A new synthetic approach to porphyrin-α-diones and a -2,3,12,13-tetraone: building blocks for laterally conjugated porphyrin arrays
Vinich
Promarak
and
Paul L.
Burn
*
The Dyson Perrins Laboratory, University of Oxford, South Parks Road, Oxford, UK OX1 3QY
First published on 11th December 2000
Abstract
We report the first use of the Dess–Martin periodinane (DMP) for the oxidation of an arylamine to an α-dione. The methodology is illustrated by the preparation of free-base and metal chelated porphyrin-α-diones in up to 52% yield by oxidation of 2-aminoporphyrins with the DMP. We also found that DMP could be used to oxidise a 2,3-diaminoporphyrin to a porphyrin-α-dione in good yield with other free-base diaminoporphyrin isomers forming a trans-porphyrintetraone in 20% yield.
Introduction
The development of synthetic porphyrin arrays has attracted much interest because of the important role porphyrins play in electron transfer processes in biological systems. Utilisation of these properties could lead to the development of new classes of organic materials for molecular wires,1–3 photovoltaic devices![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 4–8 and non-linear optics.6,9–11 Recently many types of porphyrin arrays have been reported where the porphyrin rings are covalently linked through either their meso-positions3,4,7–10,12–14 or β-pyrrolic positions.1,15–25 Of the porphyrin arrays linked through the β-pyrrolic positions most have involved condensation reactions of porphyrin-α-diones17,21–25 or tetraones1,15 with benzene-1,2,4,5-tetraamine. The traditional route to porphyrin-α-diones involved photo-oxidation of a 2-amino-5,10,15,20-tetraphenylporphyrin followed by hydrolysis of the resultant keto-imino chlorin.26 This approach was later extended and a mixture of diamino-5,10,15,20-tetrakis(3′,5′-di-tert-butylphenyl)porphyrins were photo-oxidised to form two different tetraones, the trans-2,3,12,13-tetraone and cis-2,3,7,8-tetraone.15 Free-base and metallo 2-hydroxy-5,10,15,20-tetraphenylporphyrins have also been oxidised to porphyrin-α-diones.27 However, the most facile method so far reported for preparation of porphyrin-α-diones is the oxidation of 2-hydroxyporphyrins with the Dess–Martin periodinane (DMP).28 DMP was successfully used for the preparation of porphyrin-α-diones from free-base and metal chelated 2-hydroxy-5,10,15,20-tetrakis(3′,5′-di-tert-butylphenyl)porphyrins.28 In the case of the free-base 2-hydroxyporphyrin, the reaction mechanism for the DMP oxidation was considered to be similar to the oxidation of 1,3-dicarbonyls to tricarbonyls, where the oxidation was assumed to occur through the enol form of the dicarbonyl.29 However, there was no evidence in the case of the copper and zinc 2-hydroxyporphyrins for significant keto–enol tautomerism, suggesting that DMP could oxidise “phenols” under mild conditions. In contrast to the 2-hydroxy-tetra-meso-phenylporphyrins the corresponding 2-aminoporphyrins do not show an imine tautomeric form by 1H NMR.30 We therefore considered that if porphyrin “phenols” could be oxidised by DMP then the corresponding porphyrin “anilines” might also be susceptible to DMP oxidation to a porphyrin-α-dione. In this paper, we report a new method for the preparation of porphyrin-α-diones from 2-aminoporphyrins and 2,3-diaminoporphyrins, and the extension of this methodology to the synthesis of free-base porphyrin-2,3,12,13-tetraone. For comparison with the DMP oxidation of 2-hydroxyporphyrins to porphyrin-α-diones we chose copper, zinc and free-base 2-aminoporphyrins as our substrates.
4–8 and non-linear optics.6,9–11 Recently many types of porphyrin arrays have been reported where the porphyrin rings are covalently linked through either their meso-positions3,4,7–10,12–14 or β-pyrrolic positions.1,15–25 Of the porphyrin arrays linked through the β-pyrrolic positions most have involved condensation reactions of porphyrin-α-diones17,21–25 or tetraones1,15 with benzene-1,2,4,5-tetraamine. The traditional route to porphyrin-α-diones involved photo-oxidation of a 2-amino-5,10,15,20-tetraphenylporphyrin followed by hydrolysis of the resultant keto-imino chlorin.26 This approach was later extended and a mixture of diamino-5,10,15,20-tetrakis(3′,5′-di-tert-butylphenyl)porphyrins were photo-oxidised to form two different tetraones, the trans-2,3,12,13-tetraone and cis-2,3,7,8-tetraone.15 Free-base and metallo 2-hydroxy-5,10,15,20-tetraphenylporphyrins have also been oxidised to porphyrin-α-diones.27 However, the most facile method so far reported for preparation of porphyrin-α-diones is the oxidation of 2-hydroxyporphyrins with the Dess–Martin periodinane (DMP).28 DMP was successfully used for the preparation of porphyrin-α-diones from free-base and metal chelated 2-hydroxy-5,10,15,20-tetrakis(3′,5′-di-tert-butylphenyl)porphyrins.28 In the case of the free-base 2-hydroxyporphyrin, the reaction mechanism for the DMP oxidation was considered to be similar to the oxidation of 1,3-dicarbonyls to tricarbonyls, where the oxidation was assumed to occur through the enol form of the dicarbonyl.29 However, there was no evidence in the case of the copper and zinc 2-hydroxyporphyrins for significant keto–enol tautomerism, suggesting that DMP could oxidise “phenols” under mild conditions. In contrast to the 2-hydroxy-tetra-meso-phenylporphyrins the corresponding 2-aminoporphyrins do not show an imine tautomeric form by 1H NMR.30 We therefore considered that if porphyrin “phenols” could be oxidised by DMP then the corresponding porphyrin “anilines” might also be susceptible to DMP oxidation to a porphyrin-α-dione. In this paper, we report a new method for the preparation of porphyrin-α-diones from 2-aminoporphyrins and 2,3-diaminoporphyrins, and the extension of this methodology to the synthesis of free-base porphyrin-2,3,12,13-tetraone. For comparison with the DMP oxidation of 2-hydroxyporphyrins to porphyrin-α-diones we chose copper, zinc and free-base 2-aminoporphyrins as our substrates.
