Studies on the reactivity of N-(3-thienyl)carbamates
Delphine
Brugier
,
Francis
Outurquin
and
Claude
Paulmier
*
Laboratoire de Synthèse Thio- et Séléno-organique, IRCOF-UFR Sciences, Université de Rouen, F-76821 Mont Saint Aignan Cedex, France
First published on 13th December 2000
Abstract
tert-Butyl 2-allyl- and N-allyl-3-thienylcarbamates were used as substrates for the preparation of thieno[3,2-b]pyrroles 9 and 5,6-dihydrothieno[3,2-b]pyrroles 10. Pd-catalyzed cyclization of N-allyl-(2-bromo-3-thienyl)carbamates 12 has allowed access to thienopyrroles 9. The radical route has led to the formation of a dihydrothienopyrrole 10 or a tetrahydrothienopyridine 19 according to the β-substitution of the allyl substituent. The nucleophilic participation of the tert-butoxycarbonyl group occurred during the selenium or iodine-induced cyclization of tert-butyl 2-allyl- or N-allyl-3-thienylcarbamates. The formation of the thienooxazepinone 25 and N-(3-thienyl)oxazolidinones 28 and 29 was observed.
Indole derivatives display a wide range of biological activity.1 For comparison, the study of thiophene isosteres is of great interest. The synthesis of thieno[3,2-b]pyrroles has been described but low yields were observed.2–4 The multistep methods used involve cyclization of 3-aminothiophene derivatives according to Reissert, Bischler or Fischer modified procedures. More recently, Gronowitz et al. have prepared thieno[3,2-b]pyrroles using Pd-catalyzed coupling reactions.5 Few works are devoted to 5,6-dihydrothieno[3,2-b]pyrroles
![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 5 and, in particular, to their preparation.6
5 and, in particular, to their preparation.6
We are interested in the reactivity of 3-aminothiophene 1, 3,4-diaminothiophene 2 and their carbamates: alkyl (3-thienyl)carbamates 3, tert-butyl (4-amino-3-thienyl)carbamate 4 and di-tert-butyl (thiophene-3,4-diyl)dicarbamate 57 (Scheme 1). Halogenation of these compounds was recently studied and carbamates 6–8 are now readily available.8
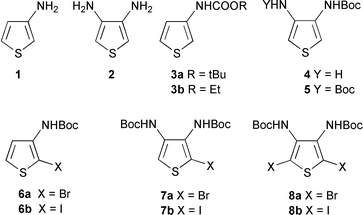 | ||
| Scheme 1 | ||
We now report our results concerning the access to 4-tert-butoxycarbonylthieno[3,2-b]pyrroles 9 and 4-tert-butoxycarbonyl 5,6-dihydrothieno[3,2-b]pyrroles 10 from 3-aminothiophene derivatives. Four routes were tested (Scheme 2). The cyclization of alkyl (2-alkynyl-3-thienyl)carbamates 11 was studied first. Methods involving Pd-catalyzed coupling reactions, radical and anionic cyclizations were then investigated. tert-Butyl N-allyl(2-bromo-3-thienyl)carbamates 12, tert-butyl (2-allyl-3-thienyl)carbamates 13 and tert-butyl N-allyl(3-thienyl)carbamates 14 were considered as potential precursors.
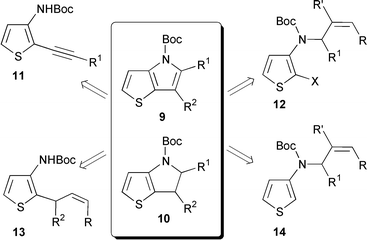 | ||
| Scheme 2 | ||
We have recently described the synthesis of alkyl (2-ethynyl-3-thienyl)carbamates 11 by Pd-catalyzed coupling reaction between acetylene compounds and the bromo carbamate 6a.8 According to recent preparations of indoles and azaindoles, we have tried to achieve the cyclization of compounds 11 in the presence of palladium(II) salts9 or copper(I) iodide,10 but the reactions were unsuccessful. Gronowitz et al. reported a two step procedure with both Pd-catalyzed carbon–carbon bond formation and cyclization.5a Our result seems to demonstrate that carbamates 11 and 15b (R = SiMe3) were not intermediates in this reaction.
The carbamate 11b (R = SiMe3) was treated with sodium ethoxide in ethanol.11 The formation of thienopyrrole was not observed and only the 2-ethynylthiophenes 11a and 15a were respectively obtained. This result shows that this method, efficient for the synthesis of indoles, cannot be transposed to the thiophene isosteres.

