The biosynthesis of plant alkaloids and nitrogenous microbial metabolites
Richard B. Herbert
School of Chemistry, University of Leeds, Leeds, UK LS2 9JT
First published on 9th January 2001
Abstract
Covering: January 1997 to December 1998. Previous review: Nat. Prod. Rep., 1999, 16, 199.
1 Introduction
As previously, reference is made to background material in earlier reports in this series as well as to a monograph.1–8 As full coverage of the literature as possible was obtained, in part, by use of CAS Online.Jasmonic acid 1 has been identified as an important compound in the elicitation of the biosynthesis of secondary metabolites in higher plants. Its biosynthesis (including enzymology), signal transduction and gene expression have been reviewed.9–11 The biosynthesis of natural products from unusual variants of the shikimate pathway have been authoritatively reviewed,12 as has the enzymology of alkaloid biosynthesis![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 13 and important oxidoreductases in the biosynthesis of indole and isoquinoline alkaloids.14 Alkaloid biosynthesis in hairy-root cultures has been surveyed.15
13 and important oxidoreductases in the biosynthesis of indole and isoquinoline alkaloids.14 Alkaloid biosynthesis in hairy-root cultures has been surveyed.15
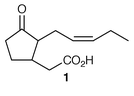
2 Pyrrolidine and piperidine alkaloids, and lunarine
2.1 Tropane alkaloids
The biosynthesis of the esterifying tropic acid 2 moiety found in some tropane alkaloids has been reviewed with authority,16 as has the biosynthesis of pyrrolidine (including tropane alkaloids) and pyrrolizidine alkaloids in root cultures.17
There have been interesting developments recently in the ongoing story of tropane alkaloid biosynthesis (ref. 7, p.199; ref. 6, p. 359). One concerns the intriguing rearrangement which must occur during the formation of the tropic acid 2 moiety found in, e.g. hyoscyamine 6, the other relates to the steps between 3 and tropinone 4. Significant new information has been reported in both areas.
Previously, evidence had been obtained that the acid 4, or its CoA ester, is a key intermediate in tropane alkaloid biosynthesis and that 8 and hygrine 7 are not precursors for these bases. The results have been convincingly confirmed in transformed root cultures of Datura stramonium.18 (R,S)-[2′,3′-13C2]hygrine, as 7, and (R,S)-[1,2-13C2,2-14C]-2-(1-methylpyrrolidin-2-yl)acetate, as 8, were found not to be significant precursors where sodium [1,2-13C2]acetate and ethyl (R,S)-[2,3-13C2,3-14C]-4-(1-methylpyrrolidin-2-yl)-3-oxobutanoate 9, were converted into tropane bases at up to 12% specific incorporation. 13C label from the acetate appeared symmetrically at C-2 and C-4, as well as being present at C-3, of (−)-hyoscyamine 6 (the observed optical activity shows that the symmetrical labelling pattern is not because the isolated alkaloid was racemic). The incorporation of 13C-labelled 9 was efficient and specific. Similar results with 9 have been obtained for (−)-scopolamine 10 and also for cocaine 11 (ref. 6, p. 359; ref. 2, p. 575). The labelling in scopolamine18 and hyoscyamine (ref. 6, p. 359) was of both C-2 and C-4 (as well as C-3) indicating curiously that both the (R)- and (S)-isomers acted as precursors.
In conclusion, questions of stereochemistry aside, 4, or CoA ester, is identified as a key precursor in the biosynthesis of cocaine 11 and of tropane alkaloids being formed by condensation of acetoacetate with 3. The failure of 8 to act as a precursor excludes stepwise addition of two acetates starting with 3. Carboxytropinone has been found in D. stramonium (see ref. 18). It is arguably the next intermediate formed by cyclisation of 4.
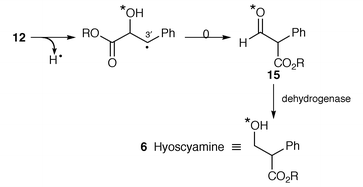
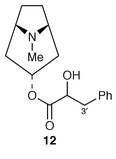
The last step in the biosynthesis of hyoscyamine 6 involves rearrangement of the esterifying group in littorine 12 to yield the tropic acid 2 fragment found in 6. The fate of individual atoms and the stereochemistry of this mutase-catalysed rearrangement has been established. A convincing argument has been made for radical intermediates beginning with loss of the 3′-pro-S proton from littorine 12 (Scheme 1) (ref. 6, p. 359; ref. 7, p. 199).
 | ||
| Scheme 1 | ||
Such hydrogen abstractions in biochemistry are achieved by P450 enzymes and also by the use of 5′-deoxyadenosyl radicals. In animals and bacteria coenzyme B12 is the major source of these latter radicals. But in plants, no trace of B12 has been found. S-Adenosylmethionine (SAM) 13 has been found to act (in Clostridium) as the source of a 5′-deoxyadenosyl radical in an aminomutase reaction, the necessary homolytic cleavage of the carbon–sulfur bond being promoted by an Fe4S4 cluster-containing enzyme.19 Plants are known to use SAM as the source of 5′-deoxyadenosyl radical in the last step of biotin biosynthesis.20,21
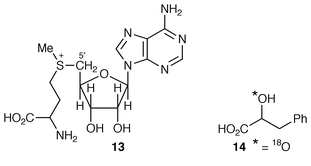
The above serves as an introduction to a very clever and successful experiment.21 A cell-free extract was prepared from cultured hairy roots of D. stramonium. When SAM was added, in addition to littorine, the rearrangement to hyoscyamine was stimulated 10–20-fold (from a very low base conversion, due presumably to endogenous SAM). Addition of SAM showed saturation kinetics with a Km of 25 μM. In preliminary experiments with [2,8,5′-3H3] SAM no incorporation of radioactivity into littorine or hyoscyamine was observed. Further results arising from these experiments are awaited with considerable interest.
As a probe into the mechanism whereby littorine 12 is transformed into hyoscyamine 6, the incorporation has been studied of (RS)-phenyl[2-18O,2-2H]-lactate 14 into 12 and 6 in root cultures of D. stramonium.22 By comparison of the isotope uptake into the two alkaloids, it was apparent that up to 29% of 18O was lost in the rearrangement of 12 into 6. The mechanism of the rearrangement was discussed22 in terms of a P450-catalysed reaction; it now will no doubt be reconsidered in terms of the above evidence for a mutase reaction.21 For both mechanisms, the reaction will proceed in outline as shown (Scheme 1), in which the aldehyde 15 is an intermediate. Its reduction by an unidentified dehydrogenase affords hyoscyamine 6. Most simply, it is at this stage that partial 18O loss could occur by exchange.
