Organophosphorus chemistry. Part 1. The synthesis of alkyl methylphosphonic acids
Christopher M.
Timperley
*,
Michael
Bird
,
Ian
Holden
and
Robin M.
Black
DERA, CBD Porton Down, Salisbury, Wiltshire, UK SP4 0JQ
First published on 11th December 2000
Abstract
Several alkyl hydrogen methylphosphonates of structure RO(HO)P(O)Me were synthesised by a three-stage route [R = i-Pr, n-Bu, i-Bu, s-Bu, pinacolyl Me3C-CH(Me)-,† cyclopentyl and cyclohexyl]. Trimethyl phosphite was transesterified with alcohols in the presence of sodium catalyst to give the mixed phosphites (MeO)2POR in 6–64% yield. Treatment of these with methyl iodide gave alkyl methyl methylphosphonates RO(MeO)P(O)Me in 66–95% yield. Selective demethylation of these compounds by bromotrimethylsilane, followed by methanolysis of the phosphorus silyl esters RO(Me3SiO)P(O)Me, gave the hydrogen phosphonates in 60–97% yield.
Introduction
The synthesis of pure alkyl hydrogen methylphosphonates of structure RO(HO)P(O)Me is challenging. These compounds are important in the verification of alleged use of chemical warfare agents, since they are stable hydrolysis products of G and V nerve agents.1,2 Most literature routes to alkyl hydrogen methylphosphonates involve alkaline hydrolysis of symmetrical or mixed phosphonates by treating them with an excess of sodium hydroxide![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 3,4 or barium hydroxide.5 Under these conditions, dialkyl alkylphosphonates hydrolyse to the mono-ester salts and the rates depend on the structure of the ester groups.6 The work-up of the reaction mixture is tedious, involving acidification and extraction, and yields of isolated products are often low. Another disadvantage is that the starting phosphonates are not readily available and have to be made in two or three steps. An improved approach to alkyl hydrogen methylphosphonates involves controlled hydrolysis of methylphosphonic dichloride to give an anhydride that, on alcoholysis, gives the desired products in moderate yields (Scheme 1).7,8
3,4 or barium hydroxide.5 Under these conditions, dialkyl alkylphosphonates hydrolyse to the mono-ester salts and the rates depend on the structure of the ester groups.6 The work-up of the reaction mixture is tedious, involving acidification and extraction, and yields of isolated products are often low. Another disadvantage is that the starting phosphonates are not readily available and have to be made in two or three steps. An improved approach to alkyl hydrogen methylphosphonates involves controlled hydrolysis of methylphosphonic dichloride to give an anhydride that, on alcoholysis, gives the desired products in moderate yields (Scheme 1).7,8
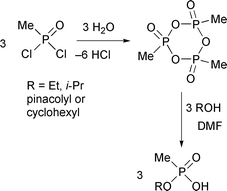 | ||
| Scheme 1 | ||
Clearly there is a need for improved syntheses of alkyl hydrogen methylphosphonates. In this paper we report a three-stage synthesis starting from commercially-available trimethyl phosphite. Its partial transesterification, followed by an Arbusov reaction with methyl iodide, gave alkyl methyl methylphosphonates in reasonable yields. Rapid reaction with bromotrimethylsilane, then methanolysis of the formed trimethylsilyl esters, provided the desired acids without resort to aqueous work-up (Scheme 2).
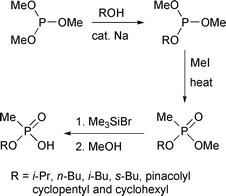 | ||
| Scheme 2 | ||
Results and discussion
Transesterification of trimethyl phosphite
A few mixed dialkyl alkyl phosphites of structure (RO)2POR′ have been prepared by reaction of alcohols with alkyl phosphorodichloridites in the presence of base (e.g. sodium methoxide or N,N-dimethylaniline)9–11 or by partial transesterification of trialkyl phosphites.12–15 In this study, trimethyl phosphite was transesterified with a number of alcohols. The reactions were carried out at temperatures slightly below the boiling points of the alcohols, with metallic sodium as catalyst, and in general proceeded smoothly. After distillation, the mixed phosphites 1a–i were isolated in low to moderate yields (Scheme 3, Table 1). These colourless liquids were stored under argon to prevent atmospheric oxidation to the corresponding phosphates and were stable for many months in a refrigerator.