We then prepared several N-allylated [2-halo or 2-phenylselanyl-3-thienyl]carbamates 12 with the goal to test the palladium-catalyzed, radical and ionic routes. Compounds 12 were easily obtained by N-allylation of (2-halo or 2-phenylselanyl-3-thienyl)carbamates 6 (Scheme 3, Table 1). The same reaction was carried out on halo dicarbamates 7b, 8a and 8b providing the bis(N-allylcarbamates) 16 and 17 in good yields (Scheme 4). Compounds 16 and 17 have given very complex 1H and 13C NMR spectra, which indicate stabilized conformers resulting from an important steric hindrance.
| No. | X | R | R′ | E∶Z | Yield (%) |
|---|---|---|---|---|---|
| 12a | Br | H | H | — | 72 |
| 12b | Br | H | Me | — | 87 |
| 12c | Br | Me | H | 90∶10 | 51 |
| 12d | Br | Ph | H | 100∶0 | 50 |
| 12e | I | H | H | — | 94 |
| 12f | SePh | H | H | — | 68 |
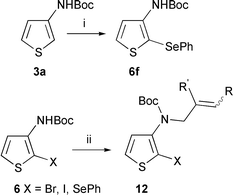 | ||
| Scheme 3 Reagents and conditions: (i) nBuLi (2.5 eq.), THF, −78 °C then PhSeSePh, −20 °C; (ii) NaH, THF then allylic halide, 20 °C. | ||
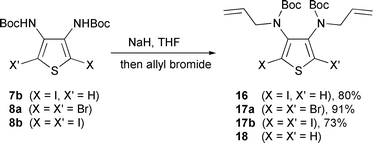 | ||
| Scheme 4 | ||
tert-Butyllithium treatment of N,N-diallylated 2-haloanilines, in a polar solvent or in the presence of a chelating agent, allows cyclization at room temperature.12 The mechanism involves the successive formation of two carbanionic species. In our hand, the lithium–bromine exchange occurred but no cyclization took place. We have observed the quantitative formation of the debrominated compounds 14a from 12a (R = R′ = R1 = H, X = Br) and 18 from 17a.
The palladium-catalyzed cyclization of a tert-butyl N-allylated (2-iodo-3-thienyl)carbamate, leading to a thieno[3,2-b]pyrrole derivative, has been described5c but the reaction was not extensively studied. We have applied this procedure to the synthesis of the 6-methyl and 6-benzylthieno[3,2-b]pyrrole derivatives 9a (R = H) and 9d (R = Ph). The iodothiophene 12e was a better substrate than the bromo analog 12a for access to 9a (Scheme 5).
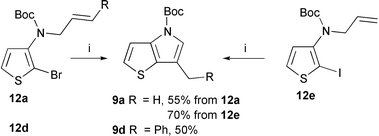 | ||
| Scheme 5 Reagents and conditions: (i) Pd(PPh3)4, K2CO3, DMF, 60 °C. | ||
The radical cyclization13 of carbamates 12 (X = Br, I, SePh) was also achieved (Scheme 6, Table 2). When R′ = H, the initial thienyl radical underwent exclusively 5-exo trigonal cyclization providing the dihydrothienopyrrole 10 with a good yield. Comparable results were observed for X = Br, I and SePh (compare entries 1, 2 and 3). According to a 6-endo process, the tetrahydrothienopyridine 19 was the major product formed from 12b (76% yield). The corresponding dihydrothienopyrrole 20 was also present as a minor product (19–20, 95∶5).
| Entry | X | R | R′ | Product | Yield (%) |
|---|---|---|---|---|---|
| 1 | Br | H | H | 10a | 68 |
| 2 | I | H | H | 10a | 73 |
| 3 | SePh | H | H | 10a | 69 |
| 4 | Br | H | Me | 19 | 76 |
| 5 | Br | Me | H | 10b | 67 |
| 6 | Br | Ph | H | 10c | 85 |
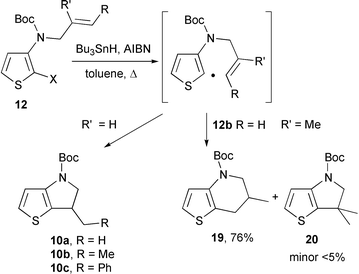 | ||
| Scheme 6 | ||
Palladium-catalyzed and radical cyclizations were unsuccessful in the case of dicarbamates 16 and 17. The cleavage of the C–X bond was never observed. Steric hindrance probably prevents palladium insertion or radical formation.