The metabolic relationship between littorine 12 and hyoscyamine 6 has been studied in transformed roots of D. stramonium.23 Whilst the former alkaloid was converted into hyoscyamine, this conversion was apparently irreversible. The elicitation of alkaloid biosynthesis in these roots has also been examined.24
It has been found25 that the demethylation of nicotine to nornicotine in Nicotiana root cultures does not involve loss of hydrogen from either C-4′ or C-5′. Induction of enzymes of nicotine biosynthesis has been examined.26
2.2 Pramanicin
The results of stable-isotope labelling experiments establish27 that pramanicin 16 is formed, in cultures of Stagonospora species, from eight intact acetate units and an intact molecule of L-serine (Scheme 2). The latter was deduced to be a primary precursor for the antibiotic; all four bonds to the α-carbon of the amino acid remain undisturbed during biosynthesis. The origin of the pyrrolidine ring from serine and acetate is notable.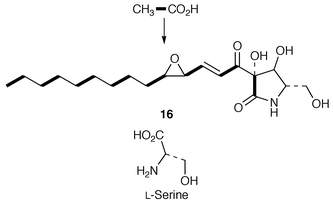 | ||
| Scheme 2 | ||
2.3 Pyrrolizidine alkaloids
Following earlier work with radioactive samples of isoleucine and 2-aminobutanoic acid 19 (a precursor for isoleucine) (for reviews see ref. 2, p. 577 and ref. 28), the incorporation has been studied of 19, labelled with deuterium and 13C, into the senecic acid portions 20 and 21 of rosmarinine 17 and of senecionine 18, respectively.29 (RS)-[3,4-13C2]-2-Aminobutanoic acid and (RS)-[3,4-2H5]-2-aminobutanoic acid, as 19, were incorporated satisfactorily into rosmarinine 17 in Senecio pleistocephalus plants and into senecionine 18 in S. vulgaris root cultures. The 13C labelling patterns in the necic acid moieties corresponded—they are shown as 20 and 21—and there was equal incorporation in both cases into each half of the senecic acid portions. The presence of deuterium at C-13 and C-20 in 21 rules out carbonyl groups at these positions in any intermediates.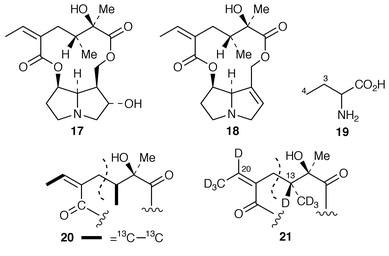
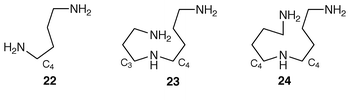
Homospermidine synthase (HSS) is a pivotal enzyme in the biosynthesis of the necine bases of pyrrolizidine alkaloids. The enzyme catalyses the conversion of two molecules of putrescine 22 into homospermidine 24. Remarkably spermidine 23 can substitute for one of the putrescine units in this conversion (ref. 5, p. 46). Short-term feeding experiments with [3H]putrescine and [14C]spermidine have shown that the aminobutyl group (C4) in spermidine 23 is directly incorporated into necine bases in root cultures of S. vulgaris.30 A similar result was obtained for homospermidine 24 that accumulated when β-hydroxyethylhydrazine was present; it is an amine oxidase inhibitor and prevents biosynthesis beyond 24. Putrescine and spermidine were incorporated with the same efficiency and affinity (identical Km and Vmax).
The turnover (not significant over 29 days) and translocation of pyrrolizidine alkaloid N-oxides in Senecio species has been examined.31
2.4 Lunarine
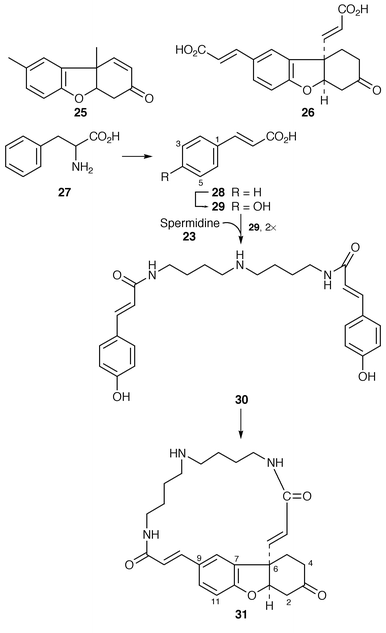
Pummerer’s ketone 25 is a synthetic compound central in the development of Barton’s ideas on oxidative phenolic coupling as a biosynthetic process.8 The naturally occurring analogue is lunarine 31, which is found in the seeds of Lunaria annua; it is optically active. Following earlier evidence that phenylalanine 27, but not tyrosine, is a (specific) precursor for lunarine 31, confirmation has come with high incorporation of L-phenylalanine in seeds of L. annua at a particular stage of development.32 Further precursors were clearly identified as cinnamic acid 28, p-coumaric acid 29, spermidine 23 and putrescine 22. All of these results were obtained with radioactive labelling. The incorporation of [1-13C]coumaric acid, as 29, with labelling of C-6 and C-9 of 31, and of [3,5-13C2]coumaric acid with labelling of C-2, C-4, C-7 and C-11 emphatically confirms a precursor role for 29 in lunarine biosynthesis.The naturally occurring spermidine derivative 30, labelled with tritum in the coumaranyl moiety and 14C in the spermidine moiety, gave results which establish it as an intact precursor of lunarine 31; the diacid 26 is therefore apparently not involved in biosynthesis. An attempt to identify a P450 oxidase for the coupling of 30 was not successful. Further results indicate that arginine is preferred over ornithine as a spermidine (putrescine) precursor. The deduced biosynthetic pathway to lunarine 31 is illustrated in Scheme 3.
 | ||
| Scheme 3 | ||
3 Isoquinoline and Amaryllidaceae alkaloids
The biosynthesis (and biological activities) of naphthylisoquinoline alkaloids has been reviewed.333.1 Benzylisoquinoline alkaloids
Out of 333 different cell suspension cultures of Papaver somniferum one has been found, for the first time, that produces the opium alkaloid thebaine 32.34 Metabolism in the cell cultures of [N-14CH3]thebaine was found to yield codeine 33, a normal opium metabolite in intact plants,8,35 tetrahydrothebaine 34, and thebainone 35. This demonstrates the presence of the enzymes necessary for O-demethylation at C-6 in the cultures. In a separate series of experiments with cell cultures of 60 different species (not including Papaver) the thebaine was found to be notably inert, with only one culture that transformed thebaine into another metabolite (by O-demethylation at C-3).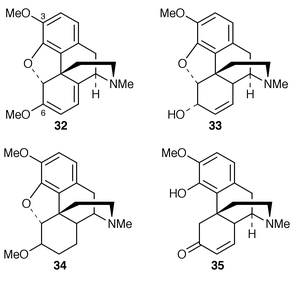
A review has been published36 on the role of cell differentiation and plant development in P. somniferum plants and tissue cultures. A temporal correlation has been made of tyramine metabolism with alkaloid and amide biosynthesis in elicited P. somniferum cell cultures.37 Expression patterns conferred by tyrosine/phenylalanine decarboxylase promoters in P. somniferum are conserved in transgenic tobacco.38