Results and discussion
5,10,15,20-Tetrakis(3′,5′-di-tert-butylphenyl)porphyrin was chosen for this study as the 3,5-di-tert-butylphenyl meso-substituents provide good solubility to the porphyrin. The synthetic pathways followed for the preparation of the porphyrin-α-diones and porphyrintetraone are illustrated in Scheme 1. To form the critical aminoporphyrin intermediates we followed literature procedures.15,26 Chelation of copper into free-base porphyrin 1 was achieved in a quantitative yield by treatment with cupric acetate monohydrate in a dichloromethane–methanol mixture heated at reflux. After the second step, the nitration, the pathways for formation of the porphyrin-α-diones and porphyrintetraone diverged. For the preparation of the porphyrin-α-diones, copper porphyrin 2 was mono-nitrated with a solution of nitrogen dioxide in light petroleum with the reaction followed closely by thin-layer chromatography. Under these conditions the mono-nitrated copper 2-nitroporphyrin 3 could be isolated in 96% yield.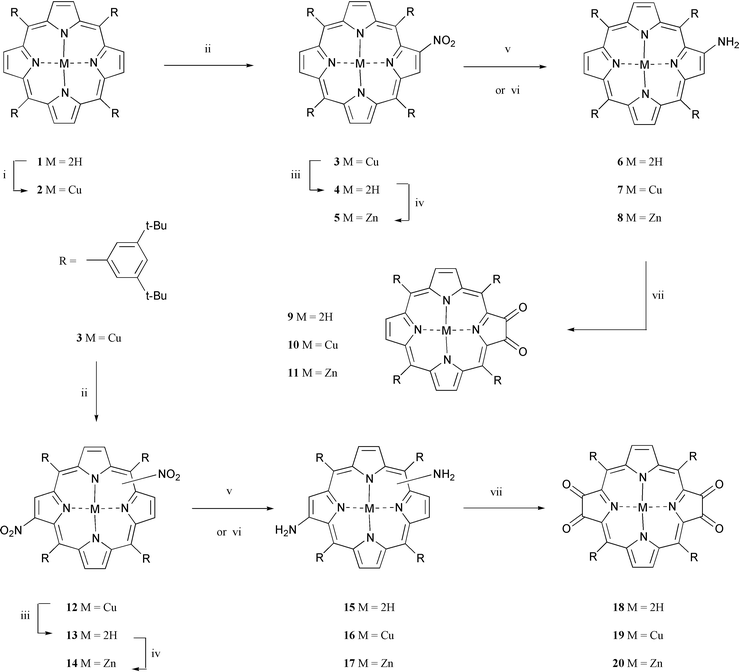 | ||
| Scheme 1 Reagents and conditions: i, Cu(OAc)2·H2O, CH2Cl2, MeOH, reflux, 1 h; ii, NO2 in light petroleum, CH2Cl2, rt; iii, conc. H2SO4, CH2Cl2, rt; iv, Zn(OAc)2·2H2O, CH2Cl2, MeOH, reflux, 1.5 h; v, SnCl2·2H2O, conc. HCl, CH2Cl2, in the dark, rt, 2–4 d; vi, NaBH4, 10% Pd–C, CH2Cl2–MeOH (4∶1), rt, 1 h; vii, DMP, CH2Cl2, rt, in the dark, then aqueous HCl. | ||
For the preparation of the porphyrin-α-diones 9, 10, and 11, the free-base and metal chelated 2-amino porphyrins were prepared from 3 in the following manner. For the preparation of the free-base 2-aminoporphyrin (6), porphyrin 3 was demetallated with concentrated sulfuric acid in dichloromethane to give the free-base 2-nitroporphyrin 4 in quantitative yield. 4 was then reduced with a mixture of tin(II) chloride dihydrate and concentrated hydrochloric acid in dichloromethane to give the crude 2-aminoporphyrin 6. The free-base 2-aminoporphyrin is very susceptible to photo-oxidation26,31 and although 1H NMR and infrared spectra could be obtained they often contained signals that corresponded to the free-base porphyrin-α-dione 9. Critically the infrared spectrum showed that there was no nitro group remaining after the reduction. Although we did not isolate 6 in a pure form before the DMP oxidation, thin-layer chromatography clearly showed that there was no porphyrin-α-dione present. The copper- and zinc-chelated 2-aminoporphyrins 7 and 8 were prepared in a slightly different manner from 6. For the zinc derivative, the free-base 2-nitroporphyrin 4 was metallated with zinc acetate dihydrate in a dichloromethane–methanol mixture heated at reflux.31 Under these conditions zinc 2-nitroporphyrin 5 was isolated in 95% yield. Reduction of 5 was achieved with sodium borohydride in the presence of 10% palladium on activated carbon in a dichloromethane–methanol mixture (4∶1) to give the zinc-chelated 2-aminoporphyrin 8 in ≈96% yield.31 As in the case of 6 we did not isolate 8 in a pure form after chromatography. However, 1H NMR and infrared spectroscopy clearly showed that 8 had been formed. In fact, it was interesting to note that, although 8 was susceptible to photo-oxidation31 on silica, the 1H NMR and infrared spectra both had little of the porphyrin-α-dione present in the samples suggesting that the zinc derivative was more photochemically stable than the free-base compound. Again the infrared spectrum showed the absence of any remaining nitro groups. The copper 2-nitroporphyrin 3 was reduced to form the corresponding copper 2-aminoporphyrin in an 89% yield. Unlike the free-base and zinc-chelated 2-aminoporphyrin derivatives we were able to isolate the copper 2-aminoporphyrin 7 as a single compound by chromatography over silica. The infrared spectrum of 7 clearly showed the presence of the amine group and the absence of absorptions from the nitro moiety. The susceptibility of 6–8 to photo-oxidation follows the same trend we observed with the photo-oxidation of the 2-hydroxyporphyrins. For the 2-hydroxyporphyrins the copper derivative was also the most resilient to oxidation with the free-base being the least.28 Nevertheless, to ensure that the formation of the porphyrin-α-diones in this study was only due to the treatment with DMP we did not purify the 2-aminoporphyrins, 6–8, before the oxidation step.