The electrophilic selenium-induced cyclization method14 was then considered. tert-Butyl (2-allyl-3-thienyl)carbamate 13a, prepared in a quantitative yield by metallation–allylation of 3a, was treated with an electrophilic selenium reagent (Scheme 7). Using PhSeBr or N-(phenylselanyl)phthalimide (NPSP), no reaction was observed even after several days. Phenylselanyl triflate† led to a complex and untractable mixture with consumption of the substrate. The generation of PhSe+ from diphenyl diselenide and PhI(OAc)215 has led to the formation of the regioisomeric addition products 21a and 22a (thermodynamic and kinetic adducts, respectively). A slow and partial isomerisation 22a → 21a occurred during the chromatographic separation. This reaction could be considered as a formal addition of “PhSeOAc” (Scheme 7). With PhSeCl, only the thermodynamic adduct 21b was formed, but was not isolated in a pure form. The cyclization occurred during silica gel chromatography. Three heterocyclic products were formed and isolated: the expected dihydrothienopyrrole 23, the tetrahydrothienopyridine 24 and the thienooxazepine 25 (ratio: 23–24–25, 50∶15∶35). The formation of 23 and 24 was the result of 5- and 7-exo cyclization processes involving the thermodynamic addition product 21b or the seleniranium ion A with respective attacks of the nitrogen and oxygen atoms of the carbamate functional group (Scheme 7).
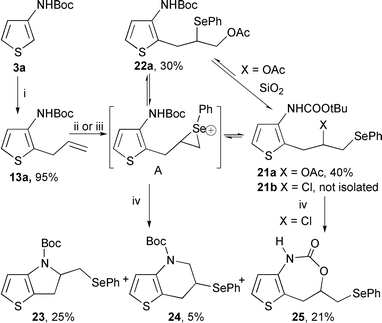 | ||
| Scheme 7 Reagents and conditions: (i) n-BuLi (2.5 eq.), THF, −78 °C then allylbromide, −10 °C; (ii) PhSeCl (1.5 eq.), CH2Cl2, Na2CO3 (1eq.); (iii) PhSeSePh (0.6 eq.), Phl(OAc)2 (excess), CH3CN; (iv) SiO2, CH2Cl2. | ||
We have previously studied the strong enamine character of β-aminothiophenes 1 and 2.7 Under acid catalysis, α-alkylation of the nucleus was efficiently achieved using an aldehyde and PhSeH as reducing agent. We were interested in evaluating the nucleophilicity of the α-carbon towards that of a seleniranium ion. We have therefore prepared the N-allylated compounds 14a, 18 (already obtained by debromination of 12a and 17a), and 27 (Scheme 8). The synthesis of the tert-butyl N-allyl-3-thienylcarbamate 14a has been previously described.16 NaH treatment of the dicarbamate 5, followed by addition of allyl bromide, afforded the dicarbamate 18 or the thienoimidazolone 26 depending on the reaction time. The monoallyl diamine derivative 27 was prepared, in good yield, from the aminocarbamate 4. In the reaction of PhSeCl with the N-allylated thienylcarbamates 14a and 18, the O-nucleophilicity of the carbamate group was also efficient and the oxazolidinones 28 and 29a were formed respectively. An analogous reactivity was observed in the reaction of N-iodosuccinimide with the dicarbamate 18. The bis(oxazolidinone) 29b was obtained albeit in a poor yield (37%).
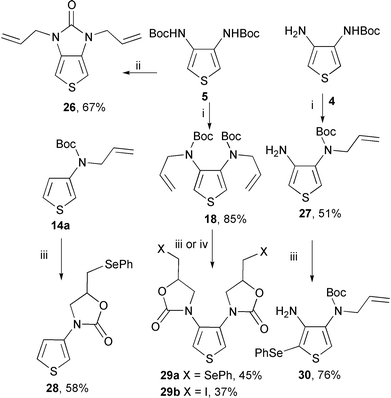 | ||
| Scheme 8 Reagents and conditions: (i) NaH, THF, allyl bromide, 5 h; (ii) NaH, THF, allyl bromide, 20 h; (iii) PhSeCl, CH2Cl2, 20 °C, 12 h; (iv) N-iodosuccinimide, CCl4, Δ, 12 h. | ||
PhSeCl treatment of the N-allylated thiophenediamine derivative 27 has led to an electrophilic aromatic substitution providing the 2-phenylselanylthiophene derivative 30 isolated in a good yield (Scheme 8).