Two alleles encoding for 3′-hydroxylation of (S)-N-methylcoclaurine 36 (for an enzyme-annotated chart of benzylisoquinoline biosynthesis which involves this compound see Scheme 8 in ref. 1, p. 512) have been isolated from a cDNA library prepared from poly(A) + RNA obtained from methyl jasmonate-induced cell-suspension cultures of Eschscholzia californica.39 The enzyme is a P450-dependent mono-oxygenase and has been characterised. A genomic clone, bbe1, has been isolated, from E. californica, that encodes the methyl jasmonate-inducible berberine bridge enzyme (ref. 6, p. 363; ref. 5, p. 48) of benzophenanthridine alkaloid biosynthesis.40 Two genes are apparently present; their characteristics have been described. The change in intracellular pH distribution as part of the signal mechanism in elicitation of benzophenanthridine alkaloid biosynthesis has been examined.41 Barbiturate-induced alkaloid formation in E. californica proceeds by gene transcript accumulation.42 Other aspects of the induction of alkaloid biosynthesis have been explored.43 The effect has been examined of P450 inhibitors on benzylisoquinoline biosynthesis in cultured roots of Stephania cepharantha and Menispermum dauricum.44
)-norprotosinomenine 37 as the key benzylisoquinoline intermediate. Furthermore, biosynthesis was believed to proceed via a late-stage symmetrical intermediate (for a summary, see ref. 8). The problem with the norprotosinomenine result is that only precursors of the coclaurine 40-reticuline type have been recognised as precursors for the multitude of benzylisoquinoline alkaloids (see ref. 8 and many of these Reports). The biosynthesis of Erythrina alkaloids has therefore been re-examined, mainly in E. crista-galli.45 As one expects of the Zenk group this has been rigorous. The results establish the sway of orthodoxy.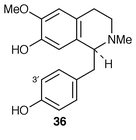
 | ||
| Scheme 4 | ||
Biosynthesis from benzylisoquinoline precursors was found only to occur in the fruit tissue of E. crista-galli; for a start there was no precursor transport in whole plants. In the fruit tissue (S)-[6-OC3H3]coclaurine, as 40, and similarly labelled (S)-norreticuline 41 were incorporated to the convincing extent of 11–12%; 3H-labelled samples of (S)-norprotosinomenine 37, (S)-nororientaline 38 and (S)-norisoorientaline 39 were not incorporated. Neither were their N-methyl derivatives or (R)-isomers incorporated and similar results were obtained with coclaurine and norreticuline. Thus, norprotosinomenine 37 was found not to be a precursor for Erythrina-type alkaloids in C. laurifolius. In confirmation of the biosynthetic roles clearly emerging for 40 and 41, (S)-[1-13C]coclaurine, as 40, and similarly labelled norreticuline 41 were efficiently and specifically incorporated into 42 in E. crista-galli with exclusive labelling of C-10. A symmetrical intermediate requires equal labelling of C-8 and C-10. There is, therefore, no symmetrical intermediate and the true Erythrina precursors are coclaurine 40 and norreticuline 41. The provenance of these alkaloids (Scheme 4) corresponds therefore in the main (cf. Scheme 8 in ref. 1, p. 512) with the legions of other benzylisoquinoline alkaloids. An intriguing mechanistic question now is how does the skeleton 41 become that of 42 and 43?
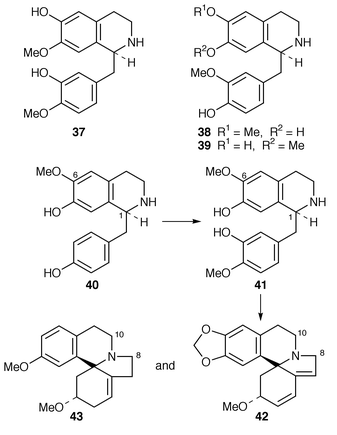
46 that (1R)-deacetylipecoside 46 is incorporated into alangiside 49 in A. lamarckii and ipecoside in Cephalis ipecacuanha while the (1S)-epimer 47 is used for the biosynthesis of emetine, 52, types of alkaloid. In support of the two epimers of deacetylipecoside (and hence their epimeric derivatives) being produced normally in vivo, a crude enzyme preparation has been obtained47 from A. lamarckii that catalyses the formation of (S)-deacetylisoipecoside 47 and its C-1 epimer 46. The presence of two enzymes in the preparation was apparent. Attempted isolation of the enzymes resulted in only deacetylipecoside synthase being obtained, i.e. only a single enzyme activity had survived purification. Importantly, it catalysed the condensation of 44 and 45 stereospecifically to give 46.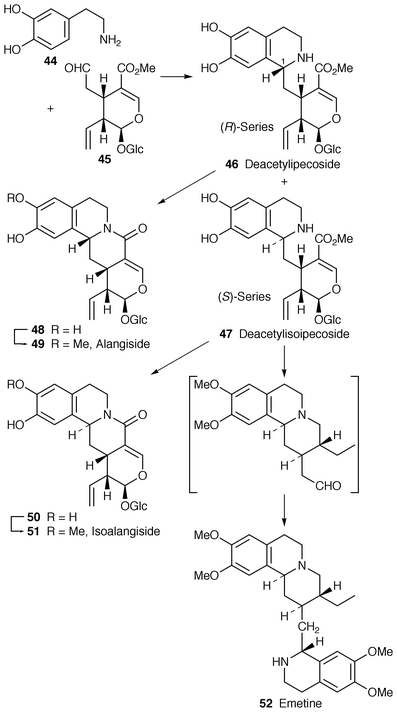 | ||
| Scheme 5 | ||
3.2 Colchicine
Because it appeared to have no structural relationship to any other alkaloid, Robert Robinson considered colchicine 56 to be sui generis.48 Subsequent research showed,8 however, that this unusual alkaloid has a whole family of structural relations, being simply a modified phenethylisoquinoline, as 60 ≡ 61, with (S)-autumnaline 53 as a late key intermediate leading to O-methylandrocymbine 55 and thence colchicine (Scheme 6) (cf., for example morphine biosynthesis35). In a sudden publication burst during the review period, much of the detail of biosynthesis has been filled in now to proclaim a well-delineated pathway. A set of full papers from Battersby and co-workers follow earlier preliminary communications.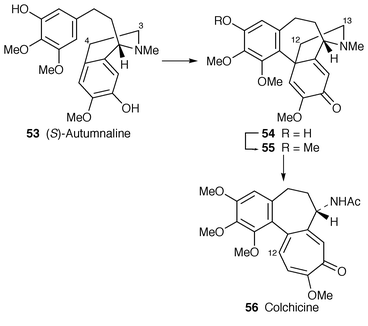 | ||
| Scheme 6 | ||
First, though, further information on the sequence of phenethylisoquinoline intermediates has been obtained which supplements earlier results.49,50 In the new work,51 consistently high, and therefore convincing, incorporations (10% or more) were obtained under optimised conditions with immature seeds of Colchicum autumnale. The first phenethylisoquinoline is 58 formed by condensation of the aldehyde 57 and dopamine 44 (Scheme 7).50N-Methylation may occur early50,51 with 5949,51 and 6051 as following biosynthetic intermediates (cf. ref. 50 where a good incorporation of the N-nor-compound related to 60 was obtained). Cinnamic acid (which leads to 5750) and autumnaline 53 are fully confirmed as colchicine precursors.51 That (S)-autumnaline 53 is involved uniquely and as an intact molecule in the biosynthesis of colchicine has been put beyond doubt.51,52
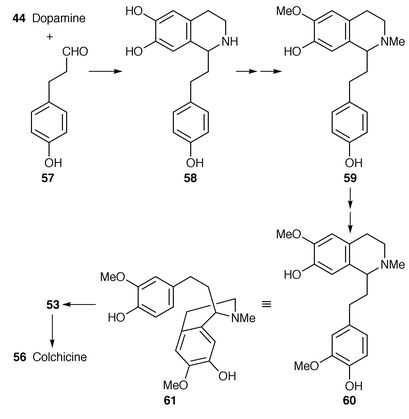 | ||
| Scheme 7 | ||
Phenol oxidative coupling on autumnaline 53 affords isoandrocymbine 54 and this coupling has been proved to be in a para–para sense as shown in Scheme 6.53 A substrate-specific microsomal-bound cytochrome P450 NADPH- and oxygen-dependent enzyme system has been obtained from C. autumnale which carries out this coupling reaction (ref. 7, p. 201).
