An approach to the synthesis of 5,5-trans-fused lactam analogues of β-lactam antibiotics
Paul W.
Smith
*,
Andrew R.
Whittington
,
Kevin N.
Cobley
,
Albert
Jaxa-Chamiec
and
Harry
Finch
Department of Medicinal Chemistry, Glaxo Wellcome Limited, Gunnels Wood Road, Stevenage, UK SG1 2NY
First published on 5th December 2000
Abstract
A racemic synthesis of two diastereoisomeric α-benzyloxycarbonylamino substituted trans-fused bicyclic lactams (4 and 5), was achieved from cyclopentene oxide. These lactams are useful intermediates to investigate the possibility of using a trans-lactam template as a replacement for the β-lactam ring found in conventional antibacterial agents. One of the intermediates (4) was further elaborated to an analogue of the antibacterial agent ceftazidime.
Introduction
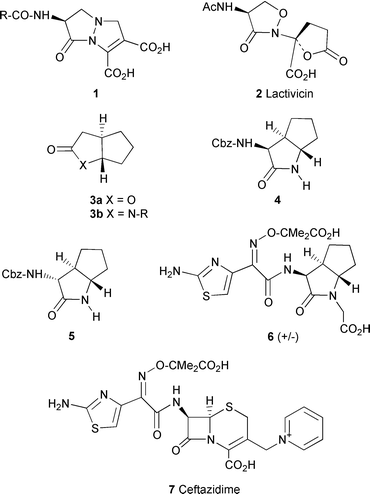
Over recent years the continuing problem of emerging resistance to chemotherapeutic agents has intensified the search for new and novel antibacterial drugs. In particular, the emergence of broad spectrum β-lactamases has reduced the effectiveness of many of the older β-lactam antibiotics and indeed rendered some of them obsolete. The situation can be retrieved by the co-administration of β-lactamase inhibitors e.g. Augmentin,1 but there are already reports of β-lactamases resistant to clavulanic acid.2 The new carbapenems such as imipenem![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 3 are potent antibacterials and useful inhibitors of β-lactamases which suggests that it is possible to combine these two properties in a single molecule. The ideal drug, however, would be a powerful inhibitor of the bacterial transpeptidases whilst displaying no affinity for β-lactamases. Towards this goal, in addition to synthesising β-lactam antibiotics with improved stability to β-lactamases, many groups have attempted to replace the β-lactam nucleus with appropriately substituted novel ring systems capable of selectively acylating the bacterial serine transpeptidases. Notable successes have been few, but the appreciable activity of the acyl hydrazides 14 and Lactivicin 2 (Fig. 1)5,6 suggests that other compounds of this type with useful antibacterial properties will be found.
3 are potent antibacterials and useful inhibitors of β-lactamases which suggests that it is possible to combine these two properties in a single molecule. The ideal drug, however, would be a powerful inhibitor of the bacterial transpeptidases whilst displaying no affinity for β-lactamases. Towards this goal, in addition to synthesising β-lactam antibiotics with improved stability to β-lactamases, many groups have attempted to replace the β-lactam nucleus with appropriately substituted novel ring systems capable of selectively acylating the bacterial serine transpeptidases. Notable successes have been few, but the appreciable activity of the acyl hydrazides 14 and Lactivicin 2 (Fig. 1)5,6 suggests that other compounds of this type with useful antibacterial properties will be found.
 | ||
| Fig. 1 | ||
The highly strained trans-fused 5,5 lactam and lactone ring systems 3a and b recently reported by co-workers at GlaxoWellcome represent novel templates for the design of compounds which can potentially acylate a wide range of serine proteases. Selective inhibitors of both thrombin and elastase have already been designed based on these templates.7–12 The current manuscript describes a useful racemic synthesis of the diastereoisomeric Cbz (CO2CH2C6H5) protected trans-lactams 4 and 5 which are suitable starting materials to investigate the potential of the 5,5-trans-lactam template to mimic the penicillin and cephalosporin β-lactam antibiotics. Transformation of compound 4 to the aminothiazolyl derivative 6, structurally related to the antibiotic ceftazidime 7, is also described.