The oxidations of 6–8 were all carried out in a similar manner. In each case DMP was added to a solution of the 2-aminoporphyrin and the mixture was stirred at room temperature in the dark. The progress of the reaction was monitored by thin-layer chromatography and when it was judged that the starting material had been consumed dilute hydrochloric acid (for 6 and 7) or water (for 8) was added. The aqueous treatment was included to hydrolyse any imine intermediates that may have formed during the oxidation as in the case of the photo-oxidation of 2-aminoporphyrins.
Therefore, the two-step procedure developed involved the reduction of the 2-nitroporphyrin to the amine and subsequent oxidation with DMP. Under these conditions 4 was converted to the free-base porphyrin-α-dione 9 in a 52% overall yield for the two steps. Moreover, the DMP oxidation of the in situ formed copper 2-aminoporphyrin 7 and zinc 2-aminoporphyrin 8 gave the corresponding copper porphyrin-α-dione 10 and zinc porphyrin-α-dione 11 in yields of 37 and 20% respectively for the two steps. The oxidation of the copper 2-aminoporphyrin was significantly slower and required more oxidant than those of the other two 2-aminoporphyrins, 6 and 8. We believe the lower yields of copper and zinc porphyrin-α-diones are due to competition between the oxidation of the 2-aminoporphyrin and the porphyrin-α-dione formed in the reaction mixture. In the case of the copper 2-aminoporphyrin 7 the sluggish nature of the oxidation leaves the formed porphyrin-α-dione in contact with the oxidant for a longer period of time and it is well known that the α-dione can undergo further oxidative degradations.26 For the zinc 2-aminoporphyrin it may be that the amine group and formed α-dione have similar rates of oxidation. It is interesting to note that in the study on the oxidation of 2-hydroxyporphyrins with DMP the zinc derivative also gave the lowest yield.28
We next investigated the oxidation reaction of diaminoporphyrins 15, 16, and 17 with DMP to give the tetraones 18, 19, and 20 respectively. Copper 2-nitroporphyrin 3 was further nitrated with nitrogen dioxide under the same conditions used for the mono-nitration. The six possible copper dinitroporphyrin 12 isomers, in contrast to the five reported in the literature,15 were isolated in a total yield of 77%. The copper dinitroporphyrin isomers could not be easily separated by column or centrifugal thin-layer chromatography over silica. As a consequence the mixture of copper dinitroporphyrins was then demetallated with concentrated sulfuric acid in dichloromethane to give 13 in a quantative yield, as a mixture of free-base dinitroporphyrins. The free-base dinitroporphyrin isomers could also not be easily separated to give the individual isomers.
To study the formation of the free-base and metal-chelated porphyrintetraones the diaminoporphyrins were synthesised following the procedures used for the 2-aminoporphyrins. The free-base diaminoporphyrins 15 were formed in ≈97% yield by reduction of 13 with stannous chloride dihydrate and hydrochloric acid. The infrared spectrum of 15 clearly showed absorptions at 3485 and 3391 cm−1 corresponding to the amine groups and no absorptions due to the nitro and carbonyl groups were observed. To form 17, 13 was first metallated with zinc acetate dihydrate in a dichloromethane–methanol mixture heated at reflux to give the zinc dinitroporphyrins 14 in 98% yield. The copper, 12, and zinc, 14, were then reduced with sodium borohydride in the presence of 10% palladium on activated carbon to the corresponding diaminoporphyrins 16 and 17. Again the complete reduction of both nitro groups was confirmed by infrared spectroscopy. There were no absorptions due to the nitro groups or indeed carbonyls and the absorptions due to the amine moieties were clearly observed. In addition, the 1H NMR of the crude 17, although complicated, was consistent with the structures. Also, as in the case of the copper 2-aminoporphyrin it was possible to isolate the copper diaminoporphyrins by chromatography over silica. Nevertheless, for all three diaminoporphyrin derivatives the DMP oxidation was carried out immediately after the reduction to avoid any photo-oxidation.
As in the case of the 2-aminoporphyrins the oxidations were carried out by the addition of aliquots of DMP and the reaction stopped when thin-layer chromatography indicated that no starting material remained. For the free-base diaminoporphyrins 15 we found that the “trans” porphyrin-2,3,12,13-tetraone 18 was formed in a yield of 20%. As only two of the six isomers can lead to the “trans” porphyrintetraone the yield of 20% corresponds to a conversion of 60% assuming that all the dinitro isomers are formed in equal amounts. The product was easily separated by column chromatography over silica gel and 1H NMR was identical, except that the NH protons were found at −1.80 rather than −1.32, to that reported in the literature.15 The yield of 18 is similar to that achieved by the photo-oxidation of free-base diaminoporphyrins. Interestingly, as in the case of the photo-oxidation of the free-base diaminoporphyrins the “cis” porphyrintetraone was not isolated. We believe that DMP might not have the right oxidation potential to oxidise the free-base diaminoporphyrins which lead to the non-aromatic “cis” porphyrin-2,3,17,18-tetraone. In the case of the photo-oxidation experiments the “cis” porphyrin-2,3,17,18-tetraone was obtained by photo-oxidising intermediary zinc aminoporphyrin-α-diones.15 We therefore wondered whether the metal-chelated diaminoporphyrins would give rise to the “cis” as well as the “trans” porphyrintetraones. To test this the oxidations of 16 and 17 were carried out by the addition of DMP to the reaction mixture until thin-layer chromatography indicated that no starting material remained. However, in both cases many compounds were found to be present by thin-layer chromatography and after aqueous acid work-up and separation by column chromatography over silica no compounds that corresponded to porphyrintetraones were obtained.