The ring-expansion of O-methylandrocymbine 55 that leads to colchicine 56 is of great, not least mechanistic, interest. The first step in establishing what happens is to discover what the intermediates are and then to look for the enzyme responsible. It is known that C-12 of O-methylandrocymbine is retained as C-12 of colchicine 56 and that C-13 is lost,8 but C-13 is retained as the formyl group of N-formyldemecolcine 62 and this compound is an efficient precursor for colchicine 56 and demecolcine 63 (with loss of the formyl group).54 Interlocking feeding results with C. autumnale, C. byzantinum and C. speciosum establish the sequence O-methylandrocymbine 55→62→63→64→65 colchicine 56 (Scheme 8). The alkaloids speciosine 67 and N-methyldemecolcine 66 derive largely from demecolcine 63. For the first time, 55 was shown to be present in C. autumnale.54
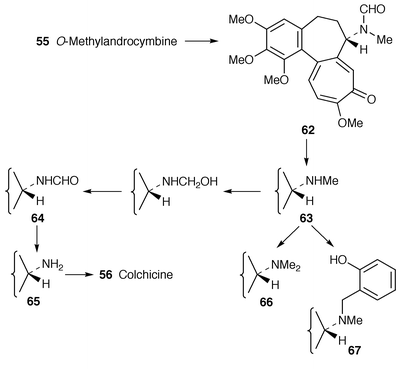 | ||
| Scheme 8 | ||
In the conversion of 55 into 62 there is a net loss of a hydrogen atom from each of C-12 and C-13 of 55. The next set of results relate to the stereochemistry associated with these losses. Loss of hydrogen from C-13 was measured on N-formyldemecolcine 62, that from C-12 on colchicine 56.
The results of experiments in C. autumnale with samples of (RS)-autumnaline, as 53, stereospecifically tritiated at C-3 (≡C-13 of O-methylandrocymbine 68 = formyl group in 62) show that in the ring expansion reaction from 55 ≡ 68 leading to 62 the 13-pro-S proton (as tritium) is lost stereospecifically (with retention of the 13-pro-R proton) (see Scheme 9).55 On the other hand, though there was a stereospecific total proton loss of the 12-pro-S proton (as tritium) in 55 ≡ 68 in the formation of colchicine 56, there was also partial loss (ca. 20%) of tritium from the HR position at C-12.56 As before, labelled autumnaline was used and it was established amongst other checks that both hydrogen labels at C-4 in 53 were retained as far as 55 ≡ 68.
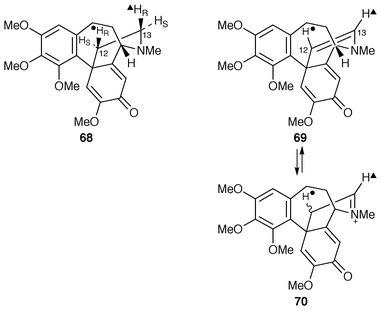 | ||
| Scheme 9 | ||
An early model8 for the ring expansion involved hydroxylation at C-12 of 55. Stereoelectronically for elimination of a hydroxy group this group should be at the HR site. The well-attested retention of configuration in such a hydroxylation is expected, i.e. loss of HR. Retention of HR, albeit partial, downgrades this possibility.
It is reasonable to suggest that what is being seen here at the C2 bridge in 68 (Scheme 9) is stereospecific dehydrogenation at C-12 and C-13 (which happens to be syn) with another event superimposed. Dehydrogenation of 68 affords the enamine 69; enamine–iminium ion 70 interconversion without stereocontrol (i.e. non-enzymatic/chemical) would result in preferential 3H over 1H retention at C-12 by a primary isotope effect. In support of such an exchange process, it was found that a synthetic sample of the enamine 69 chemically underwent deuterium incorporation at C-12 in tetradeuteriomethanol.56
This putative enamine–iminium ion intermediate could arise by enzymic syn-dehydrogenation or by amine to iminium ion oxidation (Scheme 9) followed by enzymic (stereospecific) proton removal from C-12 the first time tautomerism occurs. The observed proton exchange at C-12 could occur by the enzyme undergoing some exchange with the medium or by the enamine–iminium ion system having a significant lifetime off the enzyme before being transformed further. A new radical-based mechanism for the splendiferous ring-expansion reaction (Scheme 10) may be proposed which is electronically and sterically well-supported.56 But does the reaction involve radicals? The next piece of evidence answers that question.
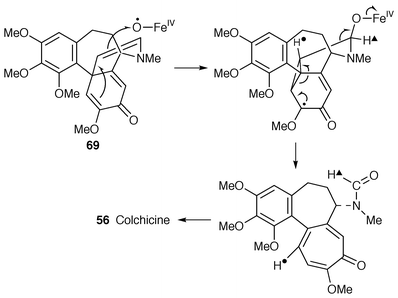 | ||
| Scheme 10 | ||
Microsomal preparations have been obtained57 from immature seeds of C. autumnale that catalyse the conversion of O-methylandrocymbine 55 ≡ 68 into demecolcine 63 in the presence of NADPH and O2. Very evidently, a P450 oxidase is involved here as it is in the coupling reaction which affords isoandrocymbine 54 (above). Not only do the coupling and ring-expansion reactions occur in the microsomal preparation, but also methylation of 54 to 55 in the presence of SAM, N-demethylation of demecolcine (by a P450; cf.Scheme 8, 63→65) and then N-acetylation of 65 to colchicine 56 in the presence of acetylcoenzyme A.
In conclusion, the two key steps of coupling and ring-expansion involve P450 enzymes and both, consequently and most reasonably, involve radicals; the simple mechanism proposed for the ring-expansion which yields colchicine 56, that enigmatic alkaloid of Robinson’s time and many years thereafter, is illustrated in Scheme 10. The mechanism is well supported by analogy (see refs. cited in ref. 56).
3.3 Amaryllidaceae alkaloids
The Amaryllidaceae alkaloid galanthamine 75 finds useful and diverse medical application. With possible biotechnological production of this alkaloid in mind, the already delineated biosynthetic pathway8 to 75 has been re-examined.58First, [OC3H3]-4′-O-methylnorbelladine, as 71, was used to identify maximal precursor incorporation in various isolated organs of Leucojum aestivum under optimal experimental conditions. The best were with sliced ovary wall tissue where high incorporations into galanthamine 75 (27%) and N-demethylgalanthamine 74 (22%) were observed. The wide exploration and optimisation of incorporation achieved here and elsewhere (see, e.g., ref. 51) by the Zenk group is notable.
Second, when labelled 4′-O-methyl-N-methylnorbelladine 76, previously deduced to be an intermediate,8 was tested as a precursor it was incorporated into galanthamine with about one-third the efficiency of the N-demethyl compound 71, and substantial labelling of the N-demethylated metabolites 71 and 74 was observed.58 On the other hand, [3,5-13C2]- and [α-13C]-4′-O-methylnorbelladine, as 71, were specifically and efficiently incorporated into 74 and galanthamine 75. Further, it was established with labelled forms of 74 and 75 that galanthamine 75 is formed from demethylgalanthamine 74 and not the other way round. Clearly the true intermediate in galanthamine biosynthesis is 71 and 76 is only utilised after demethylation.