Results and discussion
Synthesis of the lactams 4 and 5
For the synthesis of derivatives 4 and 5 we required a reliable method for the stereoselective formation of the protected amino acids 8 and 9 (see Scheme 2) which fixed the relative stereochemistry of the two adjacent chiral centres on the cyclopentane ring trans to each other. Synthesis of 8 and 9 was accomplished in 8 stages from the azido alcohol 10, which was obtained from cyclopentene oxide by ring opening with sodium azide (Scheme 1).13 Triphenylphosphine reduction of 10 and treatment with di-tert-butyl dicarbonate afforded the protected aminol 11 (41% yield) as a crystalline solid. Under Mitsunobu conditions 11 was cleanly converted to the Boc protected aziridine 12 in high yield (79%). The aziridine ring was then opened by heating 12 with the sodium salt of dimethyl malonate in DMF at 155 °C. This afforded 13 as a crystalline solid after flash chromatography in good yield (73%). On a larger scale 13 was isolated directly by crystallisation. By ring opening the aziridine in this fashion the required ring junction trans-stereochemistry has been fixed. The initial stages outlined above were readily carried out on 50 g scale affording 13 in 24% overall yield from 10 (Scheme 1).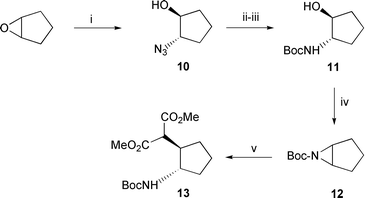 | ||
| Scheme 1 Reagents: i) NaN3, NH4Cl, aq. EtOH; ii) Ph3P; iii) Boc2O; iv) Ph3P–diisopropylazodicarboxylate; v) CH2(CO2Me)2, NaH, DMF. | ||
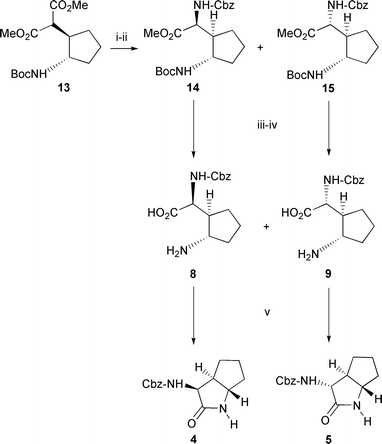
Selective hydrolysis of 13 with aqueous KOH afforded a diastereomeric mixture of monoacid esters in quantitative yield (Scheme 2). The isomers were not separated at this stage, but the mixture subjected to a Curtius rearrangement with diphenylphosphoryl azide (PhO)2P(O)N3 (DPPA) and then adding benzyl alcohol to trap the intermediate isocyanate14 affording a mixture of the Cbz protected diastereomers 14 and 15 (1∶1 ratio, 37% combined yield) (Scheme 2) which were separable by flash chromatography. Products 14 and 15 now contain both the nitrogens required for the final trans-lactam products in a differentially protected form.
Hydrolysis of the remaining methyl ester in 14 was achieved by treatment with aqueous sodium hydroxide and the Boc group subsequently removed with trifluoroacetic acid to afford the required amino acid 8 (91%). A similar sequence of reactions starting with 15 produced 9 also in high overall yield (91%). Finally 8 and 9 were cyclised to the required trans-lactams 4 and 5 respectively using DPPA in DMF (69% yield 8→4, 76% yield 9→5).6 In this reaction the initial acyl azide formed by the DPPA is trapped by the nucleophilic amine in an intramolecular reaction. The relative stereochemistry of 4 and 5 was deduced from their proton NMR spectra by examining the coupling constants and NOE’s between the H-3 proton adjacent to the amide carbonyl group and the ring junction protons H-3a and H-6a (Fig. 2). There is an NOE observed in 4 between H-3 and H-3a, but not between H-3 and H-6a, whereas in 5 there is an NOE between H-3 and H-6a but not between H-3 and H-3a. The relative stereochemistry of the earlier intermediates 14, 15, 8 and 9 was also inferred from this experiment.
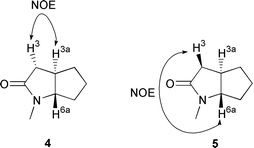 | ||
| Fig. 2 NOE’s observed in isomers 4 and 5. | ||
Design and synthesis of compound 6
In order to display antibacterial properties the β-lactam antibiotics require a suitably positioned acid moiety and an appropriately activated β-lactam nucleus.15 Once these features are in place, then the choice of the appropriate sidechain is critical in order to optimise binding affinity and specificity for the target penicillin binding proteins.16–18 Activation of the β-lactam is achieved by creating strain in the amide bond either through fusion to another ring (penicillins and cephalosporins etc.) or by introducing electron withdrawing substituents onto the amide nitrogen (monobactams such as aztreonam). An indication of ring strain can often be inferred from the IR stretching frequency of the amide carbonyl group.19,20 The carboxylic acid of the antibiotic forms a salt bridge with the lysine residue of the highly conserved Lys-X-X-Ser motif found in the penicillin binding proteins.21,22 In the initial trans-lactam target compound 6, the acetic acid extension from the amide nitrogen was chosen to allow 6 the possibility of forming this key interaction. Simple molecular modelling overlays (not shown) indicated that similar positions of the amide carbonyl groups and carboxylic acids of the trans-lactam 6 and cephalosporin nucleus of ceftazidime 7 were likely. The aminothiazolyl derived amide sidechain was chosen since this group has proved highly successful in the cephalosporin and monobactam series of antibiotics.23,24N-Alkylation of the amide in 4 was achieved with tert-butyl bromoacetate in acetonitrile with caesium carbonate25 producing 16 (79%) (Scheme 3). The Cbz group was next removed by hydrogenolysis liberating the amine 17 (94%). Conventional coupling of 17 with the acid 1826 afforded the fully protected intermediate 19 (60%). Finally, TFA deprotection of 19 gave the required target 6 (78%).