| Compound | R group | Yield (%) | Bp (°C/mmHg) |
|---|---|---|---|
| a Previously made in 78% yield by transesterification of trimethyl phosphite, but no experimental details were given (lit.,12 bp 47–48 °C/14 mmHg). b Previously made by methanolysis of butyl phosphorodichloridite BuOPCl2 in ether in the presence of N,N-dimethylaniline (lit.,10 bp 65–66 °C/18 mmHg). | |||
| 1a |
n-Pra |
21 | 46–49/10 |
| 1b | i-Pr | 12 | 37–38/8 |
| 1c |
n-Bub |
40 | 61–62/10 |
| 1d | i-Bu | 44 | 53–55/10 |
| 1e | s-Bu | 24 | 54–56/10 |
| 1f | t-Bu | 16 | 47–48/10 |
| 1g | Pinacolyl | 58 | 69–70/10 |
| 1h | Cyclopentyl | 6 | 80–81/10 |
| 1i | Cyclohexyl | 64 | 92–94/7 |
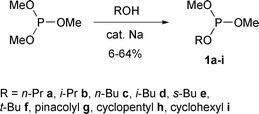 | ||
| Scheme 3 | ||
A limitation of the transesterification reaction as a preparative method is that the outgoing alcohol must have a boiling point that is not too close to that of the incoming alcohol; otherwise, the reaction cannot be forced by distilling out the originally esterified alcohol, and difficulty is encountered in the separation of the product from the starting phosphite. An example of this is the transesterification of trimethyl phosphite with i-PrOH (bp 82 °C compared to 65 °C for MeOH) which gave dimethyl isopropyl phosphite 1b in only 12% yield after five distillations. The low yield of the mixed phosphite 1h from cyclopentanol (6% after two distillations) was surprising given the high yield of phosphite 1i from cyclohexanol (64% after one distillation). The failure of the former reaction may have been due to dehydration of the alcohol, with hydrolysis of the trimethyl phosphite by the liberated water, or decomposition of the mixed phosphite thermally to cyclopentene and dimethyl phosphonate. An analogous explanation was used to account for the failure to isolate a mixed phosphite from the attempted transesterification of triethyl phosphite with tert-amyl alcohol (2-methylbuten-2-ol).13
Spectroscopic data for compounds 1a–i are given in Table 2. They have phosphorus chemical shifts between 140–135 ppm. The frequencies of the P–O–C vibrations in the infrared spectra of esters of trivalent phosphorus16 fall between 1030–1015 cm−1. The frequencies of the P–O–Me vibrations of the mixed phosphites were 1026–1014 cm−1.
| Compound | R | 1H NMR δ, J/Hz | 13C NMR δ, J/Hz | 31P NMR δ | IR ν/cm−1 |
|---|---|---|---|---|---|
| 1a | n-Pr | 3.77 (2H, dt, J = 8.3, 6.7, OCH2), 3.52 (6H, d, J = 10.4, OCH3), 1.62 (2H, qt, J = 6.5, CH2), 0.96 (3H, t, J = 7.4, CH3) | 48.7 (OCH3), 64.2 (OCH2), 24.4 (CH2), 10.3 (CH3) | 139.4 | 1462, 1389, 1257, 1180, 1016 (P–OMe), 978 (P–OCH2), 818, 729 |
| 1b | i-Pr | 4.3 (1H, dsep, J = 6, 8.5, OCH), 3.48 (6H, d, J = 10.3, OCH3), 1.23 (6H, d, J = 6.1, CH3) | 66.6 (OCH), 49 (OCH3), 24.5 (CH3) | 138.6 | 1373, 1263, 1178, 1020 (P–OMe), 974 (P–OCH), 741 |
| 1c | n-Bu | 3.8 (2H, m, OCH2), 3.52 (6H, d, J = 10.4, OCH3), 1.61 (2H, m, CH2), 1.41 (2H, m, J = 7, CH2CH3), 0.94 (3H, t, J = 7.3, CH3) | 62 (OCH2), 48.7 (OCH3), 33.1 (CH2), 18.5 (CH2CH3), 13.5 (CH3) |
139.6 | 1458, 1381, 1180, 1020 (P–OMe), 968 (P–OCH2), 881, 727 |
| 1d | i-Bu | 3.6 (2H, dd, J = 7, 12, OCH2), 3.6 (6H, d, J = 10, OCH3), 1.88 (1H, m, J = 7, CH), 0.87 (6H, d, J = 6.7, CH3) | 68.9 (OCH2), 48.9 (OCH3), 29.7 (CH), 18.7 (CH3) | 139.3 | 1469, 1392, 1367, 1180, 1018 (P–OMe), 951, 744 (P–OCH2) |