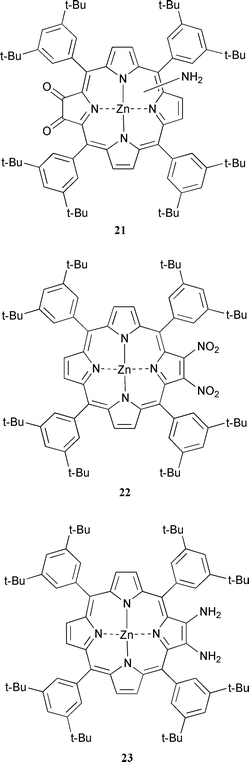
As the oxidations of the metallated 2-aminoporphyrins were sluggish and low yielding we thought that the absence of the desired porphyrintetraones might be due to over oxidation of the α-diones formed in the reaction. To investigate this we carried out the oxidation of 17 with DMP in the usual manner but stopped the reaction before all the starting material had been consumed. We chose the zinc derivative rather than the copper so that the products could be more easily identified. After work-up and purification over silica gel starting material 17, 9%, zinc porphyrin-α-dione 11, 3%, and a mixture of aminoporphyrin-α-diones 21,15 32%, were isolated. This result suggests that there is competing oxidation of the starting material and intermediate α-diones. Perhaps the most surprising result from this experiment was that zinc porphyrin-α-dione 11 was isolated as this could only come from the 2,3-diaminoporphyrin 23. To confirm this we reduced the zinc 2,3-dinitroporphyrin 2232 to 23 and oxidised 23 with DMP using the standard method. Under these conditions 11 was isolated in an excellent yield of 78% for the two steps. We believe that this is the first example of an “ortho” diamine being oxidised to an α-dione using DMP.
Conclusion
We have found that DMP can be utilised to oxidise aminoporphyrins to porphyrin-α-diones. We have utilised this new method for the preparation of free-base and metallated porphyrin-α-diones from 2-aminoporphyrins and in one case a 2,3-diaminoporphyrin. Finally, DMP has also been used to form the free-base “trans” porphyrin-2,3,12,13-tetraone. Oxidations using DMP have an advantage over the photo-oxidation route to similar compounds in that it can be carried out on larger scales more easily. However, for the formation of the simple porphyrin-α-dione, oxidation of a 2-hydroxyporphyrin with the DMP is still the preferred method.Experimental
1H NMR spectra were recorded on either Bruker AM-400 (400 MHz), Bruker AM-500 (500 MHz) or Varian Gemini 200 (200 MHz) spectrometers. Chemical shifts (δ) are reported relative to the residual solvent peak in parts per million (ppm). Infrared spectra were recorded on a Perkin-Elmer Paragon 1000 infrared spectrometer as KBr disks. UV–visible spectra were measured in spectrophotometric grade chloroform on a Perkin-Elmer Lambda 14P UV–visible spectrometer. Melting points were recorded on a Gallenkamp melting point instrument and are uncorrected. Laser desorption ionization (LDI) mass spectra were measured on a Micromass Tofspec 2E spectrometer with samples directly applied onto the MALDI target. MALDI were measured on a Micromass Tofspec 2E spectrometer using a dithranol (1,8,9-trihydroxyanthracene) matrix in reflectron (high resolution) mode. Fast atom bombardment (FAB) mass spectra were recorded on a VG Autospec Spectrometer. Due to a combination of metal chelation, molecular size, and different degrees of protonation the molecular weights observed by mass spectroscopy generally appeared as a complex isotopic cluster above the parent ion. We have quoted the mass for the monoisotopic species for each compound. Microanalyses were performed by Elemental Microanalysis Ltd, Devon, UK. All solvents for recrystallisation were distilled before use. Light petroleum ether refers to the fraction of boiling point 60–80 °C and it was routinely distilled before use. Merck AR grade methanol was used. Analytical thin-layer chromatography (TLC) was performed with Merck aluminium plates coated with silica gel 60 F254. Column chromatography was carried out using either the flash chromatography technique or gravity feed chromatography, with ICN silica gel 32-63 mesh, 60 Å. Centrifugal thin-layer chromatography was carried out using a Harrison Model 7924T Chromatotron. Where solvent mixtures are used, the proportions are given by volume. DMP was purchased from Lancaster, UK and used as obtained.[5,10,15,20-Tetrakis(3′,5′-di-tert-butylphenyl)porphyrinato]copper(II) 2
