Applications of stoichiometric transition metal complexes in organic synthesis
Anthony J. Fletcher and Steven D. R. Christie Department of Chemistry, University of Loughborough, Loughborough, UK LE11 3TU. E-mail: S.D.Christie@lboro.ac.uk
Received (in Cambridge, UK) 18th September 2000
First published on 7th December 2000
Abstract
Covering: 1st May 1999 to 30th April 2000.
1 Introduction
This review continues the series describing developments in stoichiometric transition metals applied to organic synthesis. The subdivision of the material is as outlined above, in common with the previous reviews.1,2 As always, the main aim of this article is to highlight novel or interesting organometallic mediated reactions which have particular relevance to organic synthesis.
2 Transition metal alkyl, alkenyl and allyl complexes in organic synthesis
2.1 Organozirconium-based methodology
A substantial number of zirconium mediated reactions have been reported this year; the following draws attention to some highlights. The carbozirconation of alkynes and subsequent elaboration of the resulting alkenylzirconium species is one of the most popular uses of the metal. Takahashi has shown that reaction of alkynes with diethyl zirconocene followed by a chloroformate produces α,β-unsaturated esters (Scheme 1).3 In addition, the other end of the alkene can be functionalised by quenching with various electrophiles. The initial reaction is a metallo-esterification of an alkyne, followed by quenching of the carbon–metal bond. Use of Schwartz’s reagent (Cp2ZrHCl) for the hydrozirconation of alkynes has been very popular this year. Reaction with alkynyl stannanes proceeds regioselectively to place the zirconium and the tin on the same carbon.4 This 1,1-bimetalloalkene can then be quenched with tin, selenium or tellurium halides. Huang has reported similar procedures to produce vinyl sulfones,5 vinyl selenol esters,6 vinyl phosphonates,7 and vinyl sulfides.8 A good illustration of this methodology in synthesis is the preparation of enynes by reaction of alkynes with Schwartz’s reagent to produce the functionalised alkene, followed by coupling of this with an alkynyl iodonium tosylate to produce the enyne (Scheme 2).9 Another variation on the theme is the nickel catalysed addition of vinyl zirconiums to α-bromo-α,α-difluoroesters, which produces a convenient route to allyldifluoroesters.10 Takahashi has also reported a nickel catalysed formation of aromatic rings from zirconacyclopentadienes.11
Scheme 1
Scheme 2
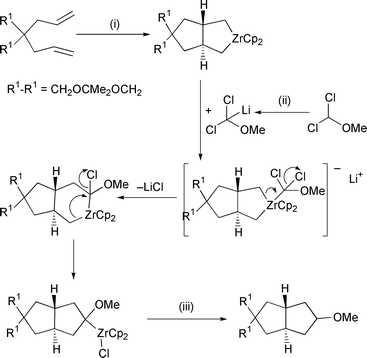
The second main area of use for zirconium is the coupling of unsaturated species to form zirconacyclopentanoid systems, and the subsequent elaboration of these to organic end products. Takahashi has used this methodology as a simple route to cyclooctatetraenes (Scheme 3).12 After a normal zirconocene mediated alkyne coupling to give a zirconacyclopentadiene, addition of two equivalents of copper chloride produces the bis(vinyl)copper species. One equivalent of NBS then facilitates dimerisation to give the cyclooctatetraene. In related chemistry, routes to spirocyclic cyclopentadienes13 and cyclobutenes14 have also been realised. Koenig has used the zirconium cyclisation to prepare a number of new polyphosphine boranes.15 Whitby has continued his work into the elaboration of zirconacycles via insertion of carbenoid species (Scheme 4).16 Cyclisation of a bis-allyl compound with zirconocene is followed by insertion of a 1-lithio-1,1-dihalo species. This firstly forms an ‘ate’ complex with the zirconium. However, since there are two leaving groups attached, first one of the carbon–zirconium bonds migrates, followed by the second producing a new ring. In this way, the zirconium facilitates formation of two rings and three carbon–carbon bonds. In addition, the regiochemistry of related carbenoid insertions has also been looked at in detail.17 A versatile approach to stereodefined unsaturated systems can also be achieved through this insertion process.18 Using this methodology, routes to stereodefined alkenes, dienes and trienes have been realised. A substantial amount of work is published in this paper, and the reader is directed to the original reference to gain a full appreciation of the scope of the reactions that can be achieved. Mori has revealed an interesting variation on the formation of zirconacycles (Scheme 5).19 A zirconium–silene complex is first made by addition of two equivalents of a silyllithium species and subsequent elimination of a silane. Addition of an alkyne to this proceeds in the normal manner to produce the silazirconacycle; however, the regiochemistry of the addition is governed by the alkyne, as shown in Scheme 5. This can then be elaborated in the usual manner to produce unsaturated silanes in good yield. Buchwald has used an interesting method to produce bi- and terphenyl compounds.20 A zirconium benzyne complex is generated in the usual manner, and then reacted with a palladium aryl species (Scheme 6). This inserts regioselectively into one of the zirconium–carbon bonds. The final zirconium–carbon bond is quenched by iodinolysis.
