Synthesis and tastant properties of disulfamates. Multisulfamation studies
Wm. J.
Spillane
*,
Caroline A.
Ryder
,
Danny G.
Concagh
and
Lorraine M.
Kelly
Chemistry Department, National University of Ireland, Galway, Ireland
First published on 5th December 2000
Abstract
Twenty four disulfamates, one trisulfamate, two tetrasulfamates and two monosulfamates have been made. The disulfamates are of two types: RN(SO3Na)2 (Type A, compounds 1–20) and NaO3S(H)NR′N(H)SO3Na (Type B, compounds 21–24) and all except three (which had not been tasted) are new materials. The positions of the –SO3Na groups in compounds 21–23 have been established by the use of model compounds (e.g. parent amines, appropriate monosulfamates) and 13C-NMR. Some multisulfamation synthesis leading to compounds 25–27 has been carried out. Taste data have been obtained for almost all the sulfamates made and the significance of these in relation to structure–taste studies for sulfamate sweeteners is discussed. In particular, the possibility that the entity ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif)
![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) CHN(R)SO3− might function as a hydrogen source in the Shallenberger–Acree, multicomponent attachment and α-helical protein receptor mechanisms has been examined.
CHN(R)SO3− might function as a hydrogen source in the Shallenberger–Acree, multicomponent attachment and α-helical protein receptor mechanisms has been examined.
There are over 30 reports in the literature dealing with disulfamates (imidodisulfonates) ranging from the initial work in 1920 of Traube and co-workers
1,2 [who made some basic compounds such as RN(SO3K)2 (R = Me, Et, Pr)1,2 and simple hydroxylamine disulfonates such as MeON(SO3K)22,3] to a current report4 where 2,2-dinitropropane-1,3-diol [HOCH2C(NO2)2CH2OH] reacts with alkylsulfamates (RNHSO3M) to give, inter alia, the disulfamates MO3SN(R)CH2C(NO2)2CH2N(R)SO3M. Most reports deal with ionic disulfamates but there are also some reports5–8 of various disulfamate esters and one9 of a ‘mixed’ disulfamate e.g. a monobactam which contains a sulfamate ester group (see Fig. 1).
 | ||
| Fig. 1 Some previously reported disulfamate esters (refs. 5–9). | ||
There are a number of reports dealing with tetra- and trisulfamates.1,10–13 Traube and Wolff1 report the preparation of the tetrasulfamate (KO3S)2NCH2CH2N(SO3K)2. Various hydrazine tri- and tetrasulfonates have been synthesized11 and in two papers12,13 Baumgarten describes the preparation of some tetra potassium and barium sulfonate salts of urea,12 a series of aminoacid disulfonates and one trisulfonate, N,N′,N′′-histidylhistidine trisulfonate.13
Few reports have focused on the syntheses of disulfamates per se apart from the work of Baumgarten,13 who synthesized seven di-/tri- amino acid sulfonates using pyridine–sulfur trioxide, and Kanetani,14,15 who used amine–SO3 adducts and amines to synthesize over 20 disulfamates including 1–3 (Fig. 2). Kanetani carried out a detailed study of the effect of the reaction time, temperature etc. on the disulfamation reaction.