A mixture of 1 (6.00 g, 5.64 mmol), copper(II) acetate monohydrate (2.00 g, 10.0 mol), dichloromethane (300 cm3), and methanol (90 cm3) was heated at reflux for 1.5 h. The reaction mixture was allowed to cool and the solvent removed. The residue was passed through a plug of silica using dichloromethane as eluent. The main fraction was collected and the solvent completely removed to give a red purple solid of 2 (6.34 g, 100%) which had similar infrared and UV–visible spectra to those reported in the literature;33 mp > 296 °C; λmax(CHCl3)/nm (log (ε/dm3 mol−1 cm−1)) 419 (5.69), 505sh (3.59), 541 (4.30), 577 (3.57), and 618 (3.18).[2-Nitro-5,10,15,20-tetrakis(3′,5′-di-tert-butylphenyl)porphyrinato]copper(II) 3
A solution of nitrogen dioxide in light petroleum (0.69 M, in total 20.5 cm3) was added in small aliquots to a solution of 2 (4.00 g, 3.55 mmol) in dichloromethane (200 cm3). The reaction was followed by thin-layer chromatography (dichloromethane–light petroleum; 1∶4). When no starting material was observed, the mixture was immediately passed through a plug of silica gel using dichloromethane as eluent. The solvent was completely removed to give a purple residue which was then purified by column chromatography over silica using a dichloromethane–light petroleum mixture (1∶4) as eluent. The main fraction was collected and the solvent completely removed to give 3 as a purple solid (3.96 g, 96%) which had identical infrared and UV–visible spectra to those reported in the literature;33 mp > 296 °C; νmax(KBr disc)/cm−1 1527 (NO2); λmax(CHCl3)/nm (log (ε/dm3 mol−1 cm−1)) 426 (5.35), 549 (4.21), and 591 (4.01).2-Nitro-5,10,15,20-tetrakis(3′,5′-di-tert-butylphenyl)porphyrin 4
A solution of 3 (100 mg, 0.08 mmol) in dichloromethane (10 cm3) was treated with concentrated sulfuric acid (0.5 cm3). The reaction mixture was stirred at room temperature for 8 min and then it was poured into an ice–water mixture (20 cm3). The aqueous layer was separated and extracted with dichloromethane (3 × 20 cm3). The combined organic layers were washed with water (20 cm3), sodium bicarbonate solution (5%, 20 cm3), water (20 cm3), dried over anhydrous sodium sulfate, filtered, and the solvent completely removed leaving a purple residue. The residue was recrystallised from a dichloromethane–methanol mixture to give 4 as a purple solid (94 mg, 100%) which had identical 1H NMR, infrared and UV–visible spectra to those reported in the literature;33νmax(KBr disc)/cm−1 3329 (NH) and 1530 (NO2); δH(200 MHz; CDCl3) −2.55 (2 H, br s, NH), 1.54 (72 H, tert-butyl H), 7.77–7.84 (4 H, m, C(4′)), 8.07–8.11 (8 H, m, C(2′)H and C(6′)H), 8.79 (2 H, β-pyrrolic H), 8.96 (3 H, β-pyrrolic H), and 9.07–9.12 (2 H, β-pyrrolic H); λmax(CHCl3)/nm (log (ε/dm3 mol−1 cm−1)) 430 (5.33), 530 (4.20), 564 (3.69), 607 (3.58), and 666 (3.99).2,3-Dioxo-5,10,15,20-tetrakis(3′,5′-di-tert-butylphenyl)chlorin 9
Tin(II) chloride dihydrate (185 mg, 0.821 mmol) and concentrated hydrochloric acid (0.5 cm3) were added to a solution of 4 (91 mg, 0.082 mmol) in dichloromethane (4 cm3). The reaction mixture was stirred under nitrogen atmosphere at room temperature in the dark. The reaction was monitored by thin-layer chromatography (dichloromethane–light petroleum 1∶4). After stirring for 3 days, dichloromethane (20 cm3) and water (20 cm3) were added. The organic layer was separated, washed with water (30 cm3), sodium bicarbonate solution (5%, 30 cm3), water (30 cm3), dried over anhydrous sodium sulfate, filtered, and the solvent completely removed to yield a brown–purple solid of 6 (≈87 mg, ≈99%); νmax(KBr disc)/cm−1 3486 (NH2), 3392 (NH2), and 3310 (NH). DMP (36 mg, 0.08 mmol) was added to a solution of 6 (≈87 mg, ≈0.08 mmol) in dichloromethane (10 cm3). The reaction mixture was stirred at room temperature in the dark for 45 min at which point thin-layer chromatography (dichloromethane–light petroleum 2∶3) showed that no 6 remained. Hydrochloric acid (1 M, 30 cm3) was added and the reaction mixture was stirred for a further 20 min. The organic layer was separated, washed with water (2 × 100 cm3), dried over anhydrous sodium sulfate, filtered, and the solvent completely removed. The residue was purified by column chromatography over silica gel using a dichloromethane–light petroleum mixture (2∶3) as eluent to give 9 (46 mg, 52%) which had identical 1H NMR28 and infrared33 spectra to those reported in the literature; νmax(KBr disc)/cm−1 3354 (NH), 1738 (C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) O), and 1727 (CO).
O), and 1727 (CO).