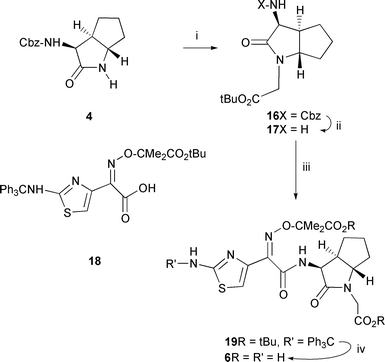 | ||
| Scheme 3 Reagents: i) BrCH2CO2tBu, Cs2CO3, MeCN; ii) H2, Pd; iii) Acid 18 + (COCl)2; iv) CF3COOH. | ||
Disappointingly compound 6 showed no whole cell antibacterial activity when evaluated in vitro against a screen of commonly encountered bacteria comprising both Gram positive and Gram negative organisms (MIC’s all > 128 μg cm3).27 Neither did it demonstrate any significant in vitro inhibition of penicillin G binding to penicillin binding proteins, or any inhibition of TEM-1 β-lactamase.28 Thus it appears likely that the trans-lactam ring in compound 6 is not sufficiently activated to acylate the bacterial enzymes. (The observed IR spectra may provide some evidence for this. Whilst the highly strained penicillins and cephalosporins typically show carbonyl stretches between 1780 and 1790 cm−1 the trans-lactam amide carbonyl group in compound 6 stretches only at 1718 cm−1.)
In conclusion, synthesis of further trans-lactams with increased activation would be of interest in order to determine whether a useful trans-lactam antibacterial agent can be achieved. Compounds 4 and 5 will be ideal starting materials to pursue this investigation.
Experimental
General methods
FTIR spectra were recorded using a Nicolet 20SXB or a Bio-Rad FTS-7. 1H NMR spectra were recorded either at 250 MHz using a Bruker AC or AM 250 or at 400 MHz with a Varian VXR 400. Mass spectra were measured on a HP Engine (thermospray positive) or VG Autospec Q (LSIMS). Routine microanalyses were performed on a Leco CHNS-932 or Carlo-Erba instrument. Fluorine analyses were carried out with a Phillips PW 9415 ion selective meter and water analyses using a Mitsubishi CA-05. Flash chromatography was performed with Merck Kieselgel 9385.trans-N-(2-Hydroxycyclopentyl)carbamic acid tert-butyl ester 11
Cyclopentene oxide (50.0 g, 0.595 mol) was dissolved in 50% aqueous ethanol (800 cm3) and ammonium chloride (31.9 g, 0.595 mol) and sodium azide (38.7 g, 0.595 mol) added. The resulting mixture was refluxed for 48 h then concentrated to a small volume and partitioned between water (200 cm3) and ethyl acetate (200 cm3). The organic layer was dried (anhydrous MgSO4) and evaporated to a yellow oil (64.4 g, 85%) which was used directly without further purification. (Crude azide νmax (CHBr3)/cm−1 3430 and 2105; δH (CDCl3): 4.2 (1H, m, H-1), 3.7 (1H, m, H-2), 2.2–2.0 (2H, m), 1.9–1.5 (5H, m).)The crude azido alcohol (33.4 g, 0.263 mol) was dissolved in toluene (500 cm3) and refluxed with triphenylphosphine (103.2 g, 0.394 mol, 1.5 eq.) for 1.5 h. The solution was cooled to 0 °C and diluted with ethyl acetate (50 cm3) and saturated sodium bicarbonate solution (50 cm3). The resulting biphasic mixture was then stirred rapidly as di-tert-butyl dicarbonate (57.3 g, 0.263 mol) in ethyl acetate (500 cm3) was added over several minutes. The mixture was allowed to warm to room temperature and stirred for 12 h. The organic phase was then separated, dried (anhydrous MgSO4) and evaporated to a yellow oil. This was treated with ether (500 cm3) to precipitate Ph3PO which was removed by filtration. The filtrate was concentrated to an oil which was purified by flash chromatography (eluted with ethyl acetate–petroleum ether 1∶1) to afford the title compound 11 (21.4 g, 41%) as a white crystalline solid. Mp 106 °C (Found C: 59.5; H, 9.25; N, 6.8. C10H19NO3 Requires C, 59.7; H, 9.5; N, 7.0%). νmax (CHBr3)/cm−1 3437, 2972, 1690, 1498 and 1366; δH (CDCl3): 4.7 (1H, br s, NH), 4.0 (2H, m, OH, H-1), 3.6 (1H, m, H-2), 2.2–1.3 (6H, m, H-3,4,5), 1.4 (9H, s, tBu).
