The X-ray structures of HOBt-based immonium-type coupling reagents and the rearrangement of benzotriazolyl esters of Nα-protected amino acids or peptides: N- vs. O-substituted forms![[hair space]](https://www.rsc.org/images/entities/h2_char_200a.gif) †
†
Peng
Li
and
Jie
Cheng Xu
*
Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 354 Feng Lin Road, Shanghai, 200032, P. R. China
First published on 5th December 2000
Abstract
The crystal structures of HOBt-based immonium salts BOMI and BDMP are reported and show that these compounds crystallize as the N-substituted form rather than as the previously assigned O-substituted form. These unexpected phenomena are explained well by semiempirical PM3 calculations; the crystal structures of other HOBt-based onium salts can also be predicted by these theoretical calculations. The reaction mechanisms for coupling reactions mediated by these HOBt-based immonium salts and the rearrangement of the benzotriazolyl ester intermediate generated during the coupling reactions were studied by both experimental observations and theoretical calculations. The crystal structures of these benzotriazolyl esters were predicted by PM3 calculations and verified by reported X-ray data.
Introduction
In previous studies, we have shown the high efficiency of the novel HOBt-derived immonium-type coupling reagents, BOMI, BDMP, BPMP and BMMP, in peptide synthesis.1,2 They can be used to synthesize various oligopeptides and biologically active peptides in both solution and the solid phase with fast reaction speeds, negligible racemization and good yields.3,4 In comparison to HOBt-based uronium- and phosphonium-type reagents, these immonium salts demonstrated much higher reactivities and lower racemization due to their unique structural features.4,5 These immonium-type reagents were designed based upon the molecular structures of HOBt-based uronium-type coupling reagents, such as HBTU,6 HBMDU,7 and HBPyU,8 by replacing one of the substituted amino groups in the uronium salts with hydrogen, alkyl or aryl. Such structural modifications cause the reaction-mediating carbons of these immonium salts to share relatively low electron density compared to the corresponding uronium salts, which results in their showing higher reactivities in nucleophilic reactions involved in peptide coupling.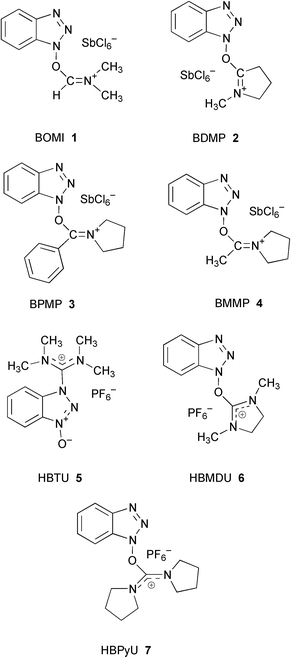
Although these HOBt-based uronium- and immonium-type coupling reagents have been commonly used in peptide synthesis, the structures of these reagents have not been determined up to now, except for reagent HBTU. X-Ray analysis revealed that HBTU crystallizes as the unusual guanidinium N-oxide (N-form),9 rather than the isomeric uronium form.6 The structures of other HOBt-derived onium salts, for which X-ray data have not been obtained, have to be arbitrarily assigned as the O-form according to general chemical knowledge since there is no general rule that can predict whether these HOBt-based onium salts are present in the O-form or the N-form. To investigate the mechanism of coupling mediated by these reagents and to study the relationship between the reactivity and structures of these reagents, as well as develop more efficient peptide coupling reagents, resolving the true structures of these onium salts is necessary and significant. In this article, we try to solve these problems by both X-ray analysis and molecular orbital calculations. Since the benzotriazolyl esters of Nα-protected amino acids or peptides, generated during peptide synthesis using HOBt-derived immonium salts, also present such a problem, the structures and tautomerization of these esters are also discussed herein.
Results and discussion
X-Ray structure determination of HOBt-derived immonium salts BOMI and BDMP![[hair space]](https://www.rsc.org/images/entities/h3_char_200a.gif) ‡
‡
These HOBt-based immonium salts were synthesized from the corresponding amides via three steps in high yields. For instance, reagents BOMI, BDMP, BPMP and BMMP can be prepared from N,N-dimethylformamide (DMF![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) ), N-methylpyrrolidone (NMP), N-benzoylpyrrolidine and N-acetylpyrrolidine, respectively, as shown in Scheme 1.