Based on these convincing results, a revised biosynthetic pathway to galanthamine 75 has been advanced (Scheme 11).58 Chemical evidence was obtained that the dienone 72 formed initially by phenol coupling would spontaneously cyclise to 73.
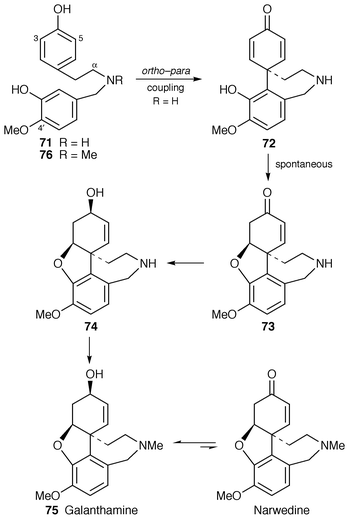 | ||
| Scheme 11 | ||
There are three different groups of Amaryllidaceae alkaloids that were previously deduced to be biosynthesised by different modes of phenol coupling on a norbelladine derivative, as 71, namely those represented by galanthamine 75, oxocrinine 78 and norpluviine 77.8 Whilst the latter two types derive directly by coupling on O-methylnorbelladine 71, the galanthamine type was previously held to be different in deriving from the N-methyl compound 76. The new evidence58 shows that the biosynthetic picture is simpler and all three types derive directly from 71 (Scheme 12).
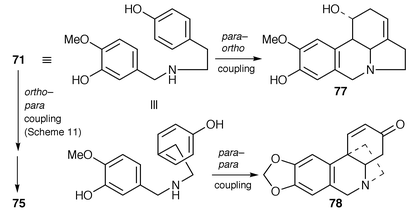 | ||
| Scheme 12 | ||
4 Metabolites derived from tryptophan
4.1 Terpenoid indole alkaloids
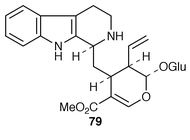
The biosynthesis of terpenoid indole alkaloids in Catharanthus roseus has been reviewed59,60 and a case has been made for its being a good model for the study of regulation.60 The application of metabolic engineering to increasing the production of secondary metabolites, e.g. terpenoid indole alkaloids, has been discussed.61 Strictosidine 79 is the first committed precursor in the biosynthesis of these alkaloids, being formed by condensation of tryptamine with the monoterpene secologanin 45, a reaction which is catalysed by strictosidine synthase. A sensitive assay has been developed for the terpene 45 based on the enzymic conversion.62 Tryptophan decarboxylase is, like strictosidine synthase, a key enzyme in the biosynthesis of terpenoid indole alkaloids. Genetic engineering has been applied to the over-expression of the two enzymes.63 The effects have been studied of a tertiary amine, N-(methoxycarbonylethyl)-N-[2-(1H-indol-3-yl)ethyl]-β-methyl alininate, on the production of alkaloids in C. roseus.64 An analysis has been made of the genes for NADH∶cytochrome P450 reductase in relation to those of terpenoid indole alkaloid biosynthesis.65 The expression of tryptophan decarboxylase and strictosidine synthase in transgenic tobacco plants has been studied66 as has the effect of precursor availability on alkaloid accumulation by a transgenic cell line of C. roseus.67 Results have been reported on the effect of dosages and exposure times of the elicitors jasmonate and pectinase on alkaloid biosynthesis in hairy root cultures,68 the modulation of alkaloid biosynthesis by jasmonate69 and the effect of synthetic auxins.70The last two steps in the biosynthesis of vindoline 80 are hydroxylation at C-4 and acetylation (ref. 6, p. 364). The gene encoding the acetyl transferase has been isolated and characterised.71 The C-4 hydroxylase has been cloned and characterised; it is a 2-oxoglutarate dependent dioxygenase.72 There is evidence of a complex mechanism of developmental and light regulation for the enzyme.73
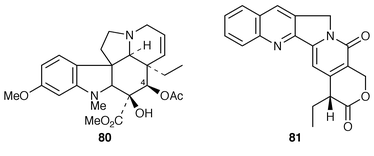
Tryptophan and tryptophan decarboxylase are implicated in the biosynthesis of camptothecin 81 as is the case for other quinoline alkaloids.8 The decarboxylase is encoded by two autonomously regulated genes in Camptotheca acuminata. They are differentially expressed during development and stress.74
4.2 Staurosporine, violacein, paraherquamide A and camalexin
Staurosporine 83, which is an antitumour antibiotic and a protein kinase inhibitor, is biosynthesised from two molecules of tryptophan and one of D-glucose (ref. 7, p. 202). Tryptamine 82 is a precursor.75 Incorporation of tryptamine 82 (tryptophan) is with loss of the two α-protons and the side-chain nitrogen atom.75 In precursor, and modified precursor, directed biosynthesis in Streptomyces staurosporeus, a number of new metabolites were produced and isolated.76 3′-Demethoxy-3′-hydroxystaurosporine-O-methyl transferase, which catalyses the conversion of 84 into 83 which is the last step in staurosporine biosynthesis, has been isolated and partially purified.77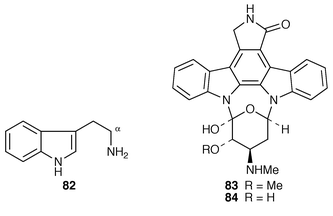
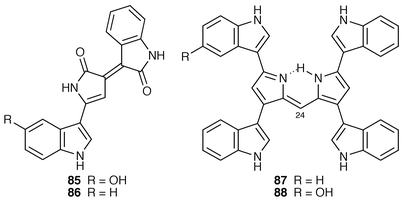
Violacein 85 and deoxyviolacein 86 are tryptophan metabolites produced by Chromobacterium violaceum (ref. 6, p. 364). When a cell-free system78 was incubated with tryptophan, two red pigments closely related to 85 and 86 were produced together with two green pigments 87 and 88. The biosynthesis79 of these latter pigments was shown to be from four molecules of tryptophan and a C1 unit (C-24) derived from the tetrahydrofolate pool ([3-13C]serine was used as labelled precursor). Attempts to correlate the biosynthesis of the six metabolites were inconclusive.
The stereochemistry of some compounds discussed in connection with the biosynthesis of paraherquamide A (ref. 7, p. 202) has been corrected.80
Camalexin 89 is a phytoalexin. It has been concluded,81 from the results of experiments in cell cultures of Arabidopsis with 14C-labelled anthranilic acid and indole, that the biosynthesis of camalexin 89 is from the tryptophan biosynthetic pathway with diversion from it at indole-3-glycerol phosphate, with indole as an intermediate.