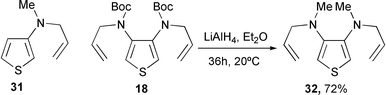
To prevent the participation of the functional group, we have applied the reaction to tertiary N-allyl amines. N-Allyl-3-(methylamino)thiophene 3116 and N,N′-diallyl-3,4-bis(methylamino)thiophene 32 were prepared by LiAlH4 reduction of the N-allylated carbamates 14a and 18, respectively (Scheme 9). In each case, the reaction with PhSeCl (1.5 eq.) has not allowed the cyclization. Complex and untractable mixtures of products were formed. The reaction was not investigated further.
 | ||
| Scheme 9 | ||
This work has shown that the synthesis of thieno[3,2-b]pyrroles from 3-aminothiophene derivatives cannot be achieved by the methods which are efficient for the preparation of indoles from 2-allyl or N-allylanilines. We succeeded, however, in the synthesis of tert-butyl 6-alkylthieno[3,2-b]pyrrole-4-carboxylates 9 by Pd-catalyzed cyclization of tert-butyl N-allyl(2-halo-3-thienyl)carbamates 12. The radical cyclization of the same substrates 12 (X = Br, I, SePh) has given an access to the 5,6-dihydrothieno[3,2-b]pyrroles 10 and 4,5,6,7-tetrahydrothieno[3,2-b]pyridine 19 according to the nature of the allyl substituent. The PhSe-induced cyclization of tert-butyl 2-allyl-3-thienylcarbamate 13a has shown competitive participation of the functional group with formation of the thienooxazepinone 25. A comparable O-nucleophilic attack by the Boc group was observed with the formation of the the oxazolidinones 28 and 29 during selenium- or iodine-induced cyclization of the N-allylated thienylcarbamates 14a and 18, respectively.
Experimental
Di-tert-butyl (thiophene-3,4-diyl)dicarbamate 5,17tert-butyl (4-amino-3-thienyl)carbamate 4,7etert-butyl (2-bromo-3-thienyl)carbamate 6a,18 the halothiophene derivatives 6–88 and acetylene compounds 11b and 15b8 were prepared as described elsewhere. All solvents were distilled before use and light petroleum refers to the fraction with bp 40–60 °C. THF was distilled over sodium–benzophenone and triethylamine was dried over sodium hydride. The chromatographic separations were achieved on silica gel (0.060–0.200 nm, pore diameter ca. 4 nm) available from ACROS. 1H and 13C NMR spectrum were recorded on Brucker AC 200 and DPX 300 instruments.
Attempted cyclization of 2-(trimethylsilylethynyl)thiophenes 11b and 15b
The carbamate 11b (0.295 g, 1 mmol) was added to a freshly prepared solution of NaOEt (5 mmol) in ethanol (10 ml). The mixture was heated on reflux for 9 h under argon. The solvent was eliminated and the residue was treated with water and extracted with CH2Cl2. The organic layer was concentrated and silica gel chromatography (eluent: AcOEt) afforded the thienylacetylene 11a. With the same treatment, 15b has led to 15a.N-Allylation of halocarbamates 6
A solution of bromocarbamate 6a (0.278 g, 1 mmol) and sodium hydride (0.120 g, 5 mmol) in THF (50 ml) was stirred for 30 min at room temperature. Allyl bromide (0.605 g, 5 mmol) was then added and the mixture was stirred for 5 h. After cooling to 0 °C, the reaction was treated with a sat. aq. NaCl solution (10 ml). The organic layer was dried and the solvent was eliminated. The oily residue was chromatographed (eluent: CH2Cl2–light petroleum, 1∶1) affording N-allyl(2-bromo-3-thienyl)carbamate 12a. The other compounds 12 were obtained in a similar way from the corresponding carbamates 6 and the appropriate allylic bromide. The N-allylated halodicarbamates 16, 17a and 17b were prepared from dicarbamates 7b, 8a and 8b respectively, using double the amount of NaH and allyl bromide.H3)3),19.6 (CH3). Anal. calc. for C13H18BrNO2S: C, 46.99; H, 5.46; N, 4.22; found: C, 46.98; H, 5.69; N, 4.07%.