cis-6-Azabicyclo[3.1.0]hexane-6-carboxylic acid tert-butyl ester 12
To a stirred solution of triphenylphosphine (114.0 g, 0.435 mol, 1.5 eq.) in THF (800 cm3) at −78 °C under nitrogen was added diisopropyl azodicarboxylate (85.6 cm3, 0.435 mol, 1.5 eq.) dropwise over 30 min. The reaction mixture was stirred for 30 min until a pale yellow suspension was formed. A solution of the alcohol 11 (58.3 g, 0.290 mol) in THF (200 cm3) was added dropwise and the reaction stirred for 60 min at −78 °C then allowed to warm to RT. After a further 21 h at RT the solvent was evaporated and the residue treated with ether. The precipitated triphenylphosphine oxide was removed by filtration and the filtrate evaporated to a pale yellow residue which was purified by flash chromatography (eluting with petroleum ether–ether, 4∶1) to give the title compound 12 (41.9 g, 79%) as a clear oil (Found C: 65.5; H, 9.35; N, 7.6. C10H17NO2 Requires C, 65.1; H, 9.2; N, 7.6%); νmax (CHBr3)/cm−1 2973, 1699 and 1367; δH (CDCl3): 2.9 (2H, s, H-1,5), 2.05 (2H, dd, H-2,4), 1.6 (3H, m, H-2,3,4), 1.4 (9H, s, tBu), 1.2 (1H, m, H-3). m/z C10H17NO2 M+ 183.26.trans-2-(2-tert-Butoxycarbonylaminocyclopentyl)malonic acid dimethyl ester 13
Dimethyl malonate (90.3 g, 0.684 mol, 3 eq.) in DMF (1.5 l) was treated with sodium hydride (60% dispersion in oil, 9.3 g, 0.232 mol, 1.02 eq.) at RT under nitrogen. After effervescence had stopped and the solution had cleared a solution of the aziridine 12 (41.8 g, 0.228 mol, 1 eq.) in DMF (25 cm3) was added. The solution was stirred with heating (155 °C) for 18 h then cooled, filtered and evaporated to a solid. The crude product was partitioned between ether (1.5 l) and water (1.5 l). The organic layer was separated and the aqueous layer further extracted with ether (1.5 l) and the combined organic extracts washed with brine (2 × 1 l) and then dried (MgSO4). Evaporation gave a solid which was triturated with petrol 40–60°C then filtered and dried in vacuo to give a white solid 13 (52.5 g, 0.167 mol, 73%). Mp 104–105 °C (Found C, 57.05; H, 8.1; N, 4.6. C15H25NO6 Requires C, 57.1; H, 8.0; N, 4.4); νmax (CHBr3)/cm−1: 3429, 2950, 1750, 1729, 1705 and 1503; δH (CDCl3): 4.5 (1H, br s, NH), 3.75 (6H, s, MeO), 3.7 (1H, m, H-2′), 3.45 (1H, d, H-2, J2,1′ 7.5 Hz), 2.3 (1H, m, H-1′), 2.15–1.2 (6H, m, H-3′,4′,5′), 1.4 (9H, s, tBu); m/z 333 (MNH4+), 316 (MH+), 216.Racemic [(1R,2S)-2-tert-butoxycarbonylaminocyclopentyl][(S![[hair space]](https://www.rsc.org/images/entities/h3_char_200a.gif) )-benzyloxycarbonylamino]acetic acid methyl ester 14 and Racemic [(1R,2S)-2-tert-butoxycarbonylaminocyclopentyl][(R)-benzyloxycarbonylamino]acetic acid methyl ester 15
)-benzyloxycarbonylamino]acetic acid methyl ester 14 and Racemic [(1R,2S)-2-tert-butoxycarbonylaminocyclopentyl][(R)-benzyloxycarbonylamino]acetic acid methyl ester 15
To a solution of the malonate 13 (10.0 g, 31.7 mmol) in methanol (150 cm3) was added potassium hydroxide (8.6 g, 153.6 mmol, 4.8 eq.) in water (60 cm3). The reaction mixture was stirred for 20 min then partitioned between ether (750 cm3) and water (750 cm3). The aqueous phase was acidified with solid potassium hydrogen sulfate and extracted with ether (2 × 750 cm3). The combined organic extracts were dried (anhydrous MgSO4) and evaporated to an off-white foam containing a mixture of the mono-acid mono-esters (9.6 g, 100%). δH (CDCl3): 5.1, 4.7 (1H, br d, NH), 3.75 (3H, s, MeO), 3.6–3.8 (2H, m, H-2,2′), 2.4–1.4 (7H, m, H-1′,3′4′5′), 1.4, 1.45 (9H, s, tBu). This material was used in the next stage without further purification.
