g1 conformer for 1–5 in a low permittivity solvent such as carbon tetrachloride (Table 1) can hardly be justified by the small (0.1 D) dipole moment difference between the two gauche rotamers of 1a. However, a close inspection of structures III and IV shows that the PO and CO dipoles are relatively close to each other in g1 and practically directly opposite each other in g2. Therefore, even the low relative permittivity but polarizable nature of carbon tetrachloride as solvent would stabilise the g1 to a greater extent than the g2 conformer.
Direct information about the relative donor/acceptor ability of the PO and SO2 groups has been obtained from an X-ray single crystal analysis of diethyl (methylsulfonyl)methyl phosphonate 18. Fig. 1 and Table 7 show that in the solid state 18 assumes a syn-clinal or quasi-gauche geometry with respect to both the α (−41.8°) and γ (−42.5°) dihedral angles. Moreover, the O(1)⋯P contact (3.18 Å) is significantly shorter than the sum of the relevant van der Waals radii (3.32 Å), while the O(3)⋯S contact (3.295 Å) is only slightly smaller. The most stable conformer of 18a from HF/6-31G** calculations has practically the same geometrical parameters as those obtained by X-ray diffraction for 18. In conclusion, this model compound shows that the O(SO2)→P(PO) charge transfer interaction between the sulfonyl oxygen (qO = −0.700 e) and the phosphoryl phosphorus (qP = 1.601 e) occurs over a shorter distance and is likely to be more pronounced than the interaction between the phosphoryl oxygen (qO = −0.743 e) and the sulfonyl sulfur (qs = 1.459 e), giving some support to the fact that O(SO2) in 6–10 is a better electron donor toward the π*CO orbital than O(PO) in 1–5.
| Dihedral anglesa/° |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | Conf.b |
α | β | γ | γ′ | P⋯O(1)/Åc |
P⋯O(2)/Åc |
S⋯O(3)/Åc |
|
| a α = O(3)–P–C(2)–S; β = P–C(2)–S–C(1); γ = P–C(2)–S–O(1); γ′ = P–C(2)–S–O(2). b Refers to the quasi-gauche conformation. c Sum of van der Waals radii = 3.32 Å. | |||||||||
| 18 | q-g | X-Ray | −41.8(2) | 74.4(2) | −42.5(2) | −170.3(2) | 3.180(2) | 4.203(2) | 3.295(2) |
| 18a | q-g | HF/6-31G** | −47.5 | 69.3 | −46.2 | −175.5 | 3.243 | 4.242 | 3.417 |
The better electron-donor ability of the sulfonyl oxygen lone pair nO(SO2) than the phosphoryl oxygen lone pair nO(PO) towards the π*CO orbital would appear to be unexpected. In fact, the basicity of the oxygen atom of the phosphoryl group evaluated from the νOH frequency shift in the diethyl ethylphosphonate–phenol complex with respect to phenol (CCl4, ΔνOH = 398 cm−1)41 is more than twice the basicity of the oxygen of the sulfonyl group estimated for the dimethyl sulfone–p-fluorophenol complex (ΔνOH = 154 cm−1),42 and the basicity trend is in line with the larger negative charge at O(PO) in 19 (−0.743 e) than at O(SO2) in 20 (−0.678 e). However, in the model compound 18/18a (structure VI), the CH2–SO angle and the SO bond length are smaller, respectively, than the CH2–PO angle and the PO bond length. Moreover, the corresponding parameters OP–CH3 (118.0°) and the PO (1.459 Å) for MeP(O)(OMe)219, and OS–CH3 (107.8°) and SO (1.435 Å) for Me2SO220 are very close to those computed for 18a. Thus, these intrinsic geometrical parameters, which allow close contact between oppositely charged atoms in 18, seem to be responsible for the abnormally stronger electron-donor ability of the sulfonyl oxygen lone pair nO(SO2) in 6–10 than the phosphoryl oxygen lone pair in 1–5.

Conclusions
The IR frequency of the single νCO band of the β-carbonylphosphonates [XC(O)CH2P(O)(OEt)2: X = Me 1, Ph 2, OEt 3, NEt24 and SEt 5] (series I), which correlates well with the νCO frequencies of the gauche rotamer of the corresponding β-carbonyl sulfones [XC(O)CH2SO2Et: X = Me 6, Ph 7, OEt 8, NEt29 and SEt 10] (series II) along with ab initio 6-31G** computations for 1a and 6a (X = Me) indicate the existence of a single gauche conformer (g1) for series I in solution.The abnormal negative carbonyl frequency shifts (ΔνCO) for both series approximately follow the electron affinities of the π*CO orbital of the parent carbonyl compounds MeC(O)X 11–15. These data suggest that the gauche conformations of series I and II should have similar geometries.