Scheme 3
Scheme 4 Reagents and conditions: (i) Cp2ZrCl2, BuLi 2 eq., −78 °C– rt, THF; (ii) MeO(Cl)2CH 1.5 eq., LDA 1.7 eq., −78 °C; (iii) MeOH, NaHCO3, −78 °C, 16 h.
Scheme 5
Scheme 6
2.2 Organotitanium-based methodology
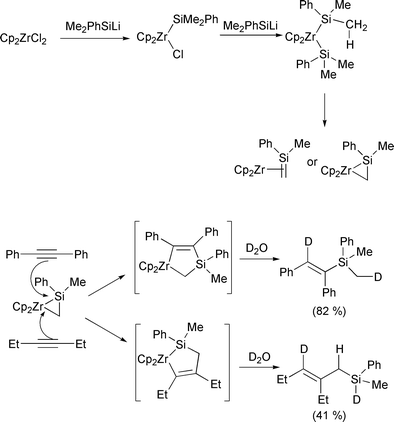
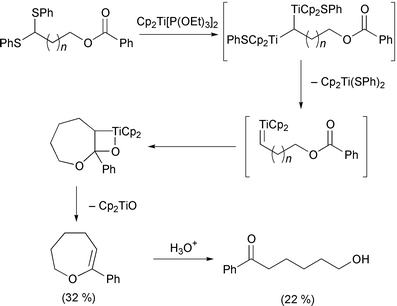
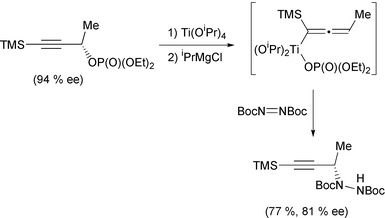
Organotitanium methodology has also continued apace in the last year. Huang has noted that titanocene hydride, prepared from the reaction of titanocene dichloride and ibutyl magnesium bromide, reacts with ditellurides to form a titanium–tellurium bond.21 This can then be further reacted to give a new tellurium–carbon bond by addition of an electrophile. Protected glycals have been prepared using titanium(III) radicals (Scheme 7).22 The anomeric bromide is abstracted by the titanium, leaving a radical in the anomeric position. The titanium then recombines, followed by elimination of the titanium and acetate. Takeda has published a few papers exploiting the reactivity of thioacetal groups with titanium(II) reagents. In the first,23 1,3-bis(phenylthio)alk-1-enes react with the titanium(II) reagent Cp2Ti[P(OEt)3]2 (Scheme 8). The proposed titanium carbene intermediate then reacts further, with a tertiary alkyl chloride to produce a new carbon–carbon bond. The regiochemistry of this last step is particularly noteworthy since it always produces the terminal olefin. More impressively, the thioacetals can also be made to undergo a tandem reaction.24 Firstly, the acetal is reduced to the carbene (Scheme 9). Reaction of this with a nitrile induces a [2 + 2] cycloaddition. Rearrangement of this adduct produces an imino–titanium species which can be hydrolysed to the ketone. Using the titanium carbene intermediate as a Tebbe type reagent has allowed formation of dihydrothiophenes.25 Finally, the same intermediate also reacts in an intramolecular manner with an ester (Scheme 10).26 Sato has continued his work into the use of the tetraisopropoxide titanium reagent. Addition of the titanium to an alkyne with a propargylic leaving group produces an allene. Reaction of this with a hydrazine gives the substituted alkyne back, with loss of the titanium (Scheme 11).27 The particularly clever part is the stereoselectivity of the two reactions: the allene formation is stereoselective, and produces a chiral allene. Subsequently, addition of a nucleophile is directed by the titanium, and gives a chiral alkyne as a product. The other use of the titanium species is as a templating agent for cyclisation reactions. By appending a chiral auxiliary onto the substrate, Sato has managed to induce a chiral cyclisation to produce a bicyclic cyclopentenone (Scheme 12).28 In a particularly clever piece of design, Sato also manages to cleave the chiral promoter from the desired product in the final step of the reaction mechanism. Finally in this section, the first synthesis of a metallated titanocyclopropene has been reported.29 Using an alkynyl chloride, reaction with a titanium(II) alkoxide allows, firstly complexation of the titanium to the alkyne, then at slightly elevated temperature, migration of the metal to the alkynyl position. This was confirmed by D2O quench. However, the intermediate can also be recomplexed by a second equivalent of titanium, and D2O quench of this, incorporates three deuterium atoms on the resulting alkene.