A mixture of acids prepared as above (17.9 g, 59.5 mmol) was dissolved in dioxane (300 cm3) and treated with triethylamine (8.3 cm3, 59.5 mmol) and diphenylphosphoryl azide (14.1 cm3, 11.8 mmol). The solution was refluxed for 4 h under nitrogen when benzyl alcohol (25 cm3) was added and refluxing continued for a further 18 h. The reaction mixture was cooled and poured into water (2 l). The aqueous solution was extracted with ether (3 × 500 cm3) then treated with brine and further extracted with ether (450 cm3). The combined organic extracts were washed with brine (500 cm3) then dried (anhydrous MgSO4) and the solvent evaporated finally in vacuo (water bath at 90 °C) to remove the benzyl alcohol. The resulting brown residue was purified by flash chromatography (eluting first with petroleum ether–ether 2∶1, then petroleum ether–ether, 1∶1) to obtain racemic 14 as a white crystalline solid (1.6 g, 3.9 mmol, 7%) eluting first, and racemic isomer 15 eluting second as an off-white gum (2.0 g, 5.0 mmol, 8%). Further 14 and 15 was obtained as a mixed fraction (5.3 g, 13.1 mmol, 22%). Thus the combined overall yield of 14 and 15 was 37%. Compound 14. (Found C, 61.8; H, 7.35; N, 7.2. C21H30N2O6 Requires C, 62.1; H, 7.4; N, 6.9%); νmax (CHBr3)/cm−1 3349 (br), 1728, 1714, 1694, 1682 and 1519; δH (CDCl3): 7.4–7.3 (5H, m, Ph), 6.4 (1H, br d, Cbz-NH), 5.1 (2H, s, PhCH2), 4.4 (2H, m, Boc-NH, H-2), 3.72 (1H, m, H-2′), 3.7 (3H, s, MeO), 2.3–1.3 (7H, m, H-1′,3′,4′,5′), 1.4 (9H, s, tBu). Compound 15. νmax (CHBr3)/cm−1 3300 (br), 1721, 1712 and 1693; δH (CDCl3): 7.4–7.3 (5H, m, Ph), 6.1 (1H, br s, Cbz-NH), 5.1 (2H, ABq, PhCH2), 4.55 (1H, br s, Boc-NH), 4.4 (1H, br t, H-2), 3.8 (1H, m, H-2′), 3.7 (3H, s, MeO), 2.2–1.1 (7H, m, H-1′,3′,4′,5′), 1.45 (9H, s, tBu); m/z 407.2182. MH+ C21H30N2O6 requires 407.2182.
Racemic [(1S,2S)-2-aminocyclopentyl][(S)-benzyloxycarbonylamino]acetic acid hydrochloride 8
The ester 14 (0.57 g, 1.4 mmol) was dissolved in methanol (10 cm3) and treated with 1 M potassium hydroxide (aq.) solution (4.2 cm3, 3 eq.). The mixture was stirred for 3 h at room temperature then partitioned between ether (150 cm3) and water (100 cm3). The aqueous layer was further washed with ether (150 cm3), then acidified to pH = 3 with cooling in an ice bath. The resulting solution was extracted with ether (2 × 100 cm3) and the combined organic extracts washed with brine (100 cm3) and dried (anhydrous MgSO4). Evaporation of the solvent gave a white foam containing the intermediate N-Boc protected acid (0.50 g, 1.3 mmol, 91.0%). This material was used in the next reaction without further purification (Found C, 60.6; H, 7.1; N, 7.2. C20H28N2O6 Requires C, 61.2; H, 7.1; N, 7.1%); νmax (KBr)/cm−1 3322 (br), 1712, 1650 and 1518; δH (CDCl3): 7.4–7.3 (5H, m, Ph), 6.85, 6.05 (1H, 2 × br d, NHCbz, rotamers), 5.1 (2H, m, PhCH2), 5.4, 4.9 (1H, 2 × br d, NHBoc, rotamers), 4.5 (1H, m, H-2), 3.75 (1H, m, H-2′), 2.25–1.5 (7H, m, H-1′,3′,4′,5′), 1.4 (9H, s, tBu).
N-Boc protected acid (1.20 g, 2.9 mmol) prepared as above was dissolved in TFA (40 cm3). After 10 min the solvent was evaporated and the residue redissolved in ethyl acetate (50 cm3) then treated with a solution of HCl in ether (1.5 M, excess). Evaporation of the solvent gave a gum which crystallized on trituration with ether. This material was dried in vacuo affording the title compound 8 (0.97 g, 2.9 mmol, 100%) as a white solid. νmax (film)/cm−1: 3487, 2960, 1715, 1669 and 1538; δH (DMSO): 8.1 (3H, br s, NH3+), 7.7 (1H, d, NH, J 7.5 Hz), 7.4–7.25 (5H, m, Ph), 5.05 (2H, s, PhCH2), 4.1 (1H, t, H-2, J 7.5 Hz), 3.4 (1H, m, H-2′), 2.25–1.3 (7H, m, H-1′, 3′, 4′, 5′); m/z 293.1500. MH+ C15H21N2O4 requires 293.1501.