), N-methylpyrrolidone (NMP), N-benzoylpyrrolidine and N-acetylpyrrolidine, respectively, as shown in Scheme 1.
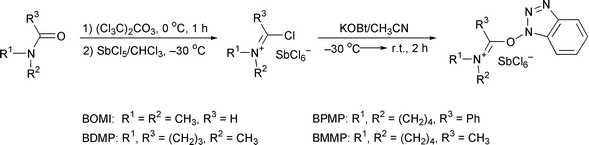 | ||
| Scheme 1 Synthesis of HOBt-derived immonium-type coupling reagents. | ||
It was of great interest that determination of the solid state structures of the representative reagents of HOBt-derived immonium salts, BOMI and BDMP, by X-ray techniques showed that these compounds crystallized as the N-substituted form (Fig. 1), rather than as the previously assigned O-substituted form.1,2 However, according to general chemical knowledge, these HOBt-derived immonium salts should be present in the O-form, hence the formation of these derivatives from the corresponding chloroimmonium salts was a typical nucleophilic substitution reaction, and the oxygen of the nucleophilic species benzotriazolyloxy anion possessed higher electron density than the nitrogen in position 3, which resulted in the nucleophilic center being at the oxygen rather than the nitrogen.
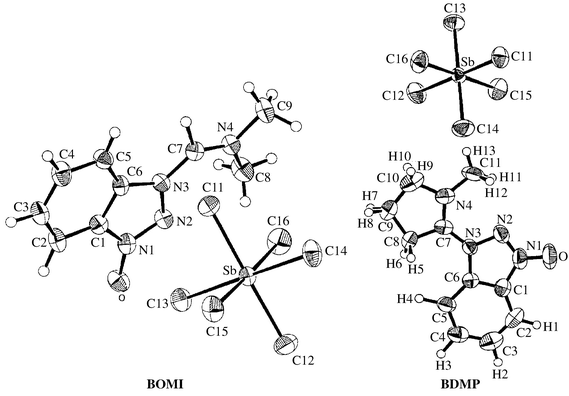 | ||
| Fig. 1 X-Ray crystal structures of HOBt-based immonium salts BOMI and BDMP. | ||
A summary of the crystal structure determination of HOBt-based immonium salts BOMI and BDMP is presented in Table 1. C–H distances of these molecules fall in the range 0.904 to 1.094 Å for BOMI and 0.930 to 1.146 Å for BDMP. Sb–Cl distances fall in the range 2.339 to 2.391 Å for BOMI and 2.357 to 2.383 Å for BDMP, with a mean value of 2.363 Å for BOMI and 2.364 Å for BDMP. Some important bond lengths and torsion angles are listed in Table 2. Comparing BOMI and BDMP with N-oxide 16, the exocyclic bonds from N3 and N1 are much shorter in BOMI (0.133 and 0.064 Å, respectively) and BDMP (0.121 and 0.067 Å, respectively), in keeping with a partial N3, C7 double-bond character due to the imine delocalization in BOMI and BDMP, with an accompanying electron shift from the N-oxide oxygen towards the triazole ring. Other bonds joining these units are also moderately adjusted in the two molecules. Among the two immonium salts, various bonds are of similar lengths except that the length of the reaction-mediating N3–C7 bond of BDMP is a little longer than that of BOMI; moreover, the intraannular C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) N+ double bond is to some extent constrained, reflected by the bond angles of BDMP. These geometric properties of BDMP may indirectly explain why BDMP demonstrates higher reactivity than BOMI in peptide synthesis.