[2-Amino-5,10,15,20-tetrakis(3′,5′-di-tert-butylphenyl)porphyrinato]copper(II) 7
10% Palladium on activated carbon (45 mg) was added to a solution of 3 (64 mg, 0.054 mmol) in a dichloromethane–methanol mixture (4∶1) (12 cm3) and the mixture was degassed with nitrogen. Sodium borohydride (52 mg, 1.75 mmol) was added in small portions over a 6 min period. The reaction mixture was stirred at room temperature under a nitrogen atmosphere in the dark for 1 h and then evaporated to dryness. The residue was passed through a plug of silica using dichloromethane as eluent. The solvent was completely removed to give a red–purple residue. For analysis the residue was purified by column chromatography over silica using a dichloromethane and light petroleum mixture (1∶4) as eluent. Care was taken to avoid photo-oxidation by keeping the silica column substantially in the dark. The main fraction was collected and the solvent completely removed. The residue was recrystallised from a dichloromethane–methanol mixture to give 7 as a purple solid (55 mg, 89%); mp > 296 °C; νmax(KBr disc)/cm−1 3486 (NH2) and 3392 (NH2); λmax(CHCl3)/nm (log (ε/dm3 mol−1 cm−1)) 423 (5.51), 544 (4.24), and 595 (3.79); m/z (LDI) 1138.8 (M+˙); C76H93N5Cu requires 1138.7.[2,3-Dioxo-5,10,15,20-tetrakis(3′,5′-di-tert-butylphenyl)chlorinato]copper(II) 10
10% Palladium on activated carbon (121 mg) was added to a solution of 3 (164 mg, 0.140 mmol) in a dichloromethane–methanol mixture (4∶1) (30 cm3) and the reaction mixture was degassed with nitrogen. Sodium borohydride (132 mg, 3.49 mmol) was added in small portions over a 10 min period. The reaction mixture was stirred at room temperature under a nitrogen atmosphere in the dark for 1 h and then evaporated to dryness. The residue was passed through a plug of silica using dichloromethane as eluent. The main fraction was collected and the solvent completely removed to give a red–purple residue of 7 (≈155 mg, ≈98%). DMP (58 mg, 0.14 mmol) was added to a solution of 7 (≈155 mg, ≈0.136 mmol) in dichloromethane (25 cm3). The reaction mixture was stirred at room temperature in the dark for 5 h. DMP (25 mg, 0.058 mmol) was added and stirred for a further 11 h. Hydrochloric acid (1 M, 30 cm3) was added and the mixture was stirred for a further 45 min. The organic layer was separated, washed with water (2 × 50 cm3), sodium bicarbonate solution (5%, 50 cm3), water (50 cm3), dried over anhydrous sodium sulfate, filtered, and the solvent completely removed. The residue was purified by column chromatography over silica gel using a dichloromethane–light petroleum mixture (2∶3–1∶1) as eluent to give 10 (58 mg, 37%) which had identical infrared28 and UV–visible spectra33 to those reported in the literature; λmax(CHCl3)/nm (log (ε/dm3 mol−1 cm−1)) 414 (4.85), 486 (4.22), 544 (3.55), 632 (3.76), and 708 (3.62).
[2,3-Dioxo-5,10,15,20-tetrakis(3′,5′-di-tert-butylphenyl)chlorinato]zinc(II) 11
33 spectra to those reported in the literature; νmax(KBr disc)/cm−1 1727 (CO); λmax(CHCl3)/nm (log (ε/dm3 mol−1 cm−1)) 410 (5.13), 489 (4.35), 633 (3.73), and 720 (3.78).
[Dinitro-5,10,15,20-tetrakis(3′,5′-di-tert-butylphenyl)porphyrinato]copper(II)s 12
A solution of nitrogen dioxide in light petroleum (0.51 M, in total 8 cm3) was added in small aliquots to a solution of 3 (1.80 g, 1.54 mmol) in dichloromethane (100 cm3). The reaction was followed by thin-layer chromatography (dichloromethane–light petroleum 1∶4). When no starting material was observed, the mixture was immediately passed through a plug of silica gel using dichloromethane as eluent. The solvent was completely removed to give a purple residue which was then purified by column chromatography over silica using a dichloromethane–light petroleum mixture (1∶4) as eluent. Six possible isomers of the copper-dinitroporphyrins were collected. The solvent was completely removed to give 1215 as a mixture of six isomers as a red–purple solid (1.44 g, 77%). A sample for analysis was recrystallised from a dichloromethane–methanol mixture; mp > 296 °C (Found: C, 74.5; H, 7.55; N, 6.85. C76H90CuN6O4 requires C, 75.1; H, 7.5; N, 6.9%); νmax(KBr disc)/cm−1 1531 (NO2); λmax(CHCl3)/nm (log (ε/dm3 mol−1 cm−1)) 438 (5.19), 559 (4.08), and 601 (4.03); m/z (MALDI) 1213.8 (M+˙); C76H90N6O4Cu requires 1213.6.
Dinitro-5,10,15,20-tetrakis(3′,5′-di-tert-butylphenyl)porphyrins 13
A solution of 12 (1.74 mg, 1.431 mmol) in dichloromethane (150 cm3) was treated with concentrated sulfuric acid (2 cm3). The reaction mixture was stirred at room temperature for 10 min and then poured into an ice–water mixture (100 cm3). The aqueous layer was separated and extracted with dichloromethane (3 × 100 cm3). The combined organic layers were washed with water (100 cm3), sodium bicarbonate solution (5%, 100 cm3), water (100 cm3), dried over anhydrous sodium sulfate, filtered, and the solvent completely removed leaving a purple residue. The residue was purified by column chromatography on silica gel using a dichloromethane–light petroleum (1∶4) mixture as eluent to give 13 as a purple solid (1.65 mg, 100%); mp > 296 °C (Found: C, 78.7; H, 8.39; N, 7.1. C76H92N6O4 requires C, 79.1; H, 8.0; N, 7.3%); νmax(KBr disc)/cm−1 3341 (NH), 3316 (NH), and 1532 (NO2), 1477, 1363, 1294, 1248, 1207, 1157, 918, and 802; λmax(CHCl3)/nm (log (ε/dm3 mol−1 cm−1)) 442 (5.32), 539 (4.13), 583 (3.96), 618sh (3.76), and 683 (4.04); δH(500 MHz; CDCl3) −2.48 to −2.40 and −2.18 to −2.09 (6 H, NH), 1.50–1.56 (72 H, tert-butyl H), 7.80–7.88 (4 H, m, C(4′)H), 8.00–8.14 (8 H, m, C(2′)H and C(6′)H), and 8.77–9.11 (6 H, m, β-pyrrolic H); m/z (MALDI) 1153.1 (M+˙) and 1154.1 (MH+); C76H93N6O4 requires 1153.7.[Dinitro-5,10,15,20-tetrakis(3′,5′-di-tert-butylphenyl)porphyrinato]zinc(II)s 14