Scheme 7
Scheme 8
Scheme 9
Scheme 10
Scheme 11
Scheme 12
2.3 Organoiron complexes
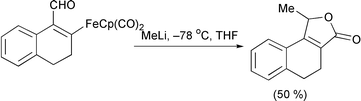
η1 Complexes of iron are still of general use as organic reagents. The synthesis of α,β-unsaturated butenolides has been reported by an intramolecular cyclocarbonylation reaction.30 Reduction of the aldehyde with a nucleophile allows the alkoxide to cyclise onto the iron group to produce the lactone (Scheme 13). The same principle has also been extended to the preparation of lactams through the reaction of amines.31 Davies has continued his use of iron acyl compounds.32 Asymmetric conjugate addition of a chiral amine to the iron α,β-unsaturated acyl complex is followed by deprotection and cyclisation with loss of the metal (Scheme 14).
Scheme 13
Scheme 14
3 Group IV transition metal carbenes in organic synthesis
Group IV carbenes remain popular in the realm of organic synthesis. Hegedus continues to be the main advocate of photochemical activation of chromium carbenes, forming β-lactams. This year, he has produced a method for tethering two carbenes together.33 Two different methods are highlighted in the paper: the one shown in Scheme 15 involves deprotonation of the carbene, then SN2 attack onto a bis-triflate. The resulting tethered bis-carbene undergoes normal photolytic reactions with imines, producing a bis-β-lactam in reasonable yield. Aumann has reported on the use of tungsten carbenes and their reaction with enamines.34 Reaction of 1-aminocyclohexenes with [(1-alkynyl)carbene] tungsten complexes gave rise to cyclopentadienes and dihydropyrroles.
Scheme 15
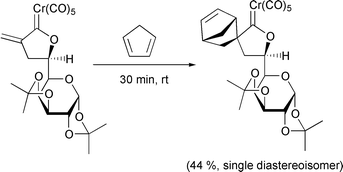
In annulation reactions of carbenes, Harrity has used alkynyl boranes as unusual substrates in a novel approach to substituted aryl boranes.35 More interestingly, when a very bulky group such as t-Bu is used, the reaction is stopped before the insertion of the phenyl group. In this way, cyclobutenone compounds can be isolated (Scheme 16). Another “unusual” annulation has been used to form methylene pyrrolidines from dimethylamino ethenyl chromium carbene complexes (Scheme 17).36 After reaction with the alkyne, the carbene moiety undergoes a C–H insertion reaction into the methyl group on the nitrogen. This leads to the chromium tricarbonyl complexes of dihydroazepine. Loss of the chromium and silicon, followed by tautomerisation and cyclisation affords the pyrrolidine. Herndon has reported surprising remote functionalisation of alkyl side chains when reacting chromium carbenes with conjugated enediynes.37 The reaction of N-methyldihydropyridines with chromium and tungsten carbenes gives rise to polyoxygenated compounds.38 The chromium carbene reaction uses three of the carbon monoxide ligands in sequence (Scheme 18), the first to form the carbene itself, the second after a redox reaction with the dihydropyridine and the third to generate a ketene which is attacked by an alkoxide to form the lactone. The tungsten carbene, however, follows a different reaction pathway. Barluenga has shown that a formal [3 + 2] cycloaddition takes place between alkenyl carbenes and enamines (Scheme 19).39 By using a chiral auxiliary on the nitrogen, the cycloaddition is stereoselective and after hydrolysis of the enol, the cyclopentenone product is isolated in good enantiomeric excess.