N+ double bond is to some extent constrained, reflected by the bond angles of BDMP. These geometric properties of BDMP may indirectly explain why BDMP demonstrates higher reactivity than BOMI in peptide synthesis.
| Compound | BOMI | BDMP |
|---|---|---|
| Crystal data | ||
| Chemical formula | C9H11N4OCl6Sb | C11H13N4OCl6Sb |
| Formula weight | 525.68 | 551.72 |
| Crystal system | Triclinic | Monoclinic |
| Space group | P1 (#2) | P21/n (#14) |
| Lattice parameters | ||
| a/Å | 9.058(2) | 9.570(3) |
| b/Å | 15.729(4) | 14.443(1) |
| c/Å | 6.598(1) | 14.261(2) |
| α/° | 94.21(2) | |
| β/° | 98.99(2) | 98.99(2) |
| γ/° | 73.38(2) | |
| V/Å3 | 889.4(4) | 1907.6(6) |
| Z | 2 | 4 |
| μ (MoKα)/cm−1 | 24.50 | 22.89 |
| Data collection | ||
| Temperature/°C | 20.0 | 20.0 |
| Reflections collected | 2943 | 3937 |
| Independent reflections | 2727 (Rint = 0.076) | 3709 (Rint = 0.020) |
| Observed reflections | 2247 [I > 3.00σ (I)] |
2355 [I > 2.50σ (I)] |
| Bond lengths | Bond angles | Torsion angles | |||||||
|---|---|---|---|---|---|---|---|---|---|
| BOMI | BDMP |
1610 |
BOMI | BDMP | BOMI | BDMP | |||
| O1–N1 | 1.251(7) | 1.248(5) | 1.315(2) | N3–C7–N4 | 127.7(7) | 125.7(4) | N2–N3–C7–C4 | 11(1) | 1.9(7) |
| N1–N2 | 1.309(8) | 1.303(5) | 1.315(2) | N4–C7–C8 | — | 112.7(4) | N1–N2–N3–C7 | 173.9(7) | −176.6(4) |
| N2–N3 | 1.375(8) | 1.397(5) | 1.341(2) | C7–N4–C10 | — | 111.9(4) | C1–C6–N3–N7 | 173.9(6) | 176.6(4) |
| N3–C6 | 1.413(9) | 1.406(5) | 1.369(2) | C7–N4–C11 | — | 130.9(4) | N3–C7–N4–C11 | — | 3.2(8) |
| C6–C1 | 1.379(10) | 1.363(6) | 1.392(3) | C7–N4–C9 | 118.3(6) | — | N4–C7–N3–C6 | −175.6(7) | −174.8(4) |
| C1–N1 | 1.423(10) | 1.407(6) | 1.369(2) | N4–C10–C9 | — | 105.4(4) | C6–N3–C7–C8 | — | 6.6(7) |
| N3–C7 | 1.348(9) | 1.360(5) | 1.481 | C8–C9–C10 | — | 106.1(4) | O–N1–N2–N3 | −177.7(6) | 179.4(4) |
| C7–N4 | 1.289(10) | 1.291(5) | — | C7–C8–C9 | — | 104.0(4) | N3–C6–C5–C4 | −179.5(8) | 179.1(5) |
Semiempirical PM3 calculations and the prediction of the structures of HOBt-based onium salts
In the X-ray studies of HOBt-based immonium-type coupling reagents, BOMI and BDMP, we found both of these compounds crystallized unexpectedly in the N-substituted form rather than the isomeric O-form. In order to explain this phenomenon and try to find a general rule that can predict the structures of these HOBt-based onium salts, molecular orbital calculations were carried out. With regard to the size and the structures of these compounds, we adopted semiempirical methods in this study, and only the carbocation moieties of these onium salts were calculated. The PF6− or SbCl6− counterions were not taken into account in order to simplify the calculation procedures. Since these anions do not participate in the rearrangement of the HOBt-based carbocations and both of the tautomers contain the same counterion, the relative stabilities and energies of these carbocation moieties reflect those of the whole molecules. In comparison to AM1 and MNDO, the PM3 semiempirical method can more accurately handle this kind of problem. The method was affirmed to be more reliable than AM1 by comparison with ab initio calculations.11 Moreover, these HOBt-based onium salts are nitrogen-rich and oxygen-containing, therefore the PM3 method can give better results than the AM1 method.12,13Consequently, the calculation results indicated that the N-substituted forms of reagents BOMI 1 and BDMP 2 were 10.5 and 14.5 kcal mol−1, respectively, more stable than the corresponding O-substituted tautomers using the PM3 method (Table 3). The N-form of the uronium/aminium salt HBTU 5 was also more stable than the corresponding O-form, which was coincident with the X-ray structure of HBTU. In the case of other HOBt-based onium salts, PM3 calculations indicate that the N-oxides are more stable than the corresponding O-forms, and the relative energies (ΔHf) are much higher than that of HBTU. Therefore, it is reasonable to predict that the HOBt-based onium salts BPMP, BMMP, HBMDU and HBPyU would crystallize as the N-oxides (N-form). We also tried using the AM1 semiempirical method to evaluate the relative stabilities of the N-forms and O-forms of these onium salts; the calculation results showed that the O-substituted forms were more stable (Table 3). These results obviously contradict the experimental observations, which was probably due to the AM1 method overestimating the instabilities of the N-oxides.