A mixture of 13 (175 mg, 0.151 mmol), zinc(II) acetate dihydrate (67 mg, 0.30 mmol), dichloromethane (30 cm−3), and methanol (8 cm−3) was heated at reflux for 1 h. The reaction mixture was allowed to cool and the solvent removed. The residue was passed through a plug of silica using dichloromethane as eluent and the main fraction was collected and the solvent completely removed. The residue was purified by column chromatography over silica using a dichloromethane–light petroleum mixture (1∶4–1∶1) as eluent to give 14 as a purple solid (181 mg, 98%); mp > 296 °C; νmax(KBr disc)/cm−1 1525 (NO2); λmax(CHCl3)/nm (log (ε/dm3 mol−1 cm−1)) 441 (5.30), 527sh (3.61), 566 (4.14), and 611 (4.06); δH(400 MHz; CDCl3) 1.53–1.55 (72 H, t-butyl H), 7.78–7.86 (4 H, C(4′)H), 7.94–8.05 (8 H, C(2′)H and C(6′)H), 8.87–9.01 (4 H, β-pyrrolic H), and 9.16–9.21 (2 H, β-pyrrolic H); m/z (MALDI) 1214.8 (M+˙); C76H90N6O4Zn requires 1214.6.2,3,12,13-Tetraoxo-5,10,15,20-tetrakis(3′,5′-di-tert-butylphenyl)porphyrin 18
Tin(II) chloride dihydrate (0.19 g, 0.84 mmol) and concentrated hydrochloric acid (0.5 cm3) were added to a solution of 13 (139 mg, 0.120 mmol) in dichloromethane (4 cm3). The reaction mixture was stirred under a nitrogen atmosphere at room temperature in the dark. The progress of the reaction was monitored by thin-layer chromatography (dichloromethane–light petroleum 1∶4). After stirring for 4 days, dichloromethane (30 cm3) and water (30 cm3) were added. The organic layer was separated, washed with water (30 cm3), sodium bicarbonate solution (5%, 30 cm3), water (30 cm3), dried over anhydrous sodium sulfate, filtered and the solvent completely removed to yield a brown–purple solid. The crude product was recrystallised from a dichloromethane–methanol mixture to give 15 as a brown–purple solid (≈127 mg, ≈97%); νmax(KBr disc)/cm−1 3485 (NH2), 3392 (NH2), and 3321 (NH). DMP (98 mg, 0.23 mmol) was added to a solution of 15 (≈127 mg, ≈0.116 mmol) in dichloromethane (15 cm3). The reaction mixture was stirred at room temperature in the dark for 35 min at which point thin-layer chromatography (dichloromethane–light petroleum 2∶3) showed that no 15 remained. Hydrochloric acid (1 M, 10 cm3) was added and the mixture was stirred for a further 30 min. The organic layer was separated, washed with water (3 × 50 cm3), dried over anhydrous sodium sulfate, filtered and the solvent completely removed. The residue was purified by column chromatography over silica gel using a dichloromethane–light petroleum (2∶3) mixture as eluent to give 18 (25 mg, 20%). The infrared and 1H NMR spectra were identical to those in the literature15 except that the NH protons in the 1H NMR were observed at −1.80 and not −1.32 as reported.
[Diamino-5,10,15,20-tetrakis(3′,5′-di-tert-butylphenyl)porphyrinato]copper(II)s 16
10% Palladium on activated carbon (93 mg) was added to a solution of 12 (57 mg, 0.05 mmol) in a dichloromethane–methanol mixture (4∶1) (12 cm−3) and the reaction mixture was degassed with nitrogen. Sodium borohydride (89 mg, 2.3 mmol) was added in small aliquots over a 10 min period. The reaction mixture was stirred at room temperature under nitrogen in the dark for 1 h and then evaporated to dryness. The residue was passed through a plug of silica using dichloromethane as eluent. The main fraction was collected and the solvent completely removed to give a purple residue. The residue was purified by column chromatography in the dark using a dichloromethane–light petroleum (2∶3) mixture as eluent. Care was taken to avoid photo-oxidation by keeping the silica column substantially in the dark. The main fraction was collected and the solvent completely removed. The residue was recrystallised from a dichloromethane–methanol mixture to give a purple solid of 16 (40 mg, 74%); mp > 296 °C (Found: C, 78.6; H, 8.2; N, 7.15. C76H94CuN6 requires C, 79.0; H, 8.0; N, 7.3%); νmax(KBr disc)/cm−1 3484 (NH2) and 3390 (NH2); λmax(CHCl3)/nm (log (ε/dm3 mol−1 cm−1)) 427 (5.18), 544 (4.08), 552sh (4.06), and 600 (3.77); m/z (LDI) 1153.9 (M+˙); C76H94N6Cu requires 1153.7.Attempted preparation of [2,3,12,13-tetraoxo-5,10,15,20-tetrakis(3′,5′-di-tert-butylphenyl)porphyrinato]zinc(II) 20
10% Palladium on activated carbon (360 mg) was added to a solution of 14 (190 mg, 0.156 mmol) in a dichloromethane–methanol mixture (4∶1) (40 cm3) and the reaction mixture was degassed with nitrogen. Sodium borohydride (295 mg, 7.80 mmol) was added in small portions over a 15 min period. The reaction mixture was stirred at room temperature under nitrogen in the dark for 1 h. The solvent was removed and the residue was passed through a plug of silica using dichloromethane as eluent. The main fraction was collected and the solvent completely removed to give a purple solid of 17 (≈170 mg, ≈94%); mp > 296 °C; νmax(KBr disc)/cm−1 3485 (NH2), 3392 (NH2); δH(400 MHz; CDCl3) 1.54–1.56 (72 H, tert-butyl H), 4.39 (2 H, br s, NH2), 7.27–7.81 (14 H, phenyl H and β-pyrrolic H), and 8.57–8.97 (4 H, β-pyrrolic H). DMP (62 mg, 0.15 mmol) was added in small aliquots to a stirred solution of 17 (≈170 mg, ≈0.146 mmol) in dichloromethane (25 cm3) at room temperature in the dark. The reaction mixture was stirred for 30 min and then water (50 cm3) was added. The reaction mixture was stirred for a further 40 min. The organic layer was separated, dried over anhydrous sodium sulfate, filtered, and the solvent completely removed. After purification by column chromatography over silica using a dichloromethane–light petroleum mixture (2∶3) as eluent, three main bands were isolated. Starting material (17) (15 mg, 9%), 11 (5 mg, 3%) (a sample of which co-chromatographed with and had an identical 1H NMR to an authentic compound), and 2115 (55 mg, 32%); νmax(KBr disc)/cm−1 3482 (NH2), 3395 (NH2), and 1726 (CO); m/z (FAB) 1170.7 (M+˙) and 1171.7 (MH+); C76H92N5O2Zn requires 1170.7.