4.3 Carquinostatin B via the mevalonate-independent pathway
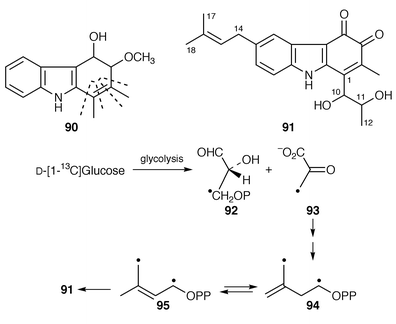
The discovery that some terpenes are formed by a pathway not involving mevalonate, but pyruvate and glyceraldehyde 3-phosphate,82 is a really significant recent event in the study of secondary metabolite biosynthesis and a telling caution against presumption. Though Streptomyces species produce few terpenes, their metabolites appear to derive from either the mevalonate or mevalonate-independent pathways. In a study with Streptomyces aeriouvifer, the organism was found to use the mevalonate-independent pathway for the production of isopentenyl pyrophosphate (IPP) during the early stages of fermentation slowly changing to the alternative pathway as fermentation proceeded.83 This change is associated with a change from the production of a primary metabolite (menaquinone) to that of a secondary one (naphterpin).Carquinostatin B 91 is produced by S. exfoliatus and is similar in structure to carbazomycin B 90 which has its provenance in tryptophan, pyruvate and acetate.84 As expected, the origins of the main skeleton of 91 were found, from the results of feeding experiments with labelled acetate and glucose, to be similar.85 Importantly, the two labelled precursors allow distinction to be made between the two pathways to the dimethylallyl side-chain in 91 with “controls” in the indole ring (shikimate pathway) and acetate derived moiety (C-1, C-10 to C-12). Thus, [1-13C]acetate was well incorporated with (expected) specific labelling of C-1 and C-11, but no labelling of C-14 to C-18 was observed. On the other hand, D-[1-13C]glucose specifically labelled, inter alia, C-14 and C-17.85 This is all as expected of the mevalonate-independent biosynthetic route82 with glycolysis of the glucose yielding [3-13C] pyruvate 93 and [3-13C]glyceraldehyde 3-phosphate 92, the precursors for IPP 94/dimethylallyl pyrophosphate (DMAPP) 95 (Scheme 13).
 | ||
| Scheme 13 | ||
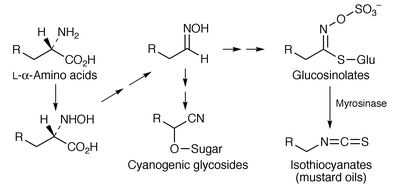
5 Glucosinolates and cyanogenic glycosides
For previous reports, see ref. 7, p. 203. Reviews have been published in this area recently: genetic and biotechnological approaches for reducing glucosinolates in Brassicas;86 wide coverage of glucosinolates including biosynthesis;87 glucosinolate biosynthesis.88Glucosinolates (mustard-oil glucosides) are, usually in association with myrosinase, plant defence compounds referred to as the “mustard oil bomb” (see Scheme 14). Parallel evolution of glucosinolates has been inferred from congruent nuclear and plastid gene phylogenies over a large number of plant species; the bomb was apparently invented twice from likely cyanogenic ancestors.89 The implication of a P450 mono-oxygenase in the biosynthesis of sinalbin 96 from Sinapis alba leads to a similar conclusion.90 This enzyme is additional to the usual biosynthetic enzymes (see ref. 7, p. 204). Examination of the cDNA of several plant species shows evolutionary conservation of enzymes in glucosinolate and cyanogenic glycoside biosynthesis (see Scheme 14).91 The biosynthesis has been explored of sinalbin 96 from [14C]tyrosine in silique walls of S. alba.92
 | ||
| Scheme 14 | ||
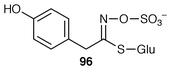
Two cytochrome P450 enzymes (CYP79 and CYP71E1) are involved in the biosynthesis of dhurrin 99 in Sorghum bicolor (Scheme 15). One of these (CYP1E1) has been isolated and partially characterised.93 When the enzyme was reconstituted in the presence of a soluble UDPG glucosyltransferase, oxime 97 was converted into dhurrin 99. The in vitro reconstitution of the entire dhurrin biosynthetic pathway from tyrosine was then achieved by the insertion of CYP79, CYP71E1 ≡ P450ox and NADPH-P450 oxidoreductase into lipid micelles in the presence of UDPG glucosyltransferase.
 | ||
| Scheme 15 | ||
The cDNA for three novel P450 enzymes have been heterologously cloned in Escherichia coli. One of the enzymes was CYP71E1. Reconstituted CYP71E1 and CYP79 were able to convert tyrosine into p-hydroxymandelonitrile 98.94 It is remarkable that these two enzymes carry out so much chemistry.
6 Other metabolites of the shikimate pathway
For alternative treatment, the reader may wish to consult the Reports in this area in Natural Product Reports currently written by A. R. Knaggs.95 A comprehensive and authoritative review on vancomycin includes a survey of the biosynthesis of this important antibiotic.966.1 Ansatrienin, ascomycin, asukamycin and manumycins
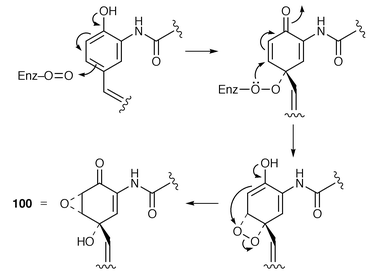
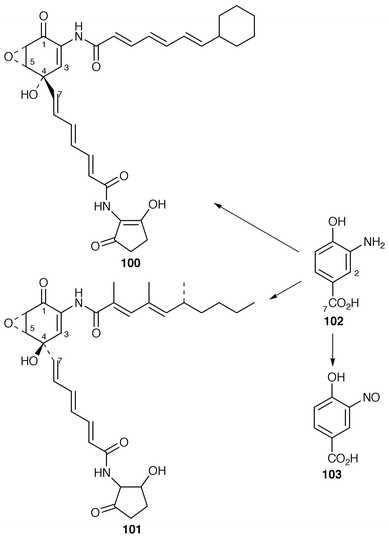
The cyclohexanecarboxylic acid and dihydroxycyclohexanecarboxylic acid moieties of ansatrienin (ref. 4, p. 456) and ascomycin (ref. 5, p. 52), respectively, are biosynthesised by deviation from the shikimate pathway. The biosynthesis of these moieties, including genetic aspects, has been reviewed.97The chemistry, biosynthesis and pharmacology of the manumycin group of antibiotics, e.g. asukamycin 100 and manumycin A 101, have been reviewed.98 These antibiotics are characterised by having two short polyketide chains linked through a mC7N moiety which serves as a starter unit for one of the pendant chains. These C7N moieties, unlike others (see below and ref. 7, p. 202), do not derive from the shikimate pathway but from a C4 dicarboxylic acid (derived from succinate; carbons 4–7) and a C3 triose phosphate (derived from glycerol; carbons 1–3) (cf. Section 8.3). The manner of biosynthesis is the same for the simpler metabolite 103. Additionally and importantly, 3-amino-4-hydroxybenzoic acid (3,4-AHBA) 102 is a specific precursor for 103 (ref. 7, p. 203). It has now been shown99 that samples of 3,4-AHBA 102, with a deuterium atom at C-2 and separately a 13C label at C-7, are efficient and specific precursors for asukamycin 100 in Streptomyces nodosus ssp. asukaensis. The mechanism by which the aromatic ring in 3,4-AHBA is transformed into that of 100 is suggested to be as shown in Scheme 16.
 | ||
| Scheme 16 | ||
[7-13C]-3,4-AHBA was also efficiently incorporated into manumycin A 101 in Streptomyces parvulus with specific labelling of C-7. The biosynthesis of 100 and 101 must be closely similar but there is a mechanistic problem associated with opposite configurations at C-4 (cf.Scheme 16).