a
| Compoundb |
Heat of formation Hf /kcal mol−1 | Total energy U/au | Compoundb |
Heat of formation Hf /kcal mol−1 | Total energy U/au | Relative energyc
ΔHf /kcal mol−1 |
Relatively stable tautomer | X-Ray structure |
|---|---|---|---|---|---|---|---|---|
| a Calculated by PM3 semiempirical method. b A++ refer to the carbocation moieties of these HOBt-based onium salts. c Relative stabilities (ΔHf) = Hf (N-form) − Hf (O-form). | ||||||||
| 1A++ (N-form) | 250.9 | −79.655 (−89.617) | 1A++ (O-form) | 261.4 | −79.638 (−89.642) | −10.5 | N-form | N-form |
| (AM1 method) | (312.6) | −89.519 (−100.072) | (297.2) | −89.496 (−100.079) | (15.4) | (O-form) | ||
| 2A++ (N-form) | 240.2 | −117.889 | 2A++ (O-form) | 254.7 | −117.872 | −14.5 | N-form | N-form |
| (AM1 method) | (296.0) | −95.013 | (291.3) | −94.999 | (4.7) | (O-form) | ||
| 3A++++ (N-form) | 267.3 | −97.162 (−109.105) | 3A++++ (O-form) | 278.0 | −97.155 (−109.187) | −10.7 | N-form | Unknown |
| 4A++++ (N-form) | 235.4 | −96.041 | 4A++++ (O-form) | 244.4 | −96.027 | −9.0 | N-form | Unknown |
| 5A++++ (N-form) | 252.6 | −116.880 | 5A++++ (O-form) | 256.6 | −116.863 | −4.0 | N-form | N-form |
| (AM1 method) | (353.0) | (301.1) | (51.9) | (O-form) | ||||
| 6A+++ (N-form) | 248.8 | 6A+++ (O-form) | 257.1 | −8.3 | N-form | Unknown | ||
| 7A+++ (N-form) | 236.9 | 7A+++ (O-form) | 247.8 | −10.9 | N-form | Unknown | ||
Although the above PM3 theoretical calculation results were consistent with experimental observations, these encouraging results should be treated with caution since they refer to gas-phase calculations. However, crystals of these reagents were obtained from solution and peptide coupling reactions mediated by these onium-type reagents were also carried out in the solution phase. Generally, The N+–O− functional group in the N3-substituted onium molecules can be strongly stabilized in a polar solvent. For example, in the polar solvent acetone, the relative permittivity of which is 20.56, the model compound, 3H-1,2,3-triazole N-oxide, was about 35 kcal mol−1 more stable than in the gas phase according to ab initio calculations using Tomasi’s SCRF procedure.11 As for these much more complicated HOBt-derived uronium- and immonium-type molecules, the quantitative calculations including the solvation effect were beyond our limitations. But the general rules developed by model compound calculations indicated that the relative energies of the N-substituted and O-substituted forms of immonium- and uronium-type reagents would increase in solution due to the solvation effect, compared to the gas phase. That means the N-substituted forms of onium salts would be even more stable than the corresponding O-substituted forms in solution.