References
- M. J. Crossley and P. L. Burn, J. Chem. Soc., Chem. Commun., 1991, 1569 RSC.
- H. L. Anderson, Chem. Commun., 1999, 2323 RSC.
- R. W. Wagner and J. S. Lindsey, J. Am. Chem. Soc., 1994, 116, 9759 CrossRef CAS.
- D. L. Officer, A. K. Burrell and D. C. W. Reid, Chem. Commun., 1996, 1657 RSC.
- J. Seth, V. Palaniappan, T. E. Johnson, S. Prathapan, J. S. Lindsey and D. F. Bocian, J. Am. Chem. Soc., 1994, 116, 10578 CrossRef CAS.
- O. Mongin, A. Schuwey, M. A. Vollet and A. Gossauer, Tetrahedron Lett., 1999, 40, 8347 CrossRef CAS.
- A. Nakano, A. Osuka, I. Yamazaki, T. Yamazaki and Y. Nishimura, Angew. Chem., Int. Ed., 1998, 37, 3023 CrossRef CAS.
- V. S. Y. Lin, S. G. Dimagno and M. J. Therien, Science, 1994, 264, 1105 CrossRef CAS.
- H. L. Anderson, S. J. Martin and D. D. C. Bradley, Angew. Chem., Int. Ed. Engl., 1994, 33, 655 CrossRef.
- M. Terazima, H. Shimizu and A. Osuka, J. Appl. Phys., 1997, 81, 2946 CrossRef CAS.
- F. Z. Henari, W. J. Blau, L. R. Milgrom, G. Yahioglu, D. Phillips and J. A. Lacey, Chem. Phys. Lett., 1997, 267, 229 CrossRef CAS.
- T. Nagata, A. Osuka and K. Maruyama, J. Am. Chem. Soc., 1990, 112, 3045.
- A. Osuka, M. Ikeda, H. Shiratori, Y. Nishimura and I. Yamazaki, J. Chem. Soc., Perkin Trans. 2, 1999, 1019 RSC.
- H. Higuchi, M. Shinbo, M. Usuki, M. Takeuchi, Y. Hasegawa, K. Tani and J. Ojiwa, Bull. Chem. Soc. Jpn., 1999, 72, 1887 CrossRef CAS.
- M. J. Crossley, L. J. Govenlock and J. K. Prashar, J. Chem. Soc., Chem. Commun., 1995, 2379 RSC.
- M. G. H. Vicente, M. T. Cancilla, C. B. Lebrilla and K. M. Smith, Chem. Commun., 1998, 2355 RSC.
- M. J. Crossley and J. A. McDonald, J. Chem. Soc., Perkin Trans. 1, 1999, 2429 RSC.
- B. Konig and H. Zieg, Synthesis, 1998, 171 CrossRef.
- L. Jaquinod, O. Sari, R. G. Khoury and K. M. Smith, Chem. Commun., 1998, 1261 RSC.
- A. K. Burrell and D. L. Officer, Synlett, 1998, 1297 CrossRef CAS.
- M. J. Crossley and P. L. Burn, J. Chem. Soc., Chem Commun., 1987, 39 RSC.
- R. Beavington and P. L. Burn, J. Chem. Soc., Perkin Trans. 1, 1999, 583 RSC.
- R. Beavington and P. L. Burn, J. Chem. Soc., Perkin Trans. 1, 2000, 605 RSC.
- R. Beavington and P. L. Burn, J. Chem. Soc., Perkin Trans. 1, 2000, 1231 RSC.
- E. J. Atkinson, A. M. Oliver and M. N. Paddon-Row, Tetrahedron, 1993, 34, 6147 CrossRef CAS.
- M. J. Crossley and L. G. King, J. Chem. Soc., Chem. Commun., 1984, 920 RSC.
- M. J. Crossley, P. L. Burn, S. J. Langford, S. M. Pyke and A. G. Stark, J. Chem. Soc., Chem. Commun., 1991, 1567 RSC.
- R. Beavington, P. A. Rees and P. L. Burn, J. Chem. Soc., Perkin Trans. 1, 1998, 2847 RSC.
- S. D. Meyer and S. L. Schreiber, J. Org. Chem., 1994, 59, 7549 CrossRef CAS.
- M. J. Crossley, M. M. Harding and S. Sternhell, J. Org. Chem., 1988, 53, 1132 CrossRef CAS.
- M. J. Crossley, L. G. King, I. A. Newsom and G. S. Sheehan, J. Chem. Soc., Perkin Trans. 1, 1996, 2675 RSC.
- V. Promarak and P. L. Burn , unpublished results. .
- P. L. Burn , PhD Thesis, Sydney University, Australia, 1989. Search PubMed.
| This journal is © The Royal Society of Chemistry 2001 |