| 1e | s-Bu | 4.12 (1H, m, OCH), 3.51 and 3.48 (6H, d, J = 10.3, OCH3), 1.5 (2H, m, CH2), 1.25 (3H, d, J = 7.9, OCCH3), 0.94 (3H, t, J = 7.3, CH3) | 71.5 (OCH), 48.3 and 48.2 (OCH3), 30.9 (CH2), 21.8 (OCCH3), 9.6 (CH3) |
139.2 | 1456, 1377, 1174, 1026 (P–OMe), 993 (P–OCH), 937, 727 |
| 1f | t-Bu | 3.48 (6H, d, J = 10.1, CH3), 1.45 (9H, s, CCH3) | 76.3 (OCCH3), 48.2 (OCH3), 31.1 (CH3) | 135.2 | 1460, 1392, 1367, 1252, 1180, 1051, 1016 (P–OMe), 943 (P–OC), 812, 742 |
| 1g | Pinacolyl | 3.77 (1H, dq, J = 7, 10, OCH), 3.51 and 3.5 (6H, d, J = 10.1 and 10.4, OCH3), 1.1 (3H, d, J = 6.4, OCCH3), 0.9 (9H, s, CH3) | 77.4 (OCH), 48.7 and 48.5 (OCH3), 35.2 (OCH), 25.7 (CH3), 17.5 (OCCH3) |
140.1 | 1479, 1375, 1180, 1082, 1043, 1018 (P–OMe), 1006, 945 (P–OCH), 930, 793, 744 |
| 1h | Cyclopentyl | 4.62 (1H, m, OCH), 3.5 (6H, d, J = 11.2, OCH3), 1.8 (8H, m, ring CH2 groups) | 75.2 (OCH), 48.4 (OCH3), 34.2 (C-2 and C-5 ring), 22.9 (C-3 and C-4 ring) | 139.0 | 1365, 1267, 1173, 1014 (P–OMe), 978 (P–OCH), 879, 849, 741 |
| 1i | Cyclohexyl | 4.04 (1H, m, J = 5.2, 9.5, OCH), 3.5 (6H, d, J = 10.4, OCH3), 2 and 1.45 (4H, m, C-2 and C-6 ring CH2), 1.75 and 1.26 (4H, m, C-3 and C-5 ring CH2), 1.51 and 1.24 (2H, m, ring CH2) | 71.9 (OCH), 48.4 (OCH3), 34.3 (C-2 and C-6 ring), 25.2 (C-3 and C-5 ring), 23.8 (C-4 ring) | 138.4 | 1450, 1373, 1180, 1016 (P–OMe), 978 (P–OCH), 856, 808, 791, 744 |
The Arbusov rearrangement of dimethyl alkyl phosphites
In its simplest form, this reaction consists of heating a trialkyl phosphite with the corresponding alkyl iodide. On distillation, the alkyl iodide is recovered unchanged but the trialkyl phosphite is transformed almost quantitatively into the isomeric dialkyl alkylphosphonate.17,18 The phosphorus atom increases its valency from three to five and the product contains a direct carbon–phosphorus bond. While Arbusov rearrangements of trialkyl phosphites have been studied in vast detail, those of mixed phosphites have received little attention. Heating (MeO)2POEt and (EtO)2POMe with methyl iodide gave the phosphonates MeO(EtO)P(O)Me and (EtO)2P(O)Me in 98% and 60% yields respectively.9 We found by GC-MS analysis that treatment of dimethyl propyl phosphites with methyl iodide gave three products: methyl propyl methylphosphonates 2a–b, dimethyl methylphosphonate 3 and methyl propyl propylphosphonates 4a–b (Scheme 4). The first two arise from breakdown of the intermediate phosphonium salt and the third from competition of the liberated propyl iodide for the starting phosphite. Therefore the reactivity of iodide ion to dealkylation of the Arbusov intermediates decreases in the substituent order OMe > OnPr > OiPr.18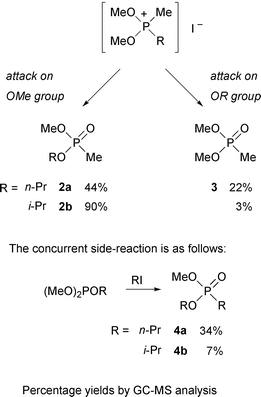 | ||
| Scheme 4 | ||
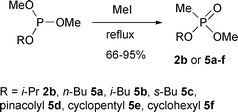
The Arbusov rearrangement of alkyl dimethyl phosphites was used to prepare alkyl methyl methylphosphonates 2b and 5a–f (Scheme 5). These stable colourless liquids were isolated in high yield (Table 3). The reactions were conducted without solvent using one molar equivalent of methyl iodide; less than one percent of dimethyl methylphosphonate was produced.