Preparation of thienopyrroles 9a and 9d
Potassium carbonate (0.512 g, 4 mmol) and Pd(PPh3)4 (0.057 g, 0.05 mmol) were successively added to a degassed solution of N-allyl(2-halo-2-thienyl)carbamate 12a (12d or 12e) (1 mmol) in DMF (3 ml). The reaction mixture was stirred for 30 min at room temperature and 5 h at 60 °C and then diluted with ether (25 ml) and water (10 ml) and filtered through a Celite pad. The organic layer was dried and evaporated. The thienopyrrole 9a (or 9d) was isolated after silica gel chromatography of the oily residue (eluent: light petroleum–CH2Cl2, 75∶25).Radical cyclization of N-allyl(2-halo-3-thienyl)carbamates 12
A degassed solution of carbamate 12 (1 mmol) in toluene (10 ml) containing AIBN (0.01 g) and tributyltin hydride (0.437 g, 1.5 mmol) was stirred under reflux for 8 h. The solvent was evaporated and silica gel chromatography (eluent: light petroleum–CH2Cl2, 80∶20, then CH2Cl2) afforded the dihydrothienopyrrole 10 (or the tetrahydrothienopyridine 19 for 12b).Preparation of tert-butyl 2-allyl-3-thienylcarbamate 13a![[hair space]](https://www.rsc.org/images/entities/h3_char_200a.gif) 16
16
The carbamate 3a (0.398 g, 2 mmol) in THF (20 ml) was treated with n-BuLi (2.5 M solution in hexane, 2 ml) at −78 °C. Allyl bromide (0.423 g, 3 mmol) was added at −10 °C. The mixture was stirred for 2 h at this temperature and quenched with sat. aq. NaCl solution. The organic layer was evaporated and the product 13a was purified by silica gel chromatography (eluent: light petroleum–CH2Cl2, 50∶50). Mp = 70 °C, yield = 95%. 1H NMR (CDCl3), δ: 7.30 (br s, 2H, H5), 7.02 (d, 1H, H4, J = 5.4 Hz), 6.50 (br s, 1H, NH), 5.80 (m, 1H, Hβ), 5.07 (m, 2H, Hγ), 3.40 (d, 2H, Hα, J = 6.5 Hz).
Formal “PhSeOAc” addition to the 2-allylthiophene 13a
Diphenyl diselenide (0.375 g, 1.2 mmol) and (diacetoxy-λ3-iodanyl)benzene (0.754 g, 2.6 mmol) were added to a solution of 13a (0.478 g, 2 mmol) in CH3CN (6 ml). The mixture was stirred overnight and treated with sat. aq. NaCl solution. The organic layer was dried and evaporated. The addition products 21a and 22a were separated by silica gel chromatography (eluent: light petroleum–CH2Cl2, 1∶1). A partial isomerization of 22a into 21a was observed during the purification.PhSeCl addition to the 2-allylthiophene 13a
The carbamate 13a (0.717 g, 3 mmol) in CH2Cl2 was treated with PhSeCl (0.862 g, 4.5 mmol) in the same solvent (50 ml). The mixture was stirred overnight and sat. aq. NaCl solution was added. The organic layer was dried and evaporated. The chloroselenide 21b was obtained as a mixture with the substrate 13a (21b–13a, 85∶15). The chromatographic separation was unsuccessful.Cyclization of the β-chloro phenylselenide 21b
A solution of the crude addition product 21b (0.862 g, 2 mmol) in CH2Cl2 was stirred overnight in the presence of silica gel (1 g). After filtration and concentration, the solid was recrystallized. The oxazepine 25 was isolated as white crystals. The solution resulting from the recrystallization was evaporated and chromatographed (eluent: CH2Cl2–light petroleum, 50∶50). The dihydrothienopyrrole 23, tetrahydrothienopyridine 24 and thienooxazepine 25 were successively isolated in 25, 5 and 21% yields respectively.N-Allylation of the amino carbamate 4 and dicarbamate 5