6.2 Rifamycins and actamycin
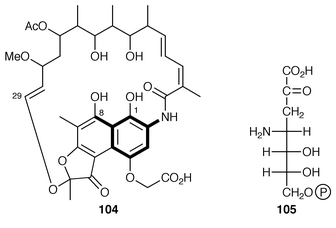
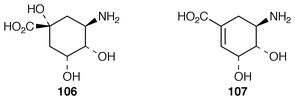
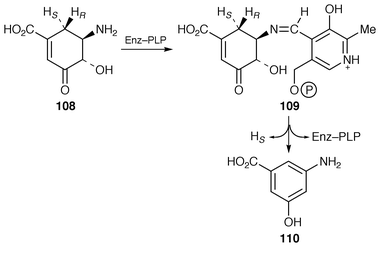
There is a mC7N unit different to the one just described that is present in ansamycin antibiotics exemplified by rifamycin B 104 (see thickened bonds). This unit derives from the shikimate pathway through amino-DAHP 105 (not DAHP) and 3-amino-5-hydroxybenzoic acid 110 (3,5-AHBA) (see ref. 7, p. 203, Scheme 9). (−)-3-Amino-3-deoxyquinic acid 106 has been synthesised and tested as an intermediate in rifamycin B biosynthesis.100 The relatively low incorporation (similar to that aminodeoxyshikimic acid 107) fails to support unequivocally its intermediacy. One possibility is that it is used in a salvage pathway.The last identified intermediate in 3,5-AHBA biosynthesis is 5-deoxy-5-amino-3-dehydroshikimic acid 108. The enzyme, 3-amino-5-hydroxybenzoic acid synthase, which catalyses the conversion of 108 into 110, has been purified to homogeneity from Amycolatopsis mediterranei. The encoding gene has been cloned, sequenced and over-expressed in E. coli. The recombinant enzyme is a dimer with one molecule of pyridoxal phosphate (PLP) per subunit. Mechanistic studies have shown that PLP forms a Schiff’s base 109 with the amino group of 108 and catalyses both an α,β-dehydration and a stereospecific 1,4-enolisation (loss of HS) of the substrate (Scheme 17).101
 | ||
| Scheme 17 | ||
A set of 34 genes associated with rifamycin biosynthesis have been found clustered around the gene for 3,5-AHBA synthase in A. mediterranei. It appears from the results obtained that rifamycin B production in this organism is governed by a single gene cluster consisting of structural, resistance and export, and regulatory genes.102 A Type I polyketide synthase gene cluster from A. mediterranei, which is implicated in rifamycin biosynthesis, has been cloned in E. coli and sequenced. The sequence has been analysed in terms of polyketide biosynthetic function.103,104
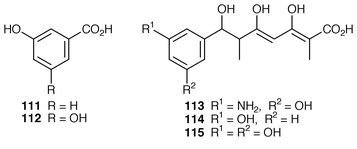
3,5-AHBA 110 is the primer for the elaboration in vivo of the polyketide handle in rifamycin B 104. A mutant of A. mediterranei which was blocked in the synthesis of 110 was found to be able de novo to make rifamycin B in its presence. With the analogues 111 and 112 as primers the stunted polyketides 114 and 115, respectively, were produced; these products are closely similar to a natural shunt compound 113. Clearly, the polyketide enzymes early in biosynthesis are more tolerant towards an unnatural primer than at least one later in the pathway.105
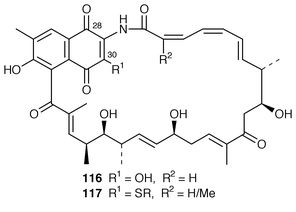
It has been shown106 that the C-1 hydroxy and C-29 vinyl ether groups in rifamycins B 104, O and S derive from molecular oxygen whilst the C-8 phenolic oxygen originates (as does C-8) in the carboxy of 110. The oxygen atoms at C-28 and C-30 in the mC7N moiety of actamycin 116 have recently been shown similarly to have their provenance in molecular oxygen in cultures of a Streptomyces species.107 This organism produces thio-substituted derivatives 117. Enhanced production of thio-derivative could be stimulated by the addition of more thiol or different thio-compounds.108
6.3 DIMBOA, phenazines and acridones
DIMBOA 118 is a pesticidal metabolite of maize. The starting point for its biosynthesis is indole (ref. 7, p. 202). It has now been demonstrated that an enzyme in maize, previously identified as a tryptophan synthase, is an indole synthase that is associated with DIMBOA biosynthesis.109 All four of the oxygen atoms in 118 have their provenance in molecular oxygen.110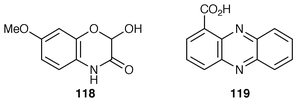
The structure and function of the operon that controls the biosynthesis of phenazine-1-carboxylic acid 119 in Pseudomonas sp. have been reported111 (for earlier work on phenazines see ref. 7, p. 203). Expression of the phenazine biosynthetic operon is controlled by N-hexanoylhomoserine lactone.112 A seven-gene locus has been identified for the synthesis of 119.113 An anthranilate synthase gene from Streptomyces venezuelae has been found most closely to resemble genes for phenazine biosynthesis in Pseudomonas.114
Information on the regulation and distribution of acridone synthase in Ruta graveolens has been published.115 For earlier work on acridone alkaloids see ref. 7, p. 202 and ref. 8.