| Compound | R group | Yield (%) | Bp (°C/mmHg)a |
|---|---|---|---|
| a Approximate oven temperatures recorded during purification by Kugelrohr distillation. b This compound has been prepared before in 55% yield by treatment of methyl methylphosphonochloridate Cl(MeO)P(O)Me with the sodium salt of pinacolyl alcohol (3,3-dimethylbutan-2-ol) in ether (lit.,4 bp 100–102 °C/18 mmHg). c This compound has been prepared similarly in 65% yield (lit.,4 bp 113 °C/10 mmHg). | |||
| 2b | i-Pr | 66 | 21–22/0.2 |
| 5a | n-Bu | 94 | 55/1 |
| 5b | i-Bu | 88 | 45/0.04 |
| 5c | s-Bu | 86 | 50/0.025 |
| 5d | Pinacolylb |
95 | 65/0.025 |
| 5e | Cyclopentyl | 87 | 60/1 |
| 5f | Cyclohexylc |
84 | 45/0.2 |
 | ||
| Scheme 5 | ||
A stoichiometric amount of methyl iodide is unnecessary as it regenerates during the isomerisation. Comparative experiments with dimethyl cyclohexyl phosphite showed that the reaction occurred to the same extent (i.e. quantitatively), but much more slowly, when 0.05 molar equivalents of methyl iodide were used.
Interaction of dimethyl tert-butyl phosphite with methyl iodide did not give the desired phosphonate on distillation. Instead an unidentified mixture of phosphorus products formed that did not contain the tert-butyl group. The thermal instability of butyl esters of methylphosphonic acid has been noted before; elimination of but-1-ene from nBuO(HO)P(O)Me occurred readily on heating.7
Spectroscopic data for methylphosphonates 2b and 5a–f are given in Table 4. Their phosphorus chemical shifts are between 32–30 ppm. In the infrared spectra, the phosphoryl vibration appears in the range 1246–1236 cm−1 and there are several strong bands in the region 1090–1060 cm−1 characteristic of different ester groups.4 During the synthesis of sec-butyl and pinacolyl phosphonate derivatives a mixture of two diastereoisomers was obtained. No attempt was made to separate these diastereoisomers and NMR and IR spectra of the mixtures were recorded.
| Compound | R | 1H NMR δ, J/Hz | 13C NMR δ, J/Hz | 31P NMR δ | IR ν/cm−1 |
|---|---|---|---|---|---|
| a Diastereoisomeric pair. | |||||
| 2b | i-Pr | 4.7 (1H, dsep, J = 6.2, 7.3, OCH), 3.71 (3H, d, J = 11.2, OCH3), 1.46 (3H, d, J = 17.1, P-CH3), 1.33 (6H, d, J = 6.4, CH3) | 70.2 (OCH), 51.9 (OCH3), 24 (CH3), 11.6 (P-CH3) | 29.7 | 1651, 1468, 1387, 1313, 1236 (P![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) O), 1180, 1107, 1051 (P–OMe), 977, 916, 811, 715 O), 1180, 1107, 1051 (P–OMe), 977, 916, 811, 715 |
| 5a | n-Bu | 4.04 (2H, dt, J = 9.2, 6.6, OCH2), 3.72 (3H, d, J = 11.1, OCH3), 1.65 (2H, m, J = 7, CH2), 1.45 (3H, d, J = 17.6, P-CH3), 1.42 (2H, m, J = 7, CH2CH3), 0.95 (3H, t, J = 7.4, CH3) | 65.4 (OCH2), 52.1 (OCH3), 32.5 (P-CH3), 18.7 (CH2), 13.6 (CH2CH3), 11.4 (CH3) |
31.1 | 1649, 1466, 1313, 1244 (PO), 1186, 1051 (P–OMe), 1028, 985, 920, 814 |
| 5b | i-Bu | 3.8 (2H, m, OCH2), 3.7 (3H, d, J = 11.1, OCH3), 1.94 (1H, tsep, J = 7, CH), 1.49 (3H, d, J = 17.8, P-CH3), 0.98 (6H, d, J = 6.8, CH3) | 71.6 (OCH2), 52 (OCH3), 29.1 (CH), 18.7 (CH3), 9.65 (P-CH3) | 31.6 | 1651, 1471, 1313, 1244 (PO), 1184, 1057 (P–OMe), 1024, 918, 827, 808, 727 |
| 5c |
s-Bua |
3.72/3.71 (3H, d, J = 11, OCH3), 4.48 (1H, m, OCH), 1.47 (3H, d, J = 17.3, P-CH3), 1.33/1.32 (3H, d, J = 6.3, OCCH3), 0.96/0.95 (3H, t, J = 7.5, CH3) | 74.5/74.4 (OCH), 51.1/51.3 (OCH3), 30/20.9 (OCCH3), 11.5/9.5 (P-CH3), 9.1/8.5 |