Racemic [(1S,2S)-2-aminocyclopentyl][(R)-benzyloxycarbonylamino]acetic acid hydrochloride 9
The methyl ester 15 (1.47 g, 3.62 mmol) was treated similarly to 14 to give the intermediate N-Boc protected acid (1.34 g, 3.41 mmol, 94%) as a white foam. This crude material (1.23 g, 3.13 mmol) was treated with TFA to give the title compound 9 (1.00 g, 3.04 mmol, 97%) as an off-white solid. νmax (Nujol)/cm−1 2923, 1700, 1521, 1455 and 1376; δH (DMSO): 8.1 (3H, br s, NH3+), 7.6 (1H, d, NH, J 8.5 Hz), 7.4–7.3 (5H, m, Ph), 5.05 (2H, s, PhCH2), 4.4 (1H, dd, H-2, J 5, 8.5 Hz), 3.3 (1H, m, H-2′), 2.4–1.5 (7H, m, H-1′,3′,4′,5′); m/z 293.1502. MH+ C15H20N2O4 requires 293.1501.
Racemic [(3S,3aS,6aS)-2-oxooctahydrocyclopenta[b]pyrrol-3-yl]carbamic acid benzyl ester 4
The amino acid hydrochloride salt 8 (0.86 g, 2.6 mmol) was dissolved in DMF (10 cm3) and added via a syringe pump to a solution of diphenylphosphoryl azide (1.12 cm3, 5.20 mmol) and triethylamine (1.26 cm3, 9.09 mmol) in DMF (200 cm3). The total addition time was 28 h. The solution was then concentrated to an oily solid and partitioned between water (300 cm3) and ether (300 cm3). The aqueous layer was further washed with ether (300 cm3) and the combined organic layers washed with brine (2 × 300 cm3). The extracts were dried (anhydrous MgSO4) and then evaporated to an oil which was triturated, filtered and dried in vacuo to give the title compound 4 as a white solid (0.49 g, 1.80 mmol, 69%) (Found C, 65.4; H, 6.6; N, 10.3. C15H18N2O3 Requires C, 65.7; H, 6.6; N, 10.2%); νmax (CHBr3)/cm−1 3418, 2974, 1712, 1506; δH (CDCl3): 7.4–7.3 (5H, m, Ph), 6.26 (1H, br s, amide NH), 5.28 (1H, br d, Cbz-NH), 5.11 (2H, ABq, PhCH2), 4.24 (1H, t, H-3), 3.21 (1H, dt, H-6a), 2.1–1.3 (7H, m, H-3a,4,5,6).
Racemic [(3R,3aS,6aS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) )-2-oxooctahydrocyclopenta[b]pyrrol-3-yl]carbamic acid benzyl ester 5
)-2-oxooctahydrocyclopenta[b]pyrrol-3-yl]carbamic acid benzyl ester 5
Using the same procedure described for preparation of 4, the amino acid hydrochloride salt 9 (0.90 g, 2.74 mmol) in DMF (10 cm3) was added via a syringe pump to a solution of diphenylphosphoryl azide (1.35 cm3, 6.26 mmol) and triethylamine (1.52 cm3, 10.96 mmol) in DMF (200 cm3) to afford 5 as a white solid (0.57 g, 76%) (Found C, 65.4; H, 6.5; N, 10.2. C15H18N2O3 Requires C, 65.7; H, 6.6; N, 10.2%); νmax (CHBr3)/cm−1 3419, 1707 and 1506; δH (CDCl3): 7.4–7.3 (5H, m, Ph), 6.2 (1H, br s, NH amide), 5.4 (1H, br s, Cbz-NH), 5.1 (2H, ABq, PhCH2), 3.97 (1H, dd, H-3), 3.09 (1H, m, H-6a), 2.1–1.3 (7H, m, H-3a,4,5,6).
Racemic [(3S,3aR,6aS)-2-oxo-3-benzyloxycarbonylaminooctahydrocyclopenta[b]pyrrol-1-yl]acetic acid tert-butyl ester 16
Cbz protected trans-lactam 4 (0.15 g, 0.58 mmol) was dissolved in acetonitrile (10 cm3) and caesium carbonate (0.47 g, 1.45 mmol) added. The resulting suspension was treated with tert-butyl bromoacetate (0.19 cm3, 1.16 mmol) and the reaction heated at 50 °C for 4 h. After cooling, filtration and evaporation of solvent a residual gum was obtained which was purified by flash chromatography (eluant ethyl acetate–cyclohexane, 1∶3) affording the title compound 16 (0.17 g, 79%) as a colourless gum. νmax (CHBr3)/cm−1 1738, 1729, 1712 (trans-lactam), 1694; δH (CDCl3): 7.4–7.3 (5H, m, Ph), 5.1 (3H, m, PhCH2, NH), 4.35 (1H, br t, H-3), 3.9 (2H, ABq, CH2CO2), 3.35 (1H, dt, H-6a), 2.1–1.3 (7H, m, H-3a,4,5,6), 1.4 (9H, s, tBu).