Scheme 16
Scheme 17
Scheme 18
Scheme 19
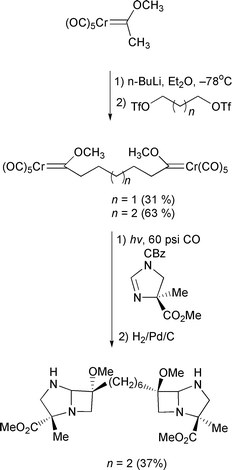
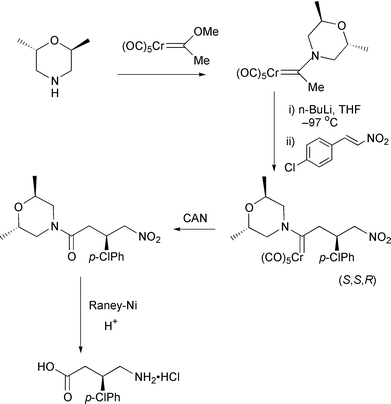
A one-step synthesis of cyclopentenediones has been achieved using chromium carbenes.40 Starting with cyclobutenediones, direct insertion of the carbene produces the five membered ring (Scheme 20). Also reported was a reaction of cyclopropenones to produce cyclobutenones. The electron accepting properties of carbenes has been used to good effect in Michael additions to unsaturated complexes this year.41 1,2- and 1,6-additions are possible. The 1,6-addition is straightforward, proceeding as expected (Scheme 21). However, when a lithium acetylide is used as the nucleophile, the 1,2-addition mode predominates. Attack at the carbene carbon is followed by a rearrangement of the metal to form an allenic substituent which produces an extended polyene on work-up. Dötz has used the carbenes from pyranosides to direct addition of electrophiles.42 The tin pyranoside is reacted with butyllithium and then chromium hexacarbonyl to produce a carbene anion (Scheme 22), which is quenched with an electrophile to produce the substituted complex. A carbene has also been used to help effect a Diels–Alder reaction (Scheme 23).43 The steric influence of the sugar structure controls the stereochemistry. Finally, an interesting application of a carbene has been reported.44 On the way to (R)-baclofen, the morpholine substituted carbene is deprotonated and reacted in a Michael reaction with an unsatuated nitro compound. Oxidation of the metal produces the ketone, which is further transformed to the natural product (Scheme 24).
Scheme 20
Scheme 21
Scheme 22
Scheme 23
Scheme 24
4 η2-Complexes in organic synthesis
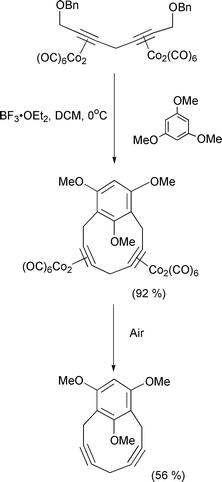
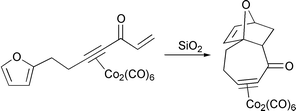
The main area of interest continues to be the use of propargylic,† or Nicholas carbocations for the formation of carbon–carbon or carbon–heteroatom bonds, aided by a cobalt–alkyne complex. By using a chiral acetal, Grée and co-workers have provided a route to chiral propargylic fluorinated alkynes.45 Reaction of the cobalt complexed propargylic alcohol with DAST gives a good yield and high stereoselectivity of the fluoride (Scheme 25). The addition of the fluoride anion is controlled by the Nicholas carbocation at the low temperature, and is not influenced by the chiral dioxolane ring. Díaz and Martin have shown that a γ-benzylated diol can provide the hydride source for the reduction of a propargylic alcohol by delivering the hydride in an intramolecular fashion.46 In natural product synthesis, Montaña has utilised a Nicholas carbocation to form the framework of the pseudoguaiane system (Scheme 26).47 The propargylic cation is an excellent trap for the trimethylsilyl enol ether, but the diastereoselectivity is negligible. Isobe has used an intramolecular cyclisation to build up the structure of the tricyclic fragment of the ciguatoxin polyether marine toxin (Scheme 27).48 Of note in this example is the stabilising effect of the cobalt carbonyl fragment on the cyclic alkyne unit. Mukai has continued his work into the use of Nicholas reactions to form large rings (Scheme 28).49 Activation of the propargylic alcohol by methanesulfonyl chloride is followed by intramolecular trapping by the pendant ether group. The final products are determined by either loss of the trimethylsilyl group, or a proton. In a double Nicholas reaction, Green has formed a ten membered ring containing two alkyne units in a single pot in a remarkable 92% yield (Scheme 29).50 The metals can be removed simply by exposure to air. In an extension of the stabilisation of adjacent positive charges, Nicholas has found that radicals in the propargylic position can also be used in organic synthesis (Scheme 30).51 The propargylic radical is formed from the corresponding propargyl bromide complex and an atom transfer cyclisation allows formation of the five membered ring.