29.5 and 29.8 | 1651, 1463, 1383, 1313, 1242 (PO), 1176, 1053 (P–OMe), 1030, 999, 960, 910, 811, 756, 723 |
| 5d | Pinacolyla |
4.26/4.23 (1H, dq, J = 6.4, 8.2, OCH), 3.74/3.71 (3H, d, J = 11.3, OCH3), 1.47 (3H, d, J = 18.4, P-CH3), 1.29/1.28 (3H, d, J = 6.4, OCCH3) | 81/80.7 (CCH3), 51.7/51.4 (OCH3), 25.5 (CH3), 16.9/16.6 (OCCH3), 11.5/10.5 (P-CH3) |
29.5 and 30.3 | 1481, 1462, 1379, 1365, 1311, 1246 (PO), 1080, 1057 (P–OMe), 1045, 1011, 976, 933, 910, 814, 737 |
| 5f | Cyclohexyl | 4.4 (1H, m, OCH), 3.7 (3H, d, J = 11.3, OCH3), 1.95/1.5 (4H, m, C-2 and C-6 ring CH2), 1.75/1.35 (4H, m, C-3 and C-5 ring CH2), 1.47 (3H, d, J = 17.7, P-CH3), 1.23 (2H, m, C-4 ring CH2) | 74.7 (OCH), 51.3 (OCH3), 33.3 (C-2 and C-6 ring), 24.6 (C-3 and C-5 ring), 23.1 (C-4 ring), 10.7 (P-CH3) | 29.6 | 1647, 1452, 1311, 1242 (PO), 1186, 1055 (P–OMe), 1034, 999, 920, 897, 806, 735 |
| 5e | Cyclopentyl | 4.9 (1H, m, OCH), 3.72 (3H, d, J = 11.9, OCH3), 1.46 (3H, d, J = 17.4, P-CH3), 1.85–1.6 (8H, m, ring CH2 groups) | 78.4 (OCH), 51.3 (OCH3), 33.6 (C-2 and C-5 ring), 21.2 (C-3 and C-4 ring), 10.6 (P-CH3) | 29.7 | 1646, 1454, 1313, 1244 (PO), 1186, 1051 (P–OMe), 999, 928, 901, 812 |
| 5f | Cyclohexyl | 4.4 (1H, m, OCH), 3.7 (3H, d, J = 11.3, OCH3), 1.95/1.5 (4H, m, C-2 and C-6 ring CH2), 1.75/1.35 (4H, m, C-3 and C-5 ring CH2), 1.47 (3H, d, J = 17.7, P-CH3), 1.23 (2H, m, C-4 ring CH2) | 74.7 (OCH), 51.3 (OCH3), 33.3 (C-2 and C-6 ring), 24.6 (C-3 and C-5 ring), 23.1 (C-4 ring), 10.7 (P-CH3) | 29.6 | 1647, 1452, 1311, 1242 (PO), 1186, 1055 (P–OMe), 1034, 999, 920, 897, 806, 735 |
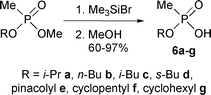
Synthesis and cleavage of phosphorus silyl esters
Often phosphorus esters undergo facile transesterification reactions with silyl halides to form the corresponding phosphorus silyl esters and alkyl halides.19,20 The silyl esters are cleaved by protic solvents under very mild conditions to yield the phosphorus acids. Overall, the equivalent of hydrolysis of the phosphorus ester is accomplished, but under conditions far milder than could be used with the parent ester. For transesterification, the reaction proceeds most rapidly with methyl esters, and much slower with more highly-substituted alkyl esters.21 The exceptional rapidity of the reaction of methyl esters permitted selective silylation of the mixed phosphonates with bromotrimethylsilane (Scheme 6). Methanol was used to effect cleavage of the silyl ester link, generating phosphonic acids 6a–g as viscous oils in high yields (Table 5).| Compound | R group | Yield (%) | Bp (°C/mmHg)a |
|---|---|---|---|
|
a
Approximate oven temperatures recorded during purification by Kugelrohr distillation.
b
Previously made in 12% yield from alcoholysis of methylphosphonic anhydride (lit.,8 bp 100–115 °C/3 mmHg) and in 60% and 41% yields respectively from hydrolysis of diisopropyl methylphosphonate with aqueous sodium hydroxide in dioxane (lit.,4 bp 115 °C/0.5 mmHg) or with aqueous barium hydroxide (lit.,5 bp 97–98 °C/0.08 mmHg).
c
Previously made in 76% yield from hydrolysis of dibutyl methylphosphonate with aqueous sodium hydroxide in dioxane (lit.,4 bp 132 °C/0.5 mmHg).
d
Previously made in 98% yield from alcoholysis of methylphosphonic anhydride (lit.,7 bp 142–143 °C/0.5 mmHg).