Racemic [(3S,3aR,6aS)-2-oxo-3-aminohexahydrocyclopenta[b]pyrrol-1-yl]acetic acid tert-butyl ester acetate salt 17
Cbz trans-lactam 16 (0.17 g, 0.44 mmol) was dissolved in acetic acid (20 cm3) and hydrogenated with 10% palladium on carbon (50 mg) at room temperature for 18 h. The solution was filtered and evaporation afforded the title compound 17 (0.13 g, 94%) as a crystalline solid. νmax (KBr)/cm−1 1745, 1710 and 1554; δH (DMSO): 4.35 (3H, br s, NH3+), 3.95 (2H, ABq, CH2CO2), 3.5 (2H, m, H-3,6a), 2.1–1.3 (7H, m, H-3a,4,5,6), 1.4 (9H, s, tBu); m/z 509 (2M + H+), 254 (MH+).
Racemic 2-[(2-Aminothiazol-4-yl)-2-[(3S,3aR,6aS)-1-carboxymethyl-2-oxooctahydrocyclopenta[b]pyrrol-3-ylcarbamoyl]methyleneaminooxy]-2-methylpropionic acid trifluoroacetate 6
Oxalyl choride (0.050 cm3, 0.57 mmol) was added to a solution of DMF (0.066 cm3, 0.85 mmol) in dichloromethane (5 cm3) at −20 °C under nitrogen. Upon completion of the addition, the solution was warmed to ice bath temperature and the acid 1818 (0.416 g, 0.73 mmol) added as a solid. After 10 min a solution of the acetate salt of the amine 17 (0.120 g, 0.38 mmol) in dichloromethane (2 cm3) was added with diisopropylethylamine (0.4 cm3, 2.29 mmol). After 24 h, the mixture was diluted with ethyl acetate and water, and the organic layer washed further with 10% aqueous citric acid solution, then saturated aqueous sodium bicarbonate and finally brine. After drying (anhydrous MgSO4) and evaporation the residue was purified by flash chromatography (eluant ethyl acetate–cyclohexane, 1∶3) affording the fully protected 19 (0.183 g, 60%) as a white solid. Compound 19: mp 103 °C; νmax (CHBr3)/cm−1 3298 (br), 1737, 1731, 1714, 1681 and 1519; δH (CDCl3): 7.4 (1H, d, NH amide), 7.35–7.25 (15H, m, 3 × Ph), 6.8 (1H, s, trityl NH), 6.75 (1H, s, thiazole H), 4.7 (1H, dd, H-3), 3.95 (2H, ABq, CH2CO2), 3.4 (1H, dt, H-6a), 1.65, 1.60 (6H, 2s, CMe2), 1.4 (18H, s, 2 × tBu), 2.1–1.3 (7H, m, H-3a,4,5,6); m/z 808 (MH+), 243.
Compound 19 (0.122 g, 0.031 mmol) was dissolved in trifluoroacetic acid (2 cm3) containing a trace of anisole. After 2.5 h, the solution was diluted with ether which precipitated the title compound 6 (0.067 g, 78%) as a white solid which was collected by filtration. Mp 141 °C; νmax (Nujol)/cm−1 1718 and 1674; δH (DMSO): 8.7 (1H, d, thiazole H), 4.45 (1H, dd, H-3), 3.9 (2H, ABq, CH2CO2H), 3.35 (1H, m, H-6a), 1.45, 1.4 (6H, 2s, CMe2), 2.0–1.3 (7H, m, H-3a,4,5,6); HPLC 95%; m/z 454.1397. C18H23N5O7S requires MH+ 454.1396.
Acknowledgements
The authors wish to express their thanks to Gavin Chung for performing the biological evaluation of compound 6.References
- D. Jackson and D. H. Staniforth, Acta Ther., 1982, 8, 367 Search PubMed.
- M. Hedberg and C. E. Nord, J. Chemother. (Florence), 1996, 8, 3 Search PubMed.
- S. P. Clissold, P. A. Todd and D. M. Campoli-Richards, Drugs, 1987, 33, 183 Search PubMed.
- L. N. Jungheim, D. B. Boyd, J. M. Indelicato, C. E. Pasini, D. A. Preston and W. E. Alborn, Jr., J. Med. Chem., 1991, 34, 1732 CrossRef CAS and references cited therein..
- Y. Nozaki, N. Katayama, H. Ono, S. Tsubotani, S. Harada, H. Okazaki and Y. Nakao, Nature, 1987, 325, 179 CrossRef CAS.
- Y. Nakao , in Recent Advances in the Chemistry of β-Lactam Antibiotics, eds. P. H. Bentley and R. Southgate, Special Publication No. 70, RSC, London, 1989, ch. 8. Search PubMed.
- M. Pass, S. Abu-Rabie, A. Baxter, R. Conroy, S. J. Coote, A. P. Craven, H. Finch, S. Hindley, H. A. Kelly, A. W. Lowdon, E. McDonald, W. L. Mitchell, N. A. Pegg, P. A. Procopiou, N. G. Ramsden, R. Thomas, D. A. Walker, N. S. Watson, H. Jhoti, C. J. Mooney, C.-M. Tang, P. J. Thomas, S. Parry and C. Patel, Bioorg. Med. Chem. Lett., 1999, 9, 1657 CrossRef CAS.