The mechanism of HOBt-based immonium salt mediated coupling reactions and the rearrangement of benzotriazolyl esters of Nα-protected amino acids and peptides
To study the influence of the isomerization of the HOBt-based immonium salts and their analogues on the coupling reaction involved in peptide synthesis, the mechanism of peptide coupling reactions mediated by these immonium-type reagents was investigated. Under basic reaction conditions, the carboxylate anion initially attacks the α-carbon atom of the immonium salt to form an unstable acyloxyimmonium intermediate with the release of benzotriazolyloxy anion (BtO−). Then, the BtO− nucleophile attacks the acyloxyimmonium intermediate via two pathways to afford the corresponding benzotriazolyl ester and a small amount of N3-acylbenzotriazole 1-oxide intermediate. The benzotriazolyl ester intermediate can spontaneously rearrange into the corresponding N-acylated isomer. If an amino component was added into the reaction system, the two forms of the active intermediates would react with the amine to give the corresponding amide. Otherwise, the intermediates can be isolated as the final products (Fig. 2). | ||
| Fig. 2 Proposed mechanism of coupling reactions mediated by the HOBt-based immonium type reagents. | ||
The arrangement of the benzotriazolyl ester was further studied by UV, IR, 1H NMR, and HPLC monitoring. For instance, in the synthesis of hindered peptide Fmoc-MeVal-Sar-OBzl by treating Fmoc-MeVal-OH and Sar-OBzl·HCl with reagent BDMP in the presence of 2,6-lutidine at −10 °C, the active intermediate Fmoc-MeVal-OBt was observed and isolated besides the dipeptide. TLC monitoring of the reaction mixture showed that the less polar O-acylated form of Fmoc-MeVal-OBt was first formed, which further rearranged into the more polar N-acylated form. When the O-form isomer was separated by preparative thin layer chromatography, the obtained product spontaneously rearranged into two forms again during storage. A similar phenomenon was also observed during the preparation of the HOBt-derived ester Z-Gly-Phe-OBt.
The UV spectra of Z-Gly-Phe-OBt indicated that this compound did exist, to some extent, in the N-acylated form, as revealed by the characteristic absorptions of N-oxide at 328 and 342 nm, besides existing as the O-isomer, characterized by the absorption at 1805–1825 cm−1 in IR spectra. Moreover, the proportion of the N-substituted form in CH3CN was higher than in CHCl3 since the more polar CH3CN can better stabilize the N-oxide isomer due to the solvation effect (Table 4). The 1H NMR spectra of Z-Gly-Phe-OBt in CDCl3 also demonstrated the existence of the two forms; however, only the N-acylated form of the compound was observed in the more polar solvent acetone-d6 which can strongly stabilize the N-oxide, and the spectrum becomes much simplified. In the case of compound Fmoc-MeVal-OBt, similar UV, IR, 1H NMR and 13C NMR spectral features were also observed, but the extent and speed of the rearrangement were smaller and slower than those of Z-Gly-Phe-OBt as shown in Table 4.

| UV | IR | |||||||
|---|---|---|---|---|---|---|---|---|
| CH3CN | CHCl3 | CH3CN | CHCl3 | |||||
| Compound | λ max/nm |
Aa |
λ max/nm |
Aa |
ν
(CO)/cm−1 |
ν
(CO)/cm−1 |
||
| a Related values based upon the characteristic absorption of N-oxide at 328 nm. | ||||||||
| 8 | 282 | O-form | 1.238 | 286 | 6.455 | 1821 (m) | O-form | 1818 (m) |
| 328 | N-form | 1.000 | 328 | 1.000 | 1729 (vs) | N-form (partly overlapped) | 1732 (vs) | |
| 342 | N-form | 0.960 | 342 | 0.876 | 1685 (m) | 1689 (vs) | ||
| 9 | 288 | O-form | 3.184 | 289 | 3.616 | 1826 (m) | O-form | 1817 (m) |
| 328 | N-form | 1.000 | 328 | 1.000 | 1708 (vs) | N-form (overlapped) | 1702 (vs) | |
| 342 | N-form | 0.984 | 344 | 0.973 | ||||
Molecular orbital calculations and the prediction of the structures of benzotriazolyl esters