e
Previously made in 88% yield from hydrolysis of methyl 3,3-dimethylbutyl methylphosphonate with aqueous sodium hydroxide in dioxane (bp not given)4 and in 18% yield from alcoholysis of methylphosphonic anhydride (lit.,8 bp 135 °C/3 mmHg).
f
Previously made in 47% yield from alcoholysis of methylphosphonic anhydride (lit.,8 bp 150 °C/0.8 mmHg).
|
|||
| 6a |
i-Prb |
97 | 80/0.04 |
| 6b |
n-Buc |
82 | 75/0.01 |
| 6c |
i-Bud |
88 | 75/0.01 |
| 6d | s-Bu | 89 | 80/0.035 |
| 6e | Pinacolyle |
90 | 90/0.02 |
| 6f | Cyclopentyl | 64 | 95/0.04 |
| 6g | Cyclohexylf |
60 | 105/0.015 |
 | ||
| Scheme 6 | ||
The formation of the silyl ester depended on the nature of the higher ester group in the starting mixed phosphonate. With the bulky sec-butyl group on phosphorus, silylation was incomplete and the corresponding acid 6d could be isolated only in 90% purity due to co-distillation of the residual starting phosphonate.
Spectroscopic data for phosphonic acids 6a–g are given in Table 6. Their phosphorus chemical shifts are between 32–30 ppm. The phosphoryl vibration appears in the range 1213–1201 cm−1 in the infrared spectra. In the absence of moisture, the phosphonic acids can be stored in a refrigerator for several years without decomposition.
| Compound | R | 1H NMR δ, J/Hz | 13C NMR δ, J/Hz | 31P NMR δ | IR ν/cm−1 |
|---|---|---|---|---|---|
| 6a | i-Pr | 11.02 (1H, br s, OH), 4.68 (1H, dsep, J = 8.2, 6.1, OCH), 1.51 (3H, d, J = 17.3, P-CH3), 1.33 (6H, d, J = 6.1, CH3) | 70.6 (OCH), 24 (CH3), 12.8 (P-CH3) | 31.4 | 2981 (OH), 2306, 1693, 1377, 1315, 1178, 1143, 1202 (PO), 1107 |
| 6b | n-Bu | 11.94 (1H, br s, OH), 4.01 (2H, dt, J = 9, 6.4, OCH2), 1.65 (2H, tt, J = 6.6, 6.8, CH2), 1.49 (3H, d, J = 16.9, P-CH3), 1.41 (2H, m, J = 7, CH2CH3), 0.94 (3H, t, J = 7.3, CH3) | 65.2 (OCH2), 32.3 (CH2), 18.6 (CH2CH3), 12.8 (CH3), 11.3 (P-CH3) |
31.6 | 2960 (OH), 2297, 1678, 1466, 1313, 1205 (PO), 1066, 1030, 997, 910, 804 |
| 6c | i-Bu | 12.1 (1H, br s, OH), 3.77 (2H, dd, J = 6.8, 7, OCH2), 1.95 (1H, m, J = 7, CH), 1.5 (3H, d, J = 17.7, P-CH3), 0.9 (6H, d, J = 6.8, CH3) | 71.1 (OCH2), 29 (CH), 18.7 (CH3), 11.5 (P-CH3) | 32.1 | 2960 (OH), 2287, 1685, 1473, 1313, 1205 (PO), 1038, 1003, 901 |
| 6d | s-Bu | 11.3 (1H, br s, OH), 4.44 (1H, m, J = 6, 7, OCH), 1.63/1.58 (2H, m, J = 7.1, 9.5, 14.4, CH2), 1.48 (3H, d, J = 17.7, P-CH3), 1.32 (3H, d, J = 6.3, OCCH3), 0.94 (3H, t, J = 6.3, CH3) | 75.3 (OCH), 30.5 (CH2), 21.5 (OCCH3), 12.4 (P-CH3), 9.6 (CH3) |
31.4 | 2973 (OH), 2306, 1689, 1459, 1383, 1313, 1209 (PO), 1174, 1034, 1001, 899, 816 |
| 6e | Pinacolyl | 11.6 (1H, br s, OH), 4.2 (1H, dq, J = 9.5, 6.5, OCH), 1.48 (3H, d, J = 17.8, P-CH3), 1.28 (3H, d, J = 6.4, OCCH3), 0.92 (9H, s, CH3) | 80.9 (OCH), 34.8 (CCH3), 25.6 (CH3), 16.8 (OCCH3), 12.3 (P-CH3) |
31.5 | 2962 (OH), 1481, 1381, 1365, 1311, 1207 (PO), 1078, 1014, 997, 933, 901 |
| 6f | Cyclopentyl | 10.9 (1H, br s, OH), 4.8 (1H, m, OCH), 1.75–1.5 (ring CH2 groups), 1.38 (3H, d, J = 18, P-CH3) | 78.7 (OCH), 34.2 (C-2 and C-5 ring), 23.1 (C-3 and C-4 ring), 12.4 (P-CH3) | 30.2 | 2960 (OH), 2289, 1678, 1452, 1313, 1213 (PO), 997, 920, 814 |
| 6g | Cyclohexyl | 11.82 (1H, br s, OH), 4.37 (1H, m, J = 4, 8, OCH), 1.9–1.5 (8H, m, ring CH2 groups), 1.3 (2H, m, C-4 ring CH2), 1.5 (3H, d, J = 17.8, P-CH3) | 75.1 (OCH), 33.6 (C-2 and C-6 ring), 25.1 (C-3 and C-4 ring), 23.6 (C-4 ring), 12.3 (P-CH3) | 31.0 | 2937 (OH), 2312, 1682, 1452, 1311, 1201 (PO), 1041, 1005, 908, 762 |
Conclusion