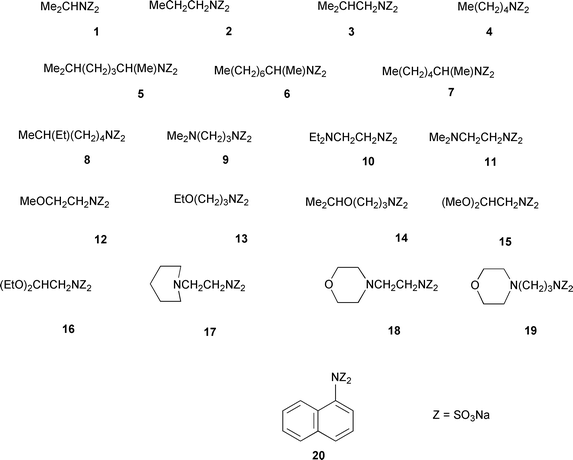 | ||
| Fig. 2 Type A disulfamates synthesized; Z = SO3Na. | ||
Disulfamates have found application in a number of areas. Thus, alkanediyl disulfamates [H2NSO2O(CH2)nOSO2NH2, n = 6, 7, 8] inhibit the growth of malignant cells in mammals,16 the monobactam sulfamates shown in Fig. 1 are reported to have in vitro antibacterial activity,9 various derivatives of 3,5-bis(sulfoamino)benzoic acid act as anti-inflammatory agents in the treatment of rheumatic fever and rheumatoid arthritis17 and a number of disulfamates are intermediates in the dyeing industry.18 Finally the disulfamate aniline-N,N-disulfonic acid, PhN(SO3H)2 has been implicated in the mechanisms of both the sulfonation of aniline in conc. sulfuric acid and the rearrangement of phenylsulfamic acid19 and in the reduction of nitro compounds with sulfite (Piria reaction).20
Disulfamates could, since they contain a sulfamate moiety, be sweet compounds and this indicates another possible use. Unfortunately, there are no taste data at all available for all the disulfamates already prepared. In this paper we explore this potential by synthesizing twenty one new disulfamates and three previously made by Kanetani14 (see Figs. 2 and 3) together with one trisulfamate, and two tetrasulfamates (see Fig. 3). The taste portfolios of 25 of the compounds together with those of two monosulfamates also made in this work for comparison have been assessed.
 | ||
| Fig. 3 Type B disulfamates (21–24) and multisulfamates (25–27) synthesized; Z = SO3Na. | ||
Results and discussion
The sweet taste elicited by a number of metallic sulfamates [RN(H)SO3−M+] has been known for about 60 years.21 Structure–taste studies have been carried out on sulfamates synthesized with alicyclic, open-chain, aromatic, heterocyclic and hetero open-chain and branched R groups, and a number of the compounds were found to be sweet.22 The cation, M+ does not appear to effect the degree of sweetness of a sulfamate.22 An intact –NHSO3− function was considered to be essential for sweetness since it provides a hydrogen and an electronegative center for the operation of the Shallenberger–Acree mechanism of sweetness.23 However, some years ago a series of heterocyclic sulfides and sulfones containing an –N(R)SO3− group were reported to be sweet,24 and their sweetness has been explained on the basis of the presence of the systemCHN(R)SO3−, in which an α-hydrogen might function as a hydrogen source in the Shallenberger–Acree mechanism.25 The reported sweetness of these compounds has more recently been disputed.26
We were interested in testing the idea that an α-hydrogen, rather than a hydrogen on nitrogen, might be able to participate in the sweetness mechanism. The synthesis of disulfamates seems to offer an ideal opportunity to test this since (i) the amino hydrogen is replaced, (ii) a second sulfamate entity is being introduced into the molecule and, given the well-established “sweet-conferring ability” of the sulfamate group, it might enhance sweetness and (iii) we have been able to show that one of the α-hydrogens in CH3N(SO3K)2 is within a distance of 3 Å of two of the oxygen atoms and it could therefore act as hydrogen source in the operation of the Shallenberger–Acree mechanism. This was established by downloading from the Cambridge Structural Database the structure for the disulfamate K2[CH3N(SO3)2] (no. 39971) (see Fig. 4) determined by Kennard et al.27 Using the ORTEX (OSCAIL) program28 we obtained the relevant interatomic distances and found that H11–O12 was 2.665 and H11–O11 was 2.916 Å. All other H–O distances were >3 Å and were not considered. It should be noted that H13 was not located in the crystallographic work of Kennard.
![Schematic representation of K2[CH3N(SO3)2] from the Cambridge Structural Database.](/image/article/2001/P2/b005475p/b005475p-f4.gif) | ||
| Fig. 4 Schematic representation of K2[CH3N(SO3)2] from the Cambridge Structural Database. | ||
Two types of disulfamate have been prepared—Type A [RN(SO3Na)2, i.e. both sulfonates on one nitrogen, N,N-disulfamates] and Type B [NaO3S(H)NR′N(H)SO3Na, i.e. the two sulfonates on different nitrogens, N,N′-disulfamates].