To explain and study the above rearrangement reactions of the benzotriazolyl esters of Nα-protected amino acids and peptides, we calculated the relative stabilities of the N-form and the O-form of these benzotriazolyl esters. Semiempirical PM3 calculations revealed that the N-oxide of compound 8 was more stable than the corresponding O-acylated form by 3.74 kcal mol−1 (Table 5). Therefore, we can infer that the O-form benzotriazolyl ester intermediate of immonium salt mediated coupling is kinetically preferred, which was proved by the TLC monitoring of the activation reactions using HOBt-derived immonium-type coupling reagents. When the reactions were carried out at low temperature (−10 °C), TLC, UV and IR analysis indicated that the O-form benzotriazolyl ester of the carboxylic component was initially generated and reacted immediately with the amino component to give the desired peptide. If the amino component was sterically hindered or was not added at all, the benzotriazolyl ester intermediate gradually rearranged into the thermodynamically stable N-oxide at room temperature, which can be monitored by UV. As for the sterically hindered Fmoc-MeVal-OBt, PM3 calculations showed that the N-acylated form was 1.47 kcal mol−1 less stable than the corresponding O-form. On the other hand, these theoretical results referred to the calculations in vacuo and the solvation effect was not taken into account while the experimental observations shown in Table 4 reflect the equilibria in solution. The N+–O− polar moieties of the N-form molecules could be further stabilized in solution, especially in a polar solvent; therefore, the N-form of compound 9 also exists in solution. Generally, the N-acylated forms of benzotriazolyl esters of Nα-protected amino acids and peptides, such as 8, 9 and 10, were thermodynamically stable in solution as shown by both calculation results and the solvation effect, as well as proved by experimental observations. a
a
| Compound | Heat of formation Hf /kcal mol−1 | Total energy U/au | Compound | Heat of formation Hf /kcal mol−1 | Total energy U/au | Relative energyb
ΔHf /kcal mol−1 |
Relatively stable tautomer | X-Ray structure |
|---|---|---|---|---|---|---|---|---|
| a Calculated by PM3 semiempirical method. b Relative stabilities (ΔHf) = Hf (N-form) − Hf (O-form). | ||||||||
| 8 (N-form) | −14.36 | −205.483 | 8 (O-form) | −10.62 | −205.477 | −3.74 | N-form | Unknown |
| 9 (N-form) | 22.68 | −199.134 | 9 (O-form) | 21.21 | −199.136 | 1.47 | O-form | Unknown |
| 10 (N-form) | −6.51 | −155.433 | 10 (N-form) | −4.27 | −155.429 | −2.24 | N-form | Unknown |
| 11 (N-form) | 69.64 | −97.779 | 11 (O-form) | 76.04 | −97.769 | −6.40 | N-form |
N-form10 |
| 12 (N-form) | 47.48 | −127.109 | 12 (O-form) | 50.56 | −127.104 | −3.08 | N-form |
N-form14 |
| 13 (N-form) | 14.25 | −91.390 | 13 (N-form) | 13.63 | −91.391 | 0.62 | O-form |
O-form14 |
| 14 (N-form) | 35.18 | −117.282 | 14 (O-form) | 36.14 | −117.281 | −0.96 | N-form |
N-form14 |
| 15 (N-form) | −10.53 | −105.402 | 15 (O-form) | −8.02 | −105.398 | −2.51 | N-form |
N-form14 |
| 16 (N-form) | −17.01 | −99.926 | 16 (O-form) | −9.34 | −99.913 | −7.67 | N-form |
N-form10 |
We investigated the possibility of theoretical calculations using the semiempirical PM3 method predicting the crystal structures of benzotriazolyl esters. The benzotriazolyl esters 11–15 and acid 16 for which X-ray structures have been reported were selected as candidates, so that we could evaluate the accuracy of the calculation results. Encouragingly, all the calculation results were coincident with the experimental observations as shown in Table 5. Therefore, a useful way to predict the X-ray structures of these kind of compounds would be by PM3 calculations. It is worth pointing out that the above calculation results should be treated with caution since they just reflect the relative stabilities in vacuo; the crystal packing contributions were not taken into account.