An improved route to alkyl hydrogen methylphosphonates has been developed. The limiting step in the synthetic sequence is the initial transesterification of trimethyl phosphite. The Arbusov reaction of the resultant mixed phosphites is reliable and high yielding. The silylation–desilylation procedure used in the last step provided a mild method of dealkylation of alkyl methyl methylphosphonates and resulted in selective methyl ester cleavage. The three-stage synthesis is an improvement on literature methods to alkyl hydrogen methylphosphonates that involve alkaline hydrolysis of symmetrical or mixed phosphonates or alcoholysis of methylphosphonic anhydride.Experimental
General details
All reagents were of commercial quality: trimethyl phosphite obtained from Aldrich Ltd (Gillingham, UK) contained a small amount of trimethyl phosphate, and was therefore distilled prior to use. Anhydrous solvents were used for reactions. NMR spectra were obtained on a JEOL Lambda 500 instrument (operating at 500 MHz for 1H, 125 MHz for 13C, 470 MHz for 19F, and 202 MHz for 31P spectra) or a JEOL Lambda 300 instrument (operating at 300 MHz for 1H, 75 MHz for 13C, 282 MHz for 19F, and 121.5 MHz for 31P spectra) as solutions in CDCl3, with internal reference SiMe4 for 1H and 13C, external CFCl3 for 19F and external (MeO)3P (δ 140 ppm) for 31P spectra. Data in Tables 2, 4 and 6 are recorded as follows: chemical shifts in ppm from reference on the δ scale, integration, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet and sep = septet; br = broad), coupling constant (J/Hz) and assignment. IR spectra were recorded as liquid films on a Nicolet SP210 instrument using Omnic software. Reaction mixtures were monitored by gas chromatography-mass spectrometry (GC-MS) using a Finnigan MAT GCQ instrument with chemical ionisation (CI) using methane as reagent gas. Molecular masses of pure phosphites and phosphonates were confirmed with methane +ve CI data. With the exception of the sec-butyl derivative 6d which was 90% pure, the alkyl hydrogen methylphosphonates were >95% pure by liquid chromatography-mass spectrometry using atmospheric pressure chemical ionisation. Their characterisation by tandem mass spectroscopic techniques has been described elsewhere.1,2The synthesis of dialkyl alkyl phosphites
This general procedure is illustrated for the synthesis of dimethyl pinacolyl phosphite. A 250 cm3 round-bottom flask was equipped with a distillation rig, a pressure-equalising dropping funnel and an argon bleed; the funnel and receiver were fitted with calcium chloride guard tubes. Pinacolyl alcohol (23.5 g, 0.23 mol) was weighed into the dropping funnel and trimethyl phosphite (49.6 g, 0.4 mol), a pea-sized piece of sodium metal, and a magnetic flea were added to the flask. The flask was heated to 100 °C and pinacolyl alcohol added dropwise with stirring over 20 min. After evolution of methanol ceased (6.4 g collected, theoretical yield 7.4 g), the residue was fractionated under reduced pressure (10 mmHg). Three fractions were collected (1) bp 22–27 °C, 18.92 g, (2) 27–68 °C, 2.4 g and (3) 68–72 °C, 28.14 g. The forerun was discarded. The second and third fractions contained 30% and 91% desired product respectively. The third fraction was redistilled to give dimethyl pinacolyl phosphite 1g as a colourless liquid (25.73 g, overall 58%); bp 69–70 °C/10 mmHg.The synthesis of dialkyl alkylphosphonates