Scheme 25
Scheme 26
Scheme 27
Scheme 28
Scheme 29
Scheme 30
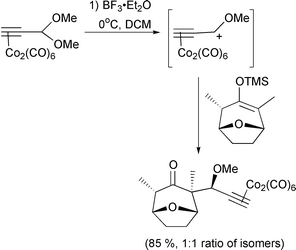
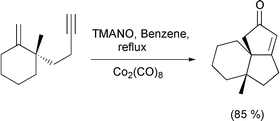
The other main use of η2-Co complexes is as cyclisation reagents for [2 + 2 + 2] cycloadditions. Vollhardt pioneered this work, and this year has published a route to the benzofuran nucleus using the methodology.52 Malacria has continued his efforts in the area by devising a chiral auxiliary that can be attached to the organic substrate and effect high levels of stereocontrol.53 This type of cycloaddition reaction has also been used in natural product synthesis by Motherwell as a means to access the steganone skeleton (Scheme 31).54 Of note in this particular example is the isolation of substantial quantities of the cyclobutadiene complex, which may be an intermediate in the formation of the arene. Finally, two references where the cobalt is not integral in the reaction itself, but instead acts as a linker or a template. Gibson has used an alkyne as a traceless linker by complexing it to a cobalt atom which itself is linked to a solid support through a phosphine ligand.55 Chemistry is carried out on the pendant chain and the alkyne then released from the support by oxidation of the metal. This is a particularly effective utilisation of transition metals and has obvious implications for solid support and combinatorial chemistry. Use of a cobalt alkyne complex as a template for a Diels–Alder reaction has also been reported.56 Complexing the alkyne to cobalt facilitates the intramolecular Diels–Alder reaction, producing the strained tricyclic system (Scheme 32). The seven membered ring containing the alkyne is stabilised by the complexed cobalt. The reaction does not proceed without the cobalt being present, which illustrates the stabilising effect of the metal, and the difference in bond angles of complexed and uncomplexed alkyne.
Scheme 31
Scheme 32
5 η3-Complexes in organic synthesis
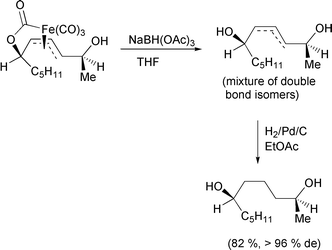
The use of η3-complexes of iron, as applied to organic synthesis, continues to be dominated by ferrilactones. Ley has continued his efforts in the field this year with the total synthesis of the cholesterol biosynthesis inhibitor 1233A.57 The vinyl epoxide serves as the precursor to the ferrilactone, which is formed in the usual way by reaction with di-iron nonacarbonyl (Scheme 33). Oxidation of the desired isomer with ceric ammonium nitrate produces the β-lactone, which is further elaborated to the natural product. The reductive decomplexation of ferrilactones has also received some interest.58 Using a hydride donor, the complex is reduced and the iron decomplexed to produce a mixture of saturated and unsaturated diols (Scheme 34). The stereochemistry of the alcohol centres is transferred from the precursor epoxide, through the iron lactone complex and retained in the reduced organic product.
Scheme 33
Scheme 34
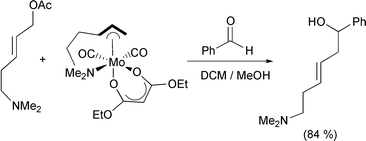
Liebeskind reports the reaction of η3-pyranyl molybdenum complexes with alkenes in a [5 + 2] cycloaddition (Scheme 35).59 The pyran complex is presumed to react in a Michael fashion to produce the intermediate η4 complex. The enolate then attacks the other end of the π-complex to form the oxabicyclo ring system. The stereochemistry is highly controlled due to the facial constraints imposed by the bulky metal and its ligands. Yields are good to excellent and enantioselectivities are always extremely high. Using allylic acetates with pendant Lewis base functionality, Krafft has shown that complexation of molybdenum produces an η3 complex (Scheme 36).60 The pendant Lewis base can then bind to the metal in an intramolecular fashion, producing a chelate. Subsequent addition of an electrophile causes reaction at the allyl group. However, because of the intramolecular coordination, one end is effectively hindered, and reaction occurs at the other end. This offers an effective method for regio- and diastereoselective reactions of π-allyl complexes of molybdenum.
Scheme 35
Scheme 36
Liu has continued his extensive studies into the use of η3 complexes of tungsten this year and applied the results to the synthesis of natural and unnatural products. The first step in the procedure tends to be displacement of a propargylic chloride with an organometallic tungsten anion. The η1 intermediate is transformed to an η3 complex by reaction with an acid. This complex can then be activated by addition of nitrosyl tetrafluoroborate, making the allyl group nucleophilic and capable of attacking an electrophile. Application of this to a natural product synthesis is outlined in Scheme 37.61 Formation of an η3 system is followed by activation and quenching with an aldehyde to give a lactone. This is further elaborated to give (+)-dihydrocanadensolide. A particularly noteworthy example is shown in Scheme 38.62 The same sequence is employed but in this case the aldehyde is exposed to the allyltungsten group in an intramolecular fashion. As shown in the scheme, the aldehyde can be deprotected under mildly acidic conditions, without compromising the organometallic group. Activation and cyclisation then occur to produce the bicyclic α-methylene butyrolactone in good yield.