- M. Pass, R. E. Bolton, S. J. Coote, H. Finch, S. Hindley, A. Lowdon, E. McDonald, J. McLaren, M. Owen, N. A. Pegg, V. J. Mooney, C.-M. Tang, S. Parry and C. Patel, Bioorg. Med. Chem. Lett., 1999, 9, 431 CrossRef CAS.
- H. Finch, N. A. Pegg, J. McLaren, A. Lowdon, R. Bolton, S. J. Coote, U. Dyer, J. G. Montana, M. R. Owen, M. Dowle, D. Buckley, B. C. Ross, C. Campbell, C. Dix, C. Mooney, C.-M. Tang and C. Patel, Bioorg. Med. Chem. Lett., 1998, 8, 2955 CrossRef CAS.
- S. J. F. Macdonald, D. J. Belton, D. M. Buckley, J. E. Spooner, M. S. Anson, L. A. Harrison, K. Mills, R. J. Upton, M. D. Dowle, R. A. Smith, C. R. Molloy and C. Risley, J. Med. Chem., 1998, 41, 3919 CrossRef CAS.
- H. A. Kelly, R. Bolton, S. A. Brown, S. J. Coote, M. Dowle, U. Dyer, H. Finch, D. Golding, A. Lowdon, J. McLaren, J. G. Montana, M. R. Owen, N. A. Pegg, B. C. Ross, R. Thomas and D. A. Walker, Tetrahedron Lett., 1998, 39, 6979 CrossRef CAS.
- S. J. F. Macdonald, G. D. E. Clarke, M. D. Dowle, L. A. Harrison, S. T. Hodgson, G. G. A. Inglis, M. R. Johnson, P. Shah, R. J. Upton and S. B. Walls, J. Org. Chem., 1999, 64, 5166 CrossRef.
- J. R. McCarthy, P. E. Wiedeman, A. J. Schuster, J. Whitten, R. J. Barbuch and J. C. Huffman, J. Org. Chem., 1985, 50, 3095 CrossRef CAS.
- T. Shioiri, K. Ninomiya and S. Yamada, J. Am. Chem. Soc., 1972, 94, 6203 CrossRef CAS.
- For a review see F. C. Neuhaus and N. Georgopapakadou , in Emerging Targets in Antibacterial and Antifungal Chemotherapy, ch. 9, eds. J. Sutcliffe and N. H. Georgopapadakou, Chapman and Hall, New York, London, 1992. Search PubMed.
- M. P. Singh, Y. I. Lee, R. Singh, R. G. Micetich and B. J. Wilkinson, J. Antimicrob. Chemother., 1991, 27, 459 CAS.
- M. Nishida and Y. Shigi , The Role of Permeability and Affinity for Penicillin-binding Proteins in the Antibacterial Activity of the Third-generation Cephalosporins, in Symp. Front. Pharmacol., 1982, vol. 1 (New β-Lactam Antibiotics), pp. 65–81. Search PubMed.
- N. A. C. Curtis, D. Orr, G. W. Ross and M. G. Boulton, Antimicrob. Agents Chemother., 1979, 16, 533 CAS.
- M. Narisada, Pure Appl. Chem., 1987, 59, 459 CAS.
- M. Takasuka, J. Nishikawa and K. Tori, J. Antibiot., 1982, 35, 1729 Search PubMed.
- J. M. Ghuysen, Annu. Rev. Microbiol., 1991, 45, 37 CrossRef CAS.
- M. Leyh-Bouille, J. Van Beeumen, S. Renier-Pirlot, B. Joris, M. Nguyen-Disteche and J. M. Ghuysen, Biochem. J., 1989, 260, 601 CAS.
- R. N. Brogden and R. C. Heel, Drugs, 1986, 31, 96 Search PubMed.
- C. E. Newall, Chem. Br., 1987, 23, 976 Search PubMed.
- D. Gala, M. Steinman and R. S. Jaret, J. Org. Chem., 1986, 51, 4488 CrossRef CAS.
- C. H. O’Callaghan , B. E. Ayres and D. G. H. Livermore , Br. Pat. Appl., 2 040 921, Oct 26, 1979. .
- Agar MIC’s were determined by a two-fold agar dilution method using Mueller–Hinton media (Becton Dickinson Ltd, USA). Exponentially growing cultures were inoculated onto the surface of agar plates using a multipoint inoculator (Denley, UK) to produce an inoculum of approximately 105 cfu per cm3 (cfu = colony forming units). The plates were incubated at 37 °C for 18 hours and the minimum inhibitory concentration (MIC) value interpreted as the lowest antibiotic concentration which completely inhibited bacterial growth..
- TEM-1 β-lactamase was pre-incubated with compound 6 before the chromogenic substrate nitrocefin (50 μg cm−3) was added. The enzymatic hydrolysis of nitrocefin was followed at 495 nm. Compound 6 failed to inhibit the activity of TEM-1 at the highest concentration used (1 mM)..
| This journal is © The Royal Society of Chemistry 2001 |