The less positive asymmetric sulfonyl frequency shifts (ΔνSO2) in comparison with the phosphoryl frequency shifts (ΔνPO) and the larger negative carbonyl gauche conformer shifts of β-carbonyl sulfones 6–10 in relation to the corresponding values of the β-carbonyl phosphonates 1–5 are in line with the greater upfield carbonyl 13C chemical shifts for series II with respect to series I. These trends are in agreement with their O(SO2)⋯C(CO) distances which are shorter than O(PO)⋯C(CO) in compounds 6a and 1a, respectively, and are discussed in terms of the Olp→π*CO charge transfer and electrostatic interactions, which are stronger for series II than for I. This unexpected behaviour indicates that the sulfonyl oxygen atom of the SO2R group is a better electron donor than the phosphoryl oxygen atom of the P(O)(OR)2 group. The intrinsic geometric parameters of the OS–CH2 and OP–CH2 moieties seem to be responsible for this behaviour. In fact, X-ray and ab initio calculations of dialkyl (methylsulfonyl)methylphosphonate MeSO2CH2P(O)(OR)2 (R = Et, 18, Me 18a) support this analysis.
Acknowledgements
The Brazilian authors thank Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) for financial support and the concession of the CAD4-Mach 3 X-ray diffractometer, and the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) for a grant (to P. R. O. and J. Z.-S.) and a scholarship (to R. R. F.). The Italian authors thank the Ministero della Ricerca Scientifica e Tecnologica for financial support.References
- Presented in part at the 18th International Symposium on the Organic Chemistry of Sulfur, Florence, Italy, 1998; summary in: Phosphorus, Sulfur Silicon Relat. Elem., 1999, 153–154, 353 Search PubMed.
- P. R. Olivato and M. G. Mondino, Phosphorus, Sulfur Silicon Relat. Elem., 1991, 59, 219 Search PubMed.
- P. R. Olivato, S. A. Guerrero and E. A. Martins, Phosphorus, Sulfur Silicon Relat. Elem., 1989, 44, 9 Search PubMed.
- P. R. Olivato, B. Wladislaw and S. A. Guerrero, Phosphorus, Sulfur Relat. Elem., 1987, 33, 135 Search PubMed.
- P. R. Olivato, E. Bonfada and R. Rittner, Magn. Reson. Chem., 1992, 30, 81 CAS.
- P. R. Olivato, B. Wladislaw, S. A. Guerrero and D. Russowsky, Phosphorus, Sulfur Relat. Elem., 1985, 24, 225 Search PubMed.
- P. R. Olivato, F. Oike and J. C. D. Lopes, Phosphorus, Sulfur Silicon Relat. Elem., 1990, 47, 391 Search PubMed.
- D. Jones, A. Modelli, P. R. Olivato, M. Dal Colle, M. de Palo and G. Distefano, J. Chem. Soc., Perkin Trans. 2, 1994, 1651 RSC.
- P. R. Olivato , M. G. Mondino and M. Sakuragi , unpublished results. .
- G. Distefano, M. Dal Colle, V. Bertolasi, P. R. Olivato, E. Bonfada and M. G. Mondino, J. Chem. Soc., Perkin Trans. 2, 1991, 1195 RSC.
- M. Dal Colle, V. Bertolasi, M. de Palo, G. Distefano, D. Jones and P. R. Olivato, J. Phys. Chem., 1995, 99, 15011 CrossRef CAS.
- P. R. Olivato, S. A. Guerrero and R. Rittner, Phosphorus, Sulfur Silicon Relat. Elem., 1997, 130, 155 Search PubMed.
- P. R. Olivato , E. Bueno , S. A. Guerrero and J. Zukerman-Schpector , 18th International Symposium on the Organic Chemistry of Sulfur, Florence, Italy, 1998, Abstract Book, p. 214. Search PubMed.
- P. R. Olivato, S. A. Guerrero and R. Rittner, Phosphorus, Sulfur Silicon Relat. Elem., 2000, 156, 255 Search PubMed.
- P. R. Olivato, S. A. Guerrero and J. Zukerman-Schpector, Acta Crystallogr., Sect. B: Struct. Sci., 2000, 56, 112 CrossRef.
- G. Distefano, M. Dal Colle, M. de Palo, D. Jones, G. Bombieri, A. Del Pra, P. R. Olivato and M. G. Mondino, J. Chem. Soc., Perkin Trans. 2, 1996, 1661 RSC.
- P. R. Olivato, M. G. Mondino, M. H. Yreijo, B. Wladislaw, L. Marzorati, M. B. Bjorklund, G. Distefano, M. Dal Colle, G. Bombieri and A. Del Pra, J. Chem. Soc., Perkin Trans. 2, 1998, 109 RSC.
- B. Solouki, H. Bock and R. Appel, Chem. Ber., 1975, 108, 897 Search PubMed.