Scheme 37
Scheme 38
6 η4-Complexes in organic synthesis
η4-Complexes are dominated by iron this year. There are many instances of iron tricarbonyl being used as a protecting group for conjugated dienes, or to facilitate reaction at adjacent centres to iron-diene units. An example of this is the synthesis of 11-Z-retinol,63 where a Peterson olefination is carried out adjacent to an iron tricarbonyl complexed diene unit. Pearson has continued his usage of iron carbonyl species in the synthesis of heptitol derivatives.64 Starting with an iron complex of cycloheptatriene, a stereocontrolled reduction of the carbonyl group is achieved (Scheme 39). After protection of the alcohol, an osmium mediated dihydroxylation of the free diene is again controlled by the iron. Depending on the conditions employed, the hydroxy ketone could also be isolated. This could then be reduced to the trans-diol. The metal is easily removed by reaction with an N-oxide. The use of iron tricarbonyl as a mobile chiral auxiliary has been put forward.65 Starting with a diene complex, Lewis acid catalysis induces loss of the adjacent leaving group and migration of the iron (Scheme 40). Attack of a nucleophile at the other end of the unsaturated system then occurs, with stereocontrol coming from the bulky organometallic group. This migration towards a cyano group has also been used to great effect for the functionalisation of a linear polyene.66 The triene is dihydroxylated, protected and then homologated as shown in Scheme 41. The lithium anion of acetonitrile then induces a migration of the iron along the triene chain, with concomitant inversion of the diene-Fe(CO)3 stereochemistry. Further hydroxylation then functionalises the newly unmasked alkene, providing access to a stereoselectively polyhydroxylated chain. The addition of nucleophiles to carbonyl functionalities adjacent to an iron complexed diene has been found to be dependant on the Lewis acid employed (Scheme 42).67 The selectivity will be dependant on the conformation of the carbonyl with respect to the diene, assuming the incoming nucleophile approaches away from the bulky –Fe(CO)3. With boron trifluoride, apparently the s-cis conformation predominates, giving rise to good stereochemical control over the new centre. However, employing titanium tetrachloride, the opposite stereochemistry is seen, requiring the s-trans conformation of the aldehyde. Addition of radicals to an η4-triene has allowed the preparation of substituted aromatic compounds.68 As shown in Scheme 43, the radical adds to the terminus of the exocyclic alkene. This creates an intermediate η5-iron radical, which loses iron to produce the aromatic system. A carboxylate substituent on the diene system has led to a synthesis of lactones;69 activation of the iron is required in order for the carboxylate to add (Scheme 44) and this is achieved by reaction with nitrosonium tetrafluoroborate. The intramolecularly appended acid then attacks the end of the diene, producing an allyl system. This is further oxidised to an unsaturated ketone by additional NOBF4. The synthesis of 4-substituted cyclohexa-2,4-dien-1-one complexes has been achieved by the oxidation of cyclohexadiene iron tricarbonyl complexes with thallium trifluoroacetate (Scheme 45).70 Interestingly, no aromatic products were isolated from this procedure, illustrating the power of the iron tricarbonyl unit at stabilising diene systems, even when an aromatic system could be realised. This stabilisation is not restricted to linear dienes. Snapper has shown that cyclobutadiene complexes can stabilise an adjacent carbocation.71 Lewis acid activation of the methyl ether generates the stabilised cation, which can be quenched with a trimethylsilyl enol ether (Scheme 46). Finally, iron tricarbonyl systems have also been utilised to help effect formation of the corannulene ring system.72
Scheme 39
Scheme 40
Scheme 41
Scheme 42
Scheme 43
Scheme 44
Scheme 45
Scheme 46
7 η5-Complexes in organic synthesis
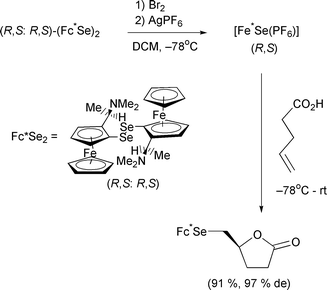
Donaldson has used the η5-pentadienyl–η4-diene interconversion of iron complexes as a means of accessing the macrolactin A structure.73 The interesting aspect of this particular paper is the unusual equilibrium between the kinetic and thermodynamic products of nucleophilic addition to the pentadienyl system (Scheme 47). The kinetic product is slowly transformed to the thermodynamic one over a period of several days through a reversal of the nucleophilic addition. The desired isomer is then taken on to the natural product. The most common type of η5-complexes is without doubt the ferrocenes. An interesting reaction published this year involves the use of chiral ferrocenyl diselenides.74 The ferrocenes contain a chiral α-methyl-dimethylamine function (Scheme 48). The diselenide is first broken down into an active form. This then acts as a template for a selenolactonisation reaction of an unsaturated carboxylic acid. The yields and enantioselectivities are good to excellent. Fluorocarbonyl ferrocene has been introduced as an efficient means of making ferrocene esters and amides. Reaction of the acid fluoride with amine or alcohols proceeds in excellent yields with a number of different substrates.75
Scheme 47
Scheme 48
8 η6-Complexes in organic synthesis
The η6-complexes of chromium have been the most popular this year. The two main uses have been the desymmetrising effect and the steric blocking of one face of the complexed metal. Simpkins has used the former with his chiral base technology to produce planarly chiral arene complexes (Scheme 49).76 Interestingly, the use of different chiral bases led to different complexes being produced. Direct deprotonation on the ring and addition to a benzophenone derivative has led to a method for the preparation of triaryl methanols.77 Deprotonation was better with t-BuLi than LDA, but diastereoselectivities were disappointingly low. Facially selective additions of sulfonamide anions to complexed benzaldehydes has been addressed.78 The stereochemistry is governed by the metal and the conformation of the arene–carbonyl bond (Scheme 50). In the cases reported, excellent selectivities are achieved. Removal of the metal and subsequent elaboration provides entry to β-sultams in excellent yields with very high stereocontrol. Finally the facially directing influence of the metal is illustrated in a stereoselective route to the mitosane skeleton.79 With the chromium complexed on one face of the ring system, selective reduction of the indolene double bond can be achieved, although the level of control is dependant on the reagents used (Scheme 51).