A mixture of amino carbamate 4 (0.214 g, 1 mmol) and NaH (0.24 g, 10 mmol) in THF (20 ml) was stirred for 30 min. Allyl bromide (1.21 g, 10 mmol) was added and the stirring continued for 5 h. The mixture was quenched at 0 °C with sat. aq. NaCl solution. The organic layer was evaporated and silica gel chromatography provided the monoallylated product 27 (eluent: light petroleum–CH2Cl2, 50∶50). The diallylated dicarbamate 18 was prepared from dicarbamate 5 using double amounts of NaH and allyl bromide, after 5 h of stirring at room temperature. The diallyl thienoimidazolone 26 was obtained after 20 h of reaction. All compounds were purified by silica gel chromatography (eluent: light petroleum–CH2Cl2, 50∶50).Reaction of N-allyl compounds 14a, 18 and 27 with PhSeCl
N-Allylthienylcarbamate 14a (0.478 g, 2 mmol) in CH2Cl2 (30 ml) was treated with PhSeCl (0.575 g, 3 mmol) dissolved in the same solvent (30 ml) containing sodium carbonate (0.848 g, 8 mmol). The reaction mixture was stirred overnight and sat. aq. NaCl solution was added. The organic layer was dried and evaporated. Silica gel chromatography afforded the oxazolidinone 28 (eluent: CH2Cl2). The same procedure gave the bis(oxazolidinone) 29a from dicarbamate 18 and the 2-(phenylselanyl)thiophene derivative 30 from the monoallyl diamine derivative 27. The chromatographic purification was achieved on silica gel (eluent: light petroleum–CH2Cl2, 50∶50).N-Iodosuccinimide-induced cyclization of di-tert-butyl N,N′-diallyl(thiophene-3,4-diyl)dicarbamate 18
NIS (0.9 g, 4 mmol) was added to N,N′-diallyldicarbamate 18 (0.394 g, 1 mmol) in CCl4 (20 ml). The mixture was stirred overnight at room temperature. The oxazolidinone 29b was isolated and purified as described for 29a.Preparation of N,N′-diallyl-3,4-bis(methylamino)thiophene 32
Dicarbamate 18 (0.394 g, 1 mmol) in dry ether (30 ml) was added to a suspension of LiAlH4 (0.8 g, 20 mmol) in the same solvent (20 ml). The mixture was stirred overnight, cooled to 0 °C and treated with sat. aq. NaCl. The aqueous solution was extracted twice with ether and the combined organic layers were dried and evaporated. Silica gel chromatography (eluent: light petroleum–CH2Cl2, 80∶20) afforded the diamine 32. Yield: 72%. 1H NMR (CDCl3), δ: 6.31 (s, 2H, H2, H5), 5.92–5.75 (m, 2H, Hβ), 5.25–5.12 (m, 4H, Hγ), 3.75 (d, 4H, Hα, J = 6.3 Hz), 2.68 (s, 6H, CH3). 13C NMR (CDCl3), δ: 145.2 (C3, C4), 135.3 (Cβ), 116.9 (Cγ), 104.7 (C2, C5), 56.1 (Cα), 39.0 (CH3). Anal. calc. for C12H18N2S: C, 64.82; H, 8.16; N, 12.60; found: C, 64.97; H, 8.22; N, 12.77%.Acknowledgements
We thank the “Conseil Régional de Haute-Normandie” for the provision of a grant (D. B.).References
- I. W. Southon and J. Buckingham , Dictionary of Alkaloids, Chapman and Hall, London, 1989. ed. J. E. Saxton, Chem. Heterocycl. Compd., 1994, 25, Suppl. IV; Search PubMed; K. C. Joshi and P. Chand, Pharmazie, 1982, 37, 1 Search PubMed.
- F. Garcia and C. Galvez, Synthesis, 1985, 143 CrossRef CAS.
- C. Paulmier, Sulfur Rep., 1996, 19, 215 Search PubMed.
- W. W. Gale, A. N. Scott and H. R. Snider, J. Org. Chem., 1964, 29, 2160 CAS; S. Soth, M. Farnier and C. Paulmier, Can. J. Chem., 1978, 56, 1429 CAS; C. Galvez and F. Garcia, J. Heterocycl. Chem., 1984, 21, 393 Search PubMed.
- (a) D. Wensbo, A. Eriksson, T. Jeschke, U. Annby, S. Gronowitz and L. A. Cohen, Tetrahedron Lett., 1993, 34, 2823 CrossRef CAS; (b) D. Wensbo, U. Annby and S. Gronowitz, Tetrahedron, 1995, 51, 10323 CrossRef CAS; (c) D. Wensbo and S. Gronowitz, Tetrahedron, 1996, 52, 14975 CrossRef CAS.