- C. Müller and A. Schweig, Tetrahedron, 1973, 29, 3973 CrossRef.
- H. D. Martin, R. Iden, H. Landen, B. Mayer, G. Distefano, A. Modelli and R. Gleiter, J. Electron Spectrosc. Relat. Phenom., 1986, 41, 385 CrossRef CAS.
- S. Chattopadway, G. Findley and S. P. McGlyn, J. Electron Spectrosc. Relat. Phenom., 1981, 24, 27 CrossRef.
- C. Hansh , A. Leo and D. Hoekman , Exploring QSAR Hydrophobic, Electronic and Steric Constants, ACS Professional Reference Book, American Chemical Society, Washington, DC, 1995. Search PubMed.
- J. A. Tossell, J. H. Moore and J. C. Giordan, Inorg. Chem., 1985, 24, 1100 CrossRef CAS.
- J. C. Giordan, J. H. Moore, J. A. Tossell and W. Kaim, J. Am. Chem. Soc., 1985, 107, 5600 CrossRef CAS.
- H. Schmidt, A. Schweig, F. Mathey and G. Mueller, Tetrahedron, 1975, 31, 1287 CrossRef CAS.
- A. N. Pudovik and V. P. Avery’anova, Zh. Obshch. Khim., 1956, 26, 1431 ( Chem. Abstr. , 1956 , 50 , 14513 ) Search PubMed.
- I. J. Borowitz, M. Anschel and S. Firstenberg, J. Org. Chem., 1967, 32, 1723 CrossRef CAS.
- A. J. Speziale and R. C. Freeman, J. Org. Chem., 1958, 23, 1883 CrossRef CAS.
- R. W. Balsiger, D. G. Jones and J. A. Montgomery, J. Org. Chem., 1959, 24, 434 CrossRef CAS.
- H. J. Liu, P. A. Rose and D. J. Sasaki, Can. J. Chem., 1991, 69, 934 CAS.
- G. H. Posner and D. J. Brunelle, J. Org. Chem., 1972, 37, 3547 CrossRef CAS.
- Gaussian 98, Revision A.6, M. J. Frisch , G. W. Trucks , H. B. Schlegel , G. E. Scuseria , M. A. Robb , J. R. Cheeseman , V. G. Zakrzewski , J. A. Montgomery, Jr. , R. E. Stratmann , J. C. Burant , S. Dapprich , J. M. Millam , A. D. Daniels , K. N. Kudin , M. C. Strain , O. Farkas , J. Tomasi , V. Barone , M. Cossi , R. Cammi , B. Mennucci , C. Pomelli , C. Adamo , S. Clifford , J. Ochterski , G. A. Petersson , P. Y. Ayala , Q. Cui , K. Morokuma , D. K. Malick , A. D. Rabuck , K. Raghavachari , J. B. Foresman , J. Cioslowski , J. V. Ortiz , B. B. Stefanov , G. Liu , A. Liashenko , P. Piskorz , I. Komaromi , R. Gomperts , R. L. Martin , D. J. Fox , T. Keith , M. A. Al-Laham , C. Y. Peng , A. Nanayakkara , C. Gonzalez , M. Challacombe , P. M. W. Gill , B. Johnson , W. Chen , M. W. Wong , J. L. Andres , M. Head-Gordon , E. S. Replogle and J. A. Pople , Gaussian Inc., Pittsburgh PA, 1998. Search PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343 CrossRef.
- G. M. Sheldrick , SHELXL97. Program for the Refinement of Crystal Structures, University of Göttingen, Germany, 1997. Search PubMed.
- M. Nardelli, J. Appl. Crystallogr., 1995, 28, 659 CrossRef CAS.
- L. J. Farrugia , WinGX. A Windows Program Package for Crystal Structure Analysis, University of Glasgow, Scotland, 2000. Search PubMed.
- L. Zsonai , ZORTEP, Molecular Graphics Program, University of Heidelberg, Germany, 1995. Search PubMed.
- In our experience, in fact, a good linear correlation is not obtained when the frequencies of the gauche rotamers of a series of compounds are plotted against the frequencies of the cis conformers of a different series..
- G. Distefano , A. Modelli and D. Jones , unpublished results. .
- P. R. Olivato and R. Rittner, Rev. Heteroat. Chem., 1996, 15, 115 Search PubMed.
- J. E. Oliveira , MSc Thesis, Instituto de Quimica, Universidade de São Paulo, Brazil, 1980. Search PubMed.
- A. Chardin, C. Laurence, M. Berthelot and D. G. Morris, J. Chem. Soc., Perkin Trans. 2, 1996, 1047 RSC.
Footnote |
| † CCDC reference number 188/279. See http://www.rsc.org/suppdata/p2/b0/b005501h/ for crystallographic files in .cif format. |
| This journal is © The Royal Society of Chemistry 2001 |