Scheme 49
Scheme 50
Scheme 51
9 Pauson–Khand reaction
The popularity of the Pauson–Khand reaction has continued in the last year, with a number of notable contributions to the area. The majority of syntheses begin with complexation of the alkyne unit to dicobalt octacarbonyl. However, a recent report uses an inorganic source, cobalt bromide, and reduces it with zinc dust in situ under an atmosphere of carbon monoxide.80 This forms the alkyne complex which can then be transformed into a cyclopentenone by reaction with an alkene. A main area of interest continues to be promoters for the reaction, i.e. reagents that in some way help to accelerate the reaction or enhance the yield. Kerr has explored the use of chiral N-oxides for promotion of an asymmetric Pauson–Khand reaction.81 For good enantioinduction, low temperatures are required and dimethoxyethane was found to be the best solvent. However, the chiral reaction appears to be restricted to the use of propargylic alcohols as substrates. Other alkynes gave virtually no enantiomeric excess. Kerr has also reported that the N-oxide promoter can be attached to a solid support.82 The amine was linked to an Argo-Gel support and then oxidised to its N-oxide. Use of this in a Pauson–Khand reaction gave good to excellent yield of the cyclopentenone product. This leads the way for the preparation of a library of N-oxide promoters that may be screened in the reaction. Other promoters that have been used this year include methyl sulfides (Scheme 52).83 A number of different methyl sulfides were screened and all gave enhanced yields of cyclopentenone products.
Scheme 52
An appealing alternative to the use of ethylene gas has been reported.84 Using vinyl esters in an intramolecular Pauson–Khand reaction, the cyclopentenone formed did not have the ester functionality present (Scheme 53). Strained alkenes are known to be good reaction partners in the Pauson–Khand reaction. Two papers this year report unsymmetrical variants and their use in determining the regiocontrol. Use of substituted 7-oxanorbornenes‡ gave reasonable regiocontrol,85 and substituted norbornenes were also found to be good substrates.86 Cazes reports the reaction of activated olefins, such as α,β-unsaturated esters (Scheme 54).87 Using an intramolecular Pauson–Khand reaction, Hoshino and co-workers have formed an angularly fused tricyclic system with control over two contiguous quaternary centres (Scheme 55).88 An interesting study on the construction of optically active bicyclo[4.3.0]nonenone derivatives has shown that changing protecting groups for adjacent hydroxy groups can have a significant effect on the stereochemistry of the annulation reaction.89 Using a bis-TBDMS protected diol, the cyclisation was biased in one direction. However, by tying the diol as a protected acetonide, the opposite stereochemistry was achieved (Scheme 56). The difference is proposed to occur through a change in the transition state geometry during the cyclisation. Brummond has continued her work in using allenes as substrates.90 In a short synthesis of the antitumour agent hydroxymethylfulvene, an allenic Pauson–Khand reaction forms the tricyclic skeleton in good yield in a remarkably short reaction time (Scheme 57). Cook has continued his work into tandem Pauson–Khand reactions.91 By building up a bis-enyne, a double, or tandem, Pauson–Khand reaction takes place to allow the construction of four new rings from an acyclic precursor in one step (Scheme 58). Use of chiral N-(ethynyl)allylglycines as substrates in an intramolecular Pauson–Khand reaction has been highlighted.92 The chirality present in the amino acid affords high levels of stereocontrol in the cyclisation step (Scheme 59). Finally, the use of the tert-butylsulfinyl group as a highly efficient chiral auxiliary has been reported.93 The chiral sulfoxide facilitates high levels of stereocontrol and is removable by reaction with ammonium chloride (Scheme 60).