Conclusions
X-Ray analysis revealed that HOBt-derived immonium-type peptide coupling reagents BOMI and BDMP crystallize as N-oxides, rather than the previously assigned O-substituted forms. Semiempirical PM3 calculations indicated that the N-forms of HOBt-based onium salts were more stable than the corresponding isomeric O-forms in vacuo. Other HOBt-derived onium salts, such as BPMP, BMMP, HBMDU and HBPyU, for which X-ray data have not been obtained, are proposed to crystallize as N-oxides as suggested by the PM3 calculation results. The reaction mechanism for coupling reactions mediated by these HOBt-derived immonium salts was investigated. The rearrangement of the benzotriazolyl ester intermediate generated during the coupling reaction was observed and studied by both experimental observations and theoretical calculations. TLC, UV, and IR monitoring of model coupling reactions showed that the key reaction mediators, benzotriazolyl esters of the carboxylic components, were initially generated as the O-acylated forms, then spontaneously rearranged into the corresponding N-substituted forms, a finding which could be explained well by PM3 calculations. The crystal structures of these benzotriazolyl esters can be predicted well by the semiempirical PM3 calculations. But the above calculation results should be treated with caution since they just reflect the relative stabilities in vacuo; the solvation effects and the crystal packing contributions were not considered.Experimental
General
Melting points were taken on a digital melting point apparatus and are uncorrected. Infrared spectra were recorded on a Shimadzu IR-440 spectrometer. Mass spectra were recorded on HP 5989A and VG QUATTRO mass spectrometers. 1H NMR spectra were recorded on Bruker AM 300 (300 MHz) and Bruker DRX-400 (400 MHz) instruments using TMS as the internal standard. Combustion analysis for elemental composition was carried out on an Italy MOD 1106 analyzer. Optical rotations were determined using a Perkin-Elmer 241 MC polarimeter. Flash column chromatography was performed with 300–400 mesh silica gel, and analytical thin layer chromatography was performed on precoated silica gel plates (60F-254) with the systems (v/v) indicated. Solvents and reagents were purified by standard methods as necessary. Amino acids were L-configuration if not otherwise stated. HOBt, SbCl5 and DCC were purchased from Aldrich Chemical Co. (Milwaukee, WI), and used without purification. Fmoc-N-Methylvaline was synthesized by the procedure of Freidinger et al.15 The synthesis and spectral properties of compounds BOMI3 and BDMP4 have been described previously.
Synthesis of compounds
N+). Anal. Calcd. for C17H17Cl6N4OSb: C, 32.52; H, 2.71; N, 8.92%. Found: C, 32.23; H, 2.61; N, 8.84%.
N+). Anal. Calcd. for C12H15Cl6N4OSb: C, 25.48; H, 2.67; N, 9.90%. Found: C, 25.33; H, 2.49; N, 9.85%.
Crystal structure determination and refinement
Single crystals of 1 and 2 were obtained from acetone–ether under argon atmosphere. Crystal data and the conditions of data collection are summarized in Table 1. The structure of BOMI was solved by heavy-atom Patterson methods16 and expanded using Fourier techniques;17 the structure of BDMP was solved by direct methods with the SHELXS 86 software package,18 and expanded using Fourier techniques. Neutral atom scattering factors were taken from Cromer and Waber.19 The values for the mass attenuation coefficients are those of Creagh and Hubbell.20 All calculations were performed using the teXsan21 crystallographic software package of Molecular Structure Corporation.21
Calculations
Semiempirical molecular orbital calculations were carried out using Hyperchem for Windows from Hypercube with PM3 or AM1 parametrizations. Geometry optimizations were first performed using standard methods of molecular dynamics with a force field MM+, then further performed by AM1 or PM3 calculations with the Polak–Ribiere conjugate gradient method. Several starting geometries were used for the geometry optimizations to ensure the optimized structures correspond to global minima.Acknowledgements
We thank the National Natural Science Foundation for the support of this work.References
- P. Li and J. C. Xu, Tetrahedron Lett., 1999, 50, 3605 CrossRef CAS.
- P. Li and J. C. Xu, Chem. Lett., 1999, 1163 CrossRef CAS.
- P. Li and J. C. Xu, J. Pept. Res., 2000, 55, 110 Search PubMed.
- P. Li and J. C. Xu, Chin. J. Chem., 2000, 18, 85 Search PubMed.
- P. Li and J. C. Xu, J. Org. Chem., 2000, 65, 2951 CrossRef CAS.
- V. Dourtoglou, J. C. Ziegler and B. Gross, Tetrahedron Lett., 1978, 1269 CrossRef CAS.
- Y. Kiso, Y. Fujiwara, T. Kimura, A. Nishitani and K. Akaji, Int. J. Pept. Protein Res., 1992, 40, 308 Search PubMed.