All the disulfamates prepared (Figs. 2 and 3) have one or two α-hydrogens at C1, except compound 20 (Fig. 2), which was included because it had not been prepared by Kanetani when he prepared a series of aryldisulfamates.15 Of the 29 compounds prepared in this work only three compounds (1–3) have been previously prepared.1,14 These were resynthesized as standards and also because their taste data was needed for comparison with their monosulfamates, two of which are sweet.
Taste portfolios for 25 compounds, together with percentage yields (for 27 compounds) are given in Table 1. From the taste data in Table 1 it is readily apparent that, though some of the compounds are multisapophoric, none of the 25 compounds tasted display sweetness, apart from a sweet aftertaste in compound 22. The pHs of most of the 0.01 M solutions for tasting were determined and these are given in Table 1. As would be expected,23 those solutions that have a predominant sour taste display a definite acidic pH, while bitter solutions give a pH in the alkaline region. The solutions found to be tasteless, viz. those of compounds 14, 19 and 23, gave pHs of 10.2, 10.3 and 5.9, respectively. In the tasting of compound 5 sourness seemed to predominate over bitterness, thus the pH of 2.3 is reasonable. The one peculiarity arises with the naphthyldisulfamate (20) which, despite being exclusively bitter, displays a solution pH of 3.00. One notes, however, that a secondary taste of mothballs was found with this compound and this may be masking a sour taste. In Table 2 data (where available) for the corresponding monosulfamates are compared with those for the disulfamates. In the case of compounds 22 and 23 taste data for the corresponding monosulfamates (28 and 29) were obtained in this work (see footnotes, Table 2). From Table 2 it is seen that for compounds 2, 3 and 23 there is a change in taste on going from the mono- to the disulfamates and for 22 there is no change. Comparison is difficult for other compounds, as when most of the monosulfamate taste data was collected between 11 and 37 years ago it was common to describe compounds that did not give a sweet taste as simply nonsweet,25,28,34 non sucre,29,33 or non dolce.35 However, for compounds 1, 4, 9, 12, 13, 15, 17–19 and 22 sweetness is absent in the monosulfamates and disulfamation does not induce sweetness. Simple meta-substituted phenylsulfamates are often sweet and hence some sweetness might be expected in 21, but not in 22, as sweetness has not been found in para- or ortho-phenylsulfamates. Some sweet taste might also be expected in 23 and in its monosulfamate 29 since some monosubstituted cyclamate derivatives are sweet,36 and in fact the monosulfamate 29 has a substantial sweet component.
 a and percentage of tasters giving the taste quality of disulfamates 1–27
a and percentage of tasters giving the taste quality of disulfamates 1–27
| Compound | Yield (%) | pH | Sour | Bitter | Salty | Tasteless | Aftertaste (% of tasters) | No. of tasters | Predominant taste (>50% tasters) |
|---|---|---|---|---|---|---|---|---|---|
| a All compounds were tasted as 0.01 M aqueous solutions and allowance was made for water of hydration when making up solutions. pH measurements were made using these solutions and an Orion pH meter model 420A buffered at pH 4.0, 7.0 and 9.2. Taste panellists did not detect an initial sweet taste in any of the solutions. b An onion-like taste was also reported for this compound. c A musty taste was also reported. d A taste of mothballs was also reported. e A taste of nuts was also reported. f Not tasted. g Taste of burnt nuts was also reported. | |||||||||