Scheme 53
Scheme 54
Scheme 55
Scheme 56
Scheme 57
Scheme 58
Scheme 59
Scheme 60
References
A. C. Comely, S. E. Gibson and S. Sur, J. Chem. Soc., Perkin Trans. 1, 2000, 109 RSC.
A. C. Comely and S. E. Gibson, J. Chem. Soc., Perkin Trans. 1, 1999, 223 RSC.
T. Takahashi, C. Xi, Y. Ura and K. Nakajima, J. Am. Chem. Soc., 2000, 122, 3228 CrossRefCAS.
C. P. Park, J. W. Sung and D. Y. Oh, Synlett, 1999, 1055 CAS.
X. Huang and D.-H. Duan, Chem. Commun., 1999, 1741 RSC.
R. P. Spencer, C. L. Cavallaro and J. Schwartz, J. Org. Chem., 1999, 64, 3987 CrossRefCAS.
T. Takeda, N. Nozaki, N. Saeki and T. Fujiwara, Tetrahedron Lett., 1999, 40, 5353 CrossRefCAS.
T. Takeda, H. Taguchi and T. Fujiwara, Tetrahedron Lett., 2000, 41, 65 CrossRefCAS.
M. Abdur Rahim, T. Fujiwara and T. Takeda, Synlett, 1999, 1029 CAS.
M. Abdur Rahim, T. Fujiwara and T. Takeda, Tetrahedron, 2000, 56, 763 CrossRefCAS.
D. K. An, K. Hirakawa, S. Okamoto and F. Sato, Tetrahedron Lett., 1999, 40, 3737 CrossRefCAS.
H. Urabe, D. Hideura and F. Sato, Org. Lett., 2000, 2, 381 CrossRefCAS.
C. Averbuj, J. Kaftanov and I. Marek, Synlett, 1999, 1939 CAS.
C. Möller, M. Mikulás, F. Wierschem and K. Rück-Braun, Synlett, 2000, 182 CAS.
M. Mikulás, S. Rust, D. Schollmeyer and K. Rück-Braun, Synlett, 2000, 185 CAS.
S. G. Davies, N. M. Garrido, P. A. McGee and J. P. Shilvock, J. Chem. Soc., Perkin Trans. 1, 1999, 3105 RSC.
E. Kuester and L. S. Hegedus, Organometallics, 1999, 18, 5318 CrossRefCAS.
R. Aumann, M. Kössmeier, C. Mück-Lichtenfeld and F. Zippel, Eur. J. Org. Chem., 2000, 37 CrossRefCAS.
M. W. Davies, C. N. Johnson and J. P. A. Harrity, Chem. Commun., 1999, 2107 RSC.
H. Schirmer, T. Labalm, B. Flynn, Y.-T. Wu and A. de Meijere, Synlett, 1999, 2004 CAS.
Y. Zhang and J. W. Herndon, Tetrahedron, 2000, 56, 2175 CrossRefCAS.
H. Rudler, A. Parlier, B. Martin-Vaca, E. Garrier and J. Vaissermann, Chem. Commun., 1999, 1439 RSC.
J. Barluenga, M. Tomás, S. Ballesteros, J. C. Brillet, S. Garelá-Granada, A. Pinera-Nicholás and J. T. Vánquez, J. Am. Chem. Soc., 1999, 121, 4516 CrossRefCAS.
M. Zora, Y.-H. Li and J. W. Herndon, Organometallics, 1999, 18, 4429 CrossRefCAS.
B. Caro, P. Le Paul, F. Robin-Le Guen, M.-C. Sénéchal-Tocquer, J. Y. Saillard, S. Kohlal, L. Ouahat and S. Golken, Eur. J. Org. Chem., 2000, 577 CrossRefCAS.
C. Jäkel and K. H. Dötz, Tetrahedron, 2000, 56, 2167 CrossRefCAS.
B. Weyerchausen, M. Nieger and K. H. Dötz, J. Org. Chem., 1999, 64, 4206 CrossRef.
E. Licandro, S. Maiorana, C. Baldoli, L. Capella and D. Berdicchia, Tetrahedron: Asymmetry, 2000, 11, 975 CrossRefCAS.
D. Grée, V. Madiot and R. Grée, Tetrahedron Lett., 1999, 40, 6399 CrossRefCAS.
D. Díaz and V. S. Martin, Tetrahedron Lett., 2000, 41, 743 CrossRefCAS.
A. M. Montaña, D. Fernández, R. Pagès, A. C. Filippou and G. Kociok-Köhn, Tetrahedron, 2000, 56, 425 CrossRefCAS.


![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 13 and cyclobutenes
13 and cyclobutenes