- S. Q. Chen and J. C. Xu, Tetrahedron Lett., 1992, 33, 647 CrossRef CAS.
- I. Abdelmoty, F. Albericio, L. A. Carpino, B. M. Foxman and S. A. Kates, Lett. Pept. Sci., 1994, 1, 57 Search PubMed.
- S. Vagarajan, S. R. Wilson and K. L. Rinehart, Jr., J. Org. Chem., 1985, 50, 2174 CrossRef.
- E. Anders, A. R. Katritzky, N. Malhotra and J. Stevens, J. Org. Chem., 1992, 57, 3698 CrossRef CAS.
- R. Karaman, J. T. L. Huang and J. L. Fry, J. Comput. Chem., 1990, 11, 1009 CrossRef CAS.
- S. Uchiyama, T. Santa and K. Imai, J. Chem. Soc., Perkin Trans. 2, 1999, 569 RSC.
- J. Singh, R. Fox, M. Wong, T. P. Kissick and J. L. Moniot, J. Org. Chem., 1988, 53, 205 CrossRef CAS.
- R. M. Freidinger, J. S. Hinkle, D. S. Perlow and B. H. Arison, J. Org. Chem., 1983, 48, 77 CrossRef CAS.
- PATTY: P. T. Beurskens, G. Admiraal, G. Beurskens, W. P. Bosman, S. Garcia-Granda, R. O. Gould, J. M. M. Smits and C. Smykalla (1992). The DIRDIF program system, Technical Report of the Crystallography Laboratory, University of Nijmegen, Netherlands. Search PubMed.
- DIRDIF92: P. T. Beurskens, G. Admiraal, G. Beurskens, W. P. Bosman, S. Garcia-Granda, R. O. Gould, J. M. M. Smits and C. Smykalla (1992). The DIRDIF program system, Technical Report of the Crystallography Laboratory, University of Nijmegen, Netherlands. Search PubMed.
- G. M. Sheldrick , Crystallographic Computing 3, G. M. Sheldrick, C. Kruger and R. Goddard, Eds., Clarendon Press, Oxford, UK, 1985, p. 175. Search PubMed.
- D. T. Cromer and J. T. Waber , International Tables for X-ray Crystallography, Vol. IV, the Kynoch Press, Birmingham, England, 1974, Table 2.2 A. Search PubMed.
- D. C. Creagh and J. H. Hubbell , International Tables for Crystallography, Vol. C, Kluwer Academic Publishers, Boston, 1992, Table 4.2.4.3, p. 200. Search PubMed.
- teXsan: Crystal Structure Analysis Package, Molecular Structure Corporation, 1992. Search PubMed.
Footnotes |
| † Nomenclature and symbols of amino acids and peptides generally follow the recommendations of the IUPAC–IUB Joint Commission of Biochemical Nomenclature in: Pure Appl. Chem., 1984, 56, 595. The following additional abbreviations are used: Aib, α-aminoisobutyric acid; BDMP, 5-(1H-benzotriazol-1-yl)-3,4-dihydro-1-methyl-2H-pyrrolium hexachloroantimonate N-oxide; BMMP, [(1H-benzotriazol-1-yloxy)methylmethylene]pyrrolidinium hexachloroantimonate; BOMI, N-(1H-benzotriazol-1-ylmethylene)-N-methylmethanaminium hexachloroantimonate N-oxide; BPMP, [1-(1H-benzotriazol-1-yloxy)phenylmethylene]pyrrolidinium hexachloroantimonate; HBMDU, O-(1H-benzotriazol-1-yl)-N,N′-dimethyl-N,N′-dimethyleneuronium hexafluorophosphate; HBPyU, O-(1H-benzotriazol-1-yl)-N,N,N′,N′-bis(tetramethylene)uronium hexafluorophosphate; HBTU, N-[(1H-benzotriazol-1-yl)(dimethylamino)methylene]-N-methylmethanaminium hexafluorophosphate N-oxide; HOBt, 1-hydroxybenzotriazole. |
| ‡ CCDC reference number 188/276. See http://www.rsc.org/suppdata/p2/b0/b003930f/ for crystallographic files in .cif format. |
| This journal is © The Royal Society of Chemistry 2001 |