- C. Galvez, F. Garcia, A. Marzal and P. Viladoms, J. Chem. Res., 1994, 12 Search PubMed; D. N. Reinhoudt, J. Geevers, W. P. Trompenaars, S. Harkema and G. J. Van Hummel, J. Org. Chem., 1981, 46, 424 CrossRef CAS.
- (a) F. Outurquin and C. Paulmier, Bull. Soc. Chim. Fr., 1983, II, 153 Search PubMedF. Outurquin and C. Paulmier, Bull. Soc. Chim. Fr., 1983, II, 159 Search PubMed; (b) F. Outurquin, P. Lerouge and C. Paulmier, Bull. Soc. Chim. Fr., 1986, 259 Search PubMedF. Outurquin, P. Lerouge and C. Paulmier, Bull. Soc. Chim. Fr., 1986, 267 Search PubMed; (c) F. Outurquin and C. Paulmier, Tetrahedron Lett., 1993, 34, 5715 CrossRef CAS; (d) M. Berkaoui, F. Outurquin and C. Paulmier, J. Heterocycl. Chem., 1996, 33, 9 Search PubMed; (e) D. Brugier, F. Outurquin and C. Paulmier, Tetrahedron, 1997, 53, 10331 CrossRef CAS.
- D. Brugier, F. Outurquin and C. Paulmier, Tetrahedron, 2000, 56, 2985 CrossRef CAS.
- M. S. Yu, L. Lopez de Leon, M. A. McGuire and G. Bothon, Tetrahedron Lett., 1998, 39, 9347 CrossRef CAS; S. Cacchi, G. Fabrizi, F. Marinelli, L. Moro and P. Pace, Synlett, 1997, 1363 CrossRef CAS; S. Cacchi, G. Fabrizi and P. Pace, J. Org. Chem., 1998, 63, 1001 CrossRef CAS.
- J. Fujiwara, Y. Fukutani, H. Sano, K. Maruoka and H. Yamamoto, J. Am. Chem. Soc., 1983, 105, 7177 CrossRef CAS; T. Nishikawa, M. Ishikawa and M. Isobe, Synlett, 1999, 123 CrossRef CAS.
- Y. Kondo, S. Kojima and T. Sakamoto, Heterocycles, 1996, 43, 2741 Search PubMed.
- (a) W. F. Bailey and X. L. Jiang, J. Org. Chem., 1996, 61, 2596 CrossRef CAS; (b) D. Zhang and L. S. Liebeskind, J. Org. Chem., 1996, 61, 2594 CrossRef CAS.
- For the preparation of indolines via radical cyclization of N-allylanilines see: D. L. Boger, R. M. Garbaccio and Q. Jin, J. Org. Chem., 1997, 62, 8875 CrossRef CAS; J. Inanaga, O. Ujikawa and M. Yamaguchi, Tetrahedron Lett., 1991, 32, 1737 CrossRef CAS; Y. Hayashi, H. Shinokubo and K. Oshima, Tetrahedron Lett., 1998, 39, 63 CrossRef CAS; J. Nakao, R. Inoue, H. Shinokubo and K. Oshima, J. Org. Chem., 1997, 62, 1910 CrossRef CAS.
- D. L. Clive, V. Farina, A. Singh, C. K. Wong, W. A. Kiel and S. M. Menchen, J. Org. Chem., 1980, 45, 2120 CrossRef CAS; K. C. Nicolaou, D. A. Claremon, W. E. Barnette and S. P. Seitz, J. Am. Chem. Soc., 1979, 103, 3703; R. R. Weeb and S. Danishefsky, Tetrahedron Lett., 1983, 24, 1357 CrossRef.
- M. Tingoli, M. Tiecco, L. Testaferi and A. Temperini, Synth. Commun., 1998, 28, 1769 CAS.
- M. Berkaoui , Thesis, Rouen, 1998. Search PubMed.
- C. Galvez, F. Garcia, J. Garcia and J. Soldevila, J. Heterocycl. Chem., 1986, 23, 1103 Search PubMed.
- F. Garcia and C. Galvez, Sulfur Lett., 1982, 1, 97 Search PubMed; E. W. Brunett and W. C. MacCarthy, J. Pharm. Sci., 1968, 57, 2003 CAS.
Footnote |
| † The IUPAC name for triflic acid is trifluoromethanesulfonic acid. |
| This journal is © The Royal Society of Chemistry 2001 |