| 1 | 42 | 2.35 | 100 | 0 | 0 | 0 | 0 | 9 | Sour |
| 2 | 15 | — | 100 | 0 | 0 | 0 | 0 | 4 | Sour |
| 3 | 12 | — | 100 | 0 | 0 | 0 | 0 | 4 | Sour |
| 4 | 45 | 6.2 | 45 | 0 | 67 | 0 | 0 | 9 | Salty |
| 5 | 41 | 2.3 | 67 | 55 | 0 | 0 | 0 | 9 | Sour/bitterb |
| 6 | 20 | 2.2 | 89 | 45 | 0 | 0 | 0 | 9 | Sour |
| 7 | 21 | 2.2 | 100 | 0 | 0 | 0 | 0 | 9 | Sour |
| 8 | 60 | 10.4 | 0 | 0 | 100 | 0 | 0 | 9 | Salty |
| 9 | 16 | 11.1 | 0 | 100 | 0 | 0 | 0 | 9 | Bitter |
| 10 | 25 | 11.3 | 22 | 78 | 0 | 0 | 0 | 9 | Bitter |
| 11 | 20 | 10.8 | 0 | 100 | 0 | 0 | Bitter (33) | 9 | Bitter |
| 12 | 45 | — | 100 | 0 | 0 | 0 | 0 | 4 | Sour |
| 13 | 20 | 8.1 | 78 | 22 | 0 | 0 | 0 | 9 | Sour |
| 14 | 68 | 10.2 | 45 | 0 | 0 | 55 | 0 | 9 | Tasteless |
| 15 | 15 | 2.5 | 78 | 0 | 0 | 22 | 0 | 9 | Sour |
| 16 | 54 | 10.5 | 0 | 55 | 0 | 45 | 0 | 9 | Bitterc |
| 17 | 33 | — | 0 | 100 | 0 | 0 | 0 | 4 | Bitter |
| 18 | 36 | 10.3 | 0 | 100 | 0 | 0 | 0 | 9 | Bitter |
| 19 | 29 | 10.3 | 0 | 0 | 0 | 100 | 0 | 9 | Tasteless |
| 20 | 30 | 3.00 | 0 | 100 | 0 | 0 | 0 | 9 | Bitterd |
| 21 | 34 | — | 0 | 100 | 0 | 0 | 0 | 4 | Bitter |
| 22 | 46 | — | 25 | 100 | 0 | 0 | Sweet (25) | 4 | Bitter |
| 23 | 15 | 5.9 | 0 | 0 | 0 | 100 | 0 | 9 | Tastelesse |
| 24 | 10 | — | — | — | — | — | — | — | —f |
| 25 | 13 | — | 100 | 0 | 0 | 0 | 0 | 9 | Sour |
| 26 | 7 | — | 0 | 100 | 0 | 0 | 0 | 9 | Bitterg |
| 27 | 10 | — | — | — | — | — | — | — | —f |
| Taste | |||
|---|---|---|---|
| Compound | Di- | Mono- | Ref.c |
| a Panellists (4) found this compound to be 100% bitter and to have 25% sweet aftertaste only—therefore its predominant taste is bitter. b Panellists (4) found this compound to be 100% bitter and 100% sweet—therefore its predominant taste is recorded as bitter/sweet. c Reference to monosulfamate. | |||
| 1 | Sour | Nonsweet | 25 |
| 2 | Sour | Sweet | 25,29,30 |
| 3 | Sour | Sweet | 25,29–31 |
| 4 | Salty | Nonsweet | 29 |
| 9 | Bitter | Bitter | 32 |
| 12 | Sour | Nonsweet | 33 |
| 13 | Sour | Nonsweet | 34 |
| 15 | Sour | Nonsweet | 33 |
| 17 | Bitter | Nonsweet | 35 |
| 18 | Bitter | Nonsweet | 35 |
| 19 | Tasteless | Nonsweet | 32 |
| 22 (di), 28 (mono) | Bitter | Bittera |
Present work |
| 23 (di), 29 (mono) | Bitter | Bitter/Sweetb |
Present work |
The di-, tri- and tetrasulfamates 24, 25 and 26 and 27, respectively, are long-chain compounds and would be unlikely to be sweet since even a C5 chain hydrocarbon sulfamate is not sweet.29
The absence of sweetness in the disulfamates does appear to underline the necessity for an imino hydrogen (–NH) in the sulfamate functionality that can actively participate in hydrogen bonding in the dual H-bond theory of Shallenberger and Acree. Substitution of an α-hydrogen, as we have tired to do with the disulfamates, does not work, even though we have shown above that the distance between the –CH– and the oxygen can be within 3 Å, the required AH–B distance of the Shallenberger–Acree theory. A C-bound hydrogen would, of course, be much less efficient in H-bonding than an –NH or –OH and this is probably an important factor too. In terms of the multicomponent attachment theory of sweetness of Nofre and Tinti,37 one might assign the alkyl or alkoxy or piperidine or morpholine rings (Fig. 2) as G sites, the α-CH as an AH site and –SO3– as a B site. On this basis the AH–G distance could be ≈3.5 Å and the B–G distance ≈5.5 Å, respectively, as required by the theory. One selects a suitable point in G to achieve this. It is difficult to assign other sites (the theory normally requires at least 8 sites) unless one takes one of the SO3− groups as a D site.