This general procedure is illustrated for the synthesis of methyl pinacolyl methylphosphonate. Dimethyl pinacolyl phosphite (9.7 g, 0.05 mol) and methyl iodide (7.1 g, 0.05 mol) were placed into a 50 cm3 round-bottom flask equipped with a condenser (fitted with a calcium chloride guard tube) and refluxed for 2 h. A portion of the reaction mixture was analysed by GC-MS and shown to comprise methyl pinacolyl methylphosphonate and a trace of dimethyl methylphosphonate. The methyl iodide was removed on a rotary evaporator. Bulb-to-bulb distillation of the residue under reduced pressure gave pure methyl pinacolyl methylphosphonate 5d as a colourless liquid (9.24 g, 94%); bp 65 °C/0.025 mmHg.The synthesis of alkyl hydrogen alkylphosphonates
This general procedure is illustrated for the synthesis of pinacolyl hydrogen methylphosphonate. Methyl pinacolyl methylphosphonate (0.97 g, 0.005 mol) was weighed into a 50 cm3 round-bottom flask to which chloroform (10 cm3) and a magnetic flea were added. The flask was stoppered with a rubber septum and purged with argon. The reaction mixture was stirred and bromotrimethylsilane (0.66 cm3, 0.005 mol) was added by syringe. After 12 h, the solvent was removed to leave a colourless oil. Methanol (2 cm3) was added and then removed on a rotary evaporator; this was repeated two more times. The resultant viscous oil was distilled using a Kugelrohr apparatus to give pinacolyl hydrogen methylphosphonate 6e as colourless liquid (0.81 g, 90%); bp 90 °C/0.02 mmHg.Acknowledgements
We thank Steve Marriott and Alison Bussey for providing the infrared spectra. The present work was funded by the Ministry of Defence UK.References
- R. M. Black, R. J. Clarke, R. W. Read and M. T. J. Reid, J. Chromatogr. A, 1994, 662, 301 CrossRef CAS.
- R. M. Black and R. W. Read, J. Chromatogr. A, 1997, 759, 79 CrossRef CAS.
- R. Rabinowitz, J. Am. Chem. Soc., 1960, 82, 4564 CrossRef CAS.
- H. Christol, M. Levy and C. Marty, J. Organomet. Chem., 1968, 12, 459 CrossRef CAS.
- E. Gryszkiewicz-Trochimowski, J. Quinchon and M. Bousquet, Bull. Soc. Chim. Fr., 1962, 1645 Search PubMed.
- R. F. Hudson and L. Keay, J. Chem. Soc., 1956, 2463 RSC.
- K. A. Petrov, R. A. Baksova, L. V. Korkhoyanu, L. P. Sinogeikina and T. V. Skudina, J. Gen. Chem. (Engl. Transl.), 1965, 35, 723 Search PubMed.
- A. Pienaar, C. M. Erasmus, M. Wentzel and E. H. Cowley, Phosphorus, Sulfur Relat. Elem., 1999, 148, 149 Search PubMed.
- S. R. Landauer and H. N. Rydon, J. Chem. Soc., 1953, 2224 RSC.
- G. Kamai and F. M. Kharrasova, J. Gen. Chem. (Engl. Transl.), 1957, 27, 1034 Search PubMed.
- M. M. Sidky, F. M. Soliman and R. Shabana, Aust. J. Chem., 1978, 31, 139 CAS.
- A. E. Arbusov and M. G. Imaev, Dokl. Chem. (Engl. Transl.), 1957, 112, 115 Search PubMed.
- F. W. Hoffmann, R. J. Ess and R. P. Usinger, J. Am. Chem. Soc., 1956, 78, 5817 CrossRef CAS.
- F. W. Hoffmann and T. R. Moore, J. Am. Chem. Soc., 1958, 80, 1150 CrossRef CAS.
- M. M. Sidky, M. N. Aboudl-Enein and R. M. Taha, Egypt. J. Chem., 1973, 43 Search PubMed.
- L. C. Thomas , The Identification of Functional Groups in Organophosphorus Compounds, Academic Press, London, 1974. Search PubMed.
- A. H. Ford-Moore and J. H. Williams, J. Chem. Soc., 1947, 1465 RSC.
- R. E. Engel , Synthesis of Carbon-Phosphorus Bonds, CRC Press Inc., Florida, 1988, p. 35. Search PubMed.
- B. Borecka, J. Chojnowski, M. Cypryk, J. Michalski and J. Zielinska, J. Organomet. Chem., 1979, 171, 17 CrossRef CAS.
- C. M. Timperley, I. J. Morton, M. J. Waters and J. L. Yarwood, J. Fluorine Chem., 1999, 96, 95 CrossRef CAS.
- R. E. Engel , Handbook of Organophosphorus Chemistry, Marcel Dekker Inc., New York, 1992, p. 178. Search PubMed.
Footnote |
| † The IUPAC name for pinacolyl is 3,3-dimethylbutan-2-yl. |
| This journal is © The Royal Society of Chemistry 2001 |