Using their α-helical protein receptor theory, Suami and Hough38 have recently examined some cyclamate derivatives and explained very well the sweetness of some and the lack of sweetness in others. In the disulfamates it is relatively easy to pick a glycophoric triad of AHs/Bs/Xs (using their notation). Thus, AHs would be the α-CH, Bs one of the –SO3− groups and Xs appropriate hydrogens in the R portion. In so far as one can see, therefore, the three main sweetness theories would indicate that the disulfamates might elicit sweetness. The failure to realize this may well be due to the weak H-bonding ability of the α-Hs.39
In summary, 29 sulfamates, mainly disulfamates, have been prepared in this work and taste profiles have been obtained for 27 of these. No sweet taste was noted in the disulfamates 1–19, all of which contain the entity CHN(SO3Na)2 (in which the α-hydrogen attached to carbon might be a source of hydrogen, in place of a hydrogen on nitrogen, for the successful operation of the Shallenberger–Acree multicomponent attachment or α-helical receptor theories of sweetness). Further, the sweetness of several monosulfamates is shown to be destroyed in the corresponding disulfamates and a sweet taste could not be “induced” in a number of nonsweet monosulfamates on their conversion to the corresponding disulfamates. In conclusion, this work is in line with the main body of evidence from earlier studies21,25,40,41 that indicates that an amino hydrogen is essential for sweetness and it appears to cast additional doubt on the 1974 report that some secondary sulfamates containing an –N(R)SO3 group were sweet.24
Experimental
Materials and methods
The 2∶1 ratio of chlorosulfonic acid to amine was stepped up to 2.5∶1 or 3∶1 in order to improve yields for the synthesis of compounds 4, 9, 11 and 18–20. If more than 10 ml of chlorosulfonic acid was used a MeOH–dry-ice bath was used to lower and maintain the temperature and to allow more rapid addition of the acid.
In Table 313C chemical shifts are given for N-phenylphenylene-1,4-diamine, the monosulfamate, 28, and the disulfamate product, 22. For the diamine spectrum the assignments have been made from calculation of theoretical 13C chemical shifts for this compound using the empirical rules and tables of data of Williams and Fleming.42 The assignments of the signals at 132.0, 144.0 and 146.9 to the three tertiary carbons were confirmed by the results of a C-J resolved spectrum. For the monosulfamate the assignments are also shown and again a C-J resolved spectrum confirmed that the three downfield peaks are due to the three tertiary carbon atoms. That the monosulfamate 22 is as shown above is evident by the fact that the C1 signal is, as expected, shifted upfield by 5 ppm, C7 is virtually unaffected and C4 shows an upfield shift of 3.4. Had sulfamation occurred at the NH between the two rings, considerable shifts in the C7 and C4 signals would have been expected. For 21 a C-J resolved spectrum again assigned the three downfield signals as shown. The disulfamate shows a strong upfield shift (6.2 ppm) at C4, whereas at C1 it is only 3.7 and this indicates that it is the N,N′- and not the N,N-disulfamate that has been formed.
a for N-phenyl-1,4-phenylenediamine, the monosulfamate 28 and the disulfamate 22 relative to Me4Si
| (CD3)2SO | C8/C12 | C3/C5 | C2/C6 | C10 | C9/C11 | C4 | C1 | C7 |
|---|---|---|---|---|---|---|---|---|
| a 270 MHz. | ||||||||
| 4-Ph(H)NC6H4NH2 | 114.0 | 115.1 | 117.3 | 122.7 | 129.2 | 132.0 | 144.0 | 146.9 |
| 28 | 115.1 | 118.4 | 118.7 | 121.1 | 130.0 | 135.4 | 139.0 | 146.7 |
| 22 | 116.8 | 117.3 | 120.3 | 123.1 | 123.6 | 129.2 | 142.7 | 143.1 |
For 23δC [D2O, 100 °C] 31.72 and 57.4; for 1,4-diaminocyclohexane δC [(CD3)2SO] 35.26 and 50.0 and for the monosulfamate, 29, δC [(CD3)2SO] 32.6, 35.1, 49.9 and 52.1. The spectrum of compound 23 had to be run in D2O to increase its solubility because it was insoluble in (CD3)2SO. Clearly again, as in the case of 21, 23 is an N,N′-disulfamate, and not an N,N-disulfamate, which would give four signals in its 13C NMR. Compound 24 gave 10 signals in the 13C NMR so it clearly has to be the N,N-disulfamate as shown in Fig. 3.
Acknowledgements
The authors thank Mr N. Ní Chonchubhair for doing the ORTEX calculations. C. A. R. and L. M. K. thank Enterprise Ireland for grants. The EU is thanked for a grant.References
- W. Traube and M. Wolff, Ber. Dtsch. Ges. Chem., 1920, 53, 1493 Search PubMed.
- W. Traube, H. Ohlendorf and H. Zander, Ber. Dtsch. Ges. Chem., 1920, 53, 1477 Search PubMed.
- It should be noted that salts of the parent compound imidodisulfonic acid (aquo-ammonosulfuric acid) have been known since 1834. See L. F. Audrieth, M. Sveda, H. H. Sisler and M. J. Butler, Chem. Rev., 1940, 26, 49 CrossRef CAS for a review..
- V. A. Tartakovsky, A. S. Ermakov and O. N. Varfolomeeva, Russ. Chem. Bull., 1999, 48, 1385 Search PubMed.
- M. Goehring and H. K. A. Zahn, Chem. Ber., 1956, 89, 179 Search PubMed.
- M. Hedayatullah and J. F. Brault, Phosphorus Sulfur, 1981, 11, 303 Search PubMed.
- N. S. Zefirov, N. V. Zyk and A. G. Kutateladze and Yu. A. Lapin, Zh. Org. Khim., 1987, 23, 229 Search PubMed.
- M. A. Pudovik, L. K. Kibardina and A. N. Pudovik, Russ. J. Gen. Chem., 1993, 63, 267 Search PubMed.
- G. Guanti, R. Riva, G. Cascio, E. Manghisi, G. Morandotti, G. Satta and R. Sperning, Farmaco, 1998, 53, 173 CrossRef CAS; G. Guanti, R. Riva, G. Cascio, E. Manghisi, G. Morandotti, G. Satta and R. Sperning, Chem. Abstr., 1998, 129, 1754465 Search PubMed.
- Salts of the nitrilosulfonates, N(SO3H)3 have been known since the last century. See Audrieth et al., reference 3..
- A. Meuwsen and H. Tischer, Z. Anorg. Allg. Chem., 1958, 294, 282 CrossRef CAS.
- P. Baumgarten and I. Marggraff, Ber. Dtsch. Ges. Chem., 1931, 64, 301 Search PubMed.
- P. Baumgarten, I. Marggraf and E. Dammann, Z. Physiol. Chem., 1932, 209, 145 Search PubMed.
- F. Kanetani and H. Yamaguchi, Bull. Chem. Soc. Jpn., 1974, 47, 2713 CAS.
- F. Kanetani, Bull. Chem. Soc. Jpn., 1986, 59, 952 CAS.
- D. M. Bailey, Sterling Drug Co., U.S. Patent 4 829 084, 1989; ; D. M. Bailey, Chem. Abstr., 1989, 111, 120951. Search PubMed.
- UCLAF, Br. Patent 879 050, 1961; Search PubMed; Chem. Abstr., 1962, 56, 14173b. Search PubMed.
- (a) R. Lantz, Bull. Soc. Chim. Fr., 1948, 489 Search PubMed; (b) F. Ackerman, U.S. Patent 2 567 796, 1951; Search PubMed F. Ackerman, Chem. Abstr., 1952, 46, 754; Search PubMed (c) R. Lantz and P. M. J. Obellianne, Br. Patent 753 983, 1956; Search PubMed R. Lantz and P. M. J. Obellianne, Chem. Abstr., 1957, 51, 5826e; Search PubMed (d) R. F. M. Sureau and P. M. J. Obellianne, U.S. Patent 2 853 359, 1958; Search PubMed R. F. M. Sureau and P. M. J. Obellianne, Chem. Abstr., 1959, 53, 3721h. Search PubMed.
- Z. Vrba and Z. J. Allan, Tetrahedron Lett., 1968, 4507 CrossRef CAS.
- Z. Vbra and Z. J. Allan, Collect. Czech. Chem. Commun., 1968, 33, 431 CAS.
- L. F. Audrieth and M. Sveda, J. Org. Chem., 1944, 9, 89 CrossRef CAS.
- W. J. Spillane, C. A. Ryder, M. R. Walsh, P. J. Curran, D. G. Concagh and S. N. Wall, Food Chem., 1996, 56, 255 CrossRef CAS.
- R. S. Shallenberger, Taste Chemistry, Blackie, Academic & Professional, New York, 1993. Search PubMed.
- G. R. Wendt and M. W. Winkley, U.S. Patent 3 787 442, 1974; Search PubMed; G. R. Wendt and M. W. Winkley, Chem. Abstr., 1974, 80, 82669 Search PubMed.
- G. A. Benson and W. J. Spillane, J. Med. Chem., 1976, 19, 869 CrossRef CAS.
- See W. J. Spillane, C. A. Ryder, P. J. Curran, S. N. Wall, L. M. Kelly, B. G. Feeney and J. Newell, J. Chem. Soc., Perkin Trans. 2, 2000, 1369 Search PubMed for a discussion of this matter..
- A. J. Morris, C. H. L. Kennard and J. R. Hall, Inorg. Chim. Acta, 1982, 62, 247 CrossRef CAS.
- P. McArdle, J. Appl. Crystallogr., 1995, 28, 65 CrossRef.
- C. Nofre and F. Pautet, Bull. Soc. Chim. Fr., 1975, 686 Search PubMed.
- H. Yamaguchi, Nippo Chemical Co., Jpn. Patent 13,056, 1963; Search PubMed; H. Yamaguchi, Chem. Abstr., 1964, 60, 2771a. Search PubMed.
- B. Unterhalt and L. Böschemeyer, Z. Lebensm. Unters. Forsch., 1972, 149, 227 CAS.
- W. J. Spillane, G. McGlinchey, I. Ó Muircheartaigh and G. A. Benson, J. Pharm. Sci., 1983, 72, 852 CAS.
- F. Pautet and M. Daudon, Pharmazie, 1985, 40, 428 Search PubMed.
- W. J. Spillane and M. B. Sheahan, J. Chem. Soc., Perkin Trans. 2, 1989, 741 RSC.
- M. DeNardo, C. Ruuti and F. Ulian, Farmaco, Ed. Sci., 1984, 39, 125 Search PubMed; M. DeNardo, C. Ruuti and F. Ulian, Chem. Abstr., 1984, 100, 137568 Search PubMed.
- W. J. Spillane and G. McGlinchey, J. Pharm. Sci., 1981, 70, 933 CAS.
- J. M. Tinti and C. Nofre, in Sweeteners Molecular Design and Chemoreception, ed. D. E. Walters, F. T. Orthoefer and G. E. DuBois, ACS Symposium Series 450, American Chemical Society, Washington, DC, 1991, ch. 15. Search PubMed.
- T. Suami, L. Hough, T. Machinami, T. Saito and K. Nakamura, Food Chem., 1998, 63, 391 CrossRef CAS.
- G. A. Jeffrey and W. Saenger, Hydrogen Bonding in Biological Structures, Springer Verlag, 1994, Study Edition, ch. 10. Search PubMed.
- F. F. Blicke, H. E. Millson and N. J. Doorenbos, J. Am. Chem. Soc., 1954, 76, 2498 CrossRef CAS.
- C. D. Hurd and N. Kharasch, J. Am. Chem. Soc., 1946, 68, 653 CrossRef CAS.
- D. H. Williams and I. Fleming, Spectroscopic Methods in Organic Chemistry, McGraw-Hill, London, 4th edn., 1987, pp. 125–149. Search PubMed.
| This journal is © The Royal Society of Chemistry 2001 |