6.4 Betalains
The gene encoding a type of aromatic ring-cleaving dioxygenase not previously described, namely the DOPA-dioxygenase involved in betalain biosynthesis,8 has been identified in Amanita muscaria and cloned.116 The dioxygenase catalyses the formation of two products with DOPA 120 as the substrate.117 One of the products is betalamic acid 121, the betalain precursor formed by 4,5-ring-cleavage. Muscflavin 122 which occurs naturally in A. muscaria and in mushrooms of the Hygrocybe family, but not in the betalain-containing plants of the order Caryophyllales, is the other product. It is formed by 2,3-cleavage. Thus the (recombinant) enzyme has two sorts of dioxygenase activity and this argues against formation of 121 and 122 by two distinct enzymes in vivo. The dioxygenase has been expressed in the petals of white Portulaca grandiflora plants.118 Transformation resulted in the formation of yellow and violet spots that contained betalain pigments and muscflavin. The formation of muscflavin in plants is unknown and it is clear, therefore, that there are differences in the structure and specificity of plant and A. muscaria dioxygenases.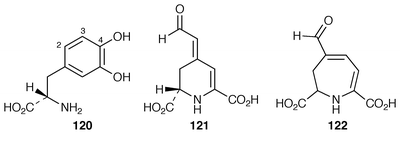
7 β-Lactams
The tale of the phenomenon, and understanding, of the stimulation of the production of cephalosporin C by methionine in Cephalosporium acremonium cultures has been told in detail.119 β-Lactams have been the subjects of reviews which include biosynthetic and genetic aspects.120–124 The chemistry and biosynthesis of clavulanic acid has been surveyed.125 The enzymes involved in the formation of the ACV tripeptide 126 from the three L-amino acid precursors and the oxidative transformation of 126 into isopenicillin N 127 have been the subject of ranging review.126 The applications of peptide synthetases in the synthesis of peptide analogues, which include penicillins, have been reviewed.127Lysine ε-aminotransferase, the first enzyme of cephalosporin biosynthesis in Actinomycetes, has been the subject of review.128 The transferase from Streptomyces clavuligerus has been purified partially, characterized and its nitrogen regulation studied.129
Regulation of penicillin biosynthesis has been studied.130 The energy requirement (3ATP molecules, as expected, under favourable circumstances) has been defined for the synthesis of the ACV tripeptide 126 from the adenylates of the L-amino acids, α-aminoadipate 123, cysteine 124 and valine in Acremonium chrysogenum.131 The sequence of assembly of the amino acids by the multifunctional ACV synthetase in this organism has been demonstrated to involve first the ATP-driven formation of the enzyme-bound dipetidyl-thioester δ-(L-α-aminoadipyl)-L-cysteine 125. Valine is deduced to be the third unit added (Scheme 18).132
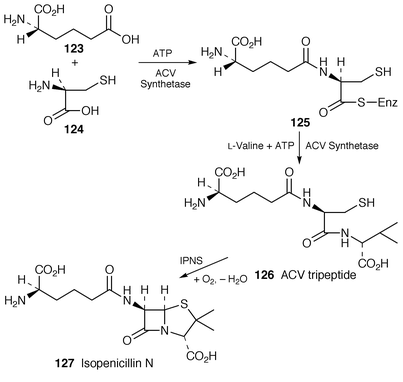 | ||
| Scheme 18 | ||
Δ1-Piperideine-6-carboxylate dehydrogenase that catalyses the conversion of δ1-piperideine-6-carboxylate into the penicillin/cephalosporin precursor α-aminoadipic acid has been isolated from Streptomyces clavuligerus, purified to homogeneity and charcterised;133S. clavuligerus produces the cephalosporin, cephamycin C. Further sequence analysis of the cephamycin gene cluster has identified new gene products relevant to antibiotic biosynthesis.134 A ccAR regulatory gene has been shown to be essential for the biosynthesis of cephamycin, clavulanic acid and non-clavulanic acid clavams.134 Its amplification in S. clavuligerus and the overproduction of cephamycin and clavulanic acid have been reported.135
7.1 Isopenicillin N synthase
A signal event in this reviewing period has been the publication136 of the X-ray crystal structure of isopenicillin N synthase (IPNS) complexed to Fe(II) and the substrate ACV. This follows upon the crystal structure of IPNS complexed to manganese which now provides a valuable reference structure for the synthase minus substrate. The new structure naturally allows refinement of the previous biosynthetic mechanism (ref. 6, p. 368). It is proposed136 that in this conversion ACV is ligated via its cysteinyl sulfur to the iron centre which creates a vacant iron coordination site into which dioxygen can bind. The crystal structure of the enzyme complex with the dioxygen analogue, NO, bound with ACV to the active site supports the manner of oxygen binding. Subsequent to dioxygen binding, iron–dioxygen and iron–oxo species remove the requisite hydrogens from ACV without direct assistance. The events associated with the marvellous transformation of ACV 126 into isopenicillin N 127 are summarized in Scheme 19. (The reader is wise to consult the paper for the intricate detail of binding and reaction.) Previous, notably spectroscopic, evidence is consistent with the proposed mechanism of binding and reaction.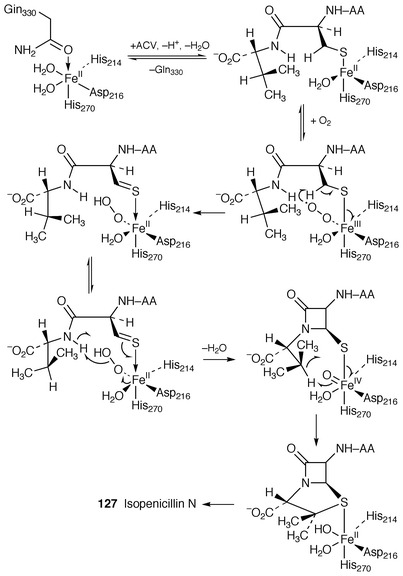 | ||
| Scheme 19 | ||
Current knowledge on oxygen-activating non-haeme iron enzymes, including IPNS, has been reviewed137 (see also ref. 136). Associated with the IPNS work are results relating to 1-aminocyclopropane-1-carboxylate oxidase which is a member of the non-haeme iron(II) dependent family of oxygenases and oxidases.138 The importance of various amino acid residues in IPNS have been examined: glutamine,139 serine,140 arginine,141 aspartate,142 and histidine.143 The cloning of the IPNS gene from Lysobacter lactamgenus has been reported.144
In on-going research, a number of analogues of ACV 126 have been synthesised and examined as substrates for IPNS.145
7.2 Clavulanic acid, tabtoxinine, nocardicins and related metabolites
The potent β-lactamase inhibitor clavulanic acid 129146 is biosynthesised in S. clavuligerus from pyruvate147 and arginine. These latter compounds serve as primary sources for the skeleton of 129 by way of clavaminic acid 128 which is a late biosynthetic intermediate (ref. 6, p. 366). It has now been demonstrated148 that [2,3-13C2]clavaminic acid, as 128, is an intact and efficient precursor for valclavam 130 and 2-(2-hydroxyethyl)clavam 131 in Streptomyces antibioticus. This result, plus those from an 18O2 experiment, has led to the conclusion that, after a long common pathway, 128 is the final intermediate shared by clavulanic acid 129 and the antipodal clavams exemplified by 130 and 131.148Clavaminate synthase is one member of a large family of non-haem iron enzymes that require α-ketoglutarate as co-substrate. The synthase, with its co-substrates 132 and α-ketoglutarate, has been subject to spectroscopic scrutiny.149 The coupled mechanism of substrate 132 hydroxylation (to give 133) and α-ketoglutarate decarboxylation has been further examined and it has been concluded150 that iron at the active site is converted into a five-coordinate species which can react with dioxygen to form an active oxygen intermediate. Subsequent to hydroxylation, the synthase catalyses the cyclisation of 134 to afford clavaminic acid 128 (ref. 6, p. 367, Scheme 12).
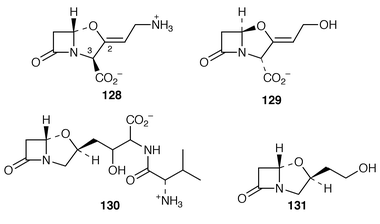
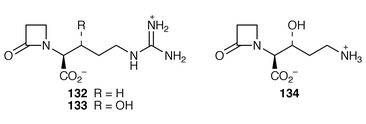
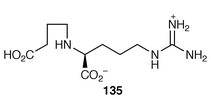
An open reading frame within the clavulanate biosynthetic cluster has been noted, which, with ensuing biomolecular experimentation, has led two groups of workers151,152 to the identification and characterisation of an enzyme, β-lactam synthetase, which, in the presence of Mg2+/ATP, catalyses a missing, key step in clavulanate biosynthesis, namely the conversion of 135 into the β-lactam 132. The approaches and experimental evidence of the two groups is satisfyingly complementary.
Further genetic work on the regulation of clavulanate biosynthesis has been reported.153 Clavulanic acid production is enhanced by redox-cycling agents.154
Gene expression products associated with tabtoxinine and 1-carbpen-2-em-3-carboxylic acid biosynthesis have been reported,155 as has work on putative transferases associated with nocardicin biosynthesis.156
8 Miscellaneous metabolites
The biosynthetic pathway to thiopeptide antibiotics (ref. 7, p. 205) has been authoritatively reviewed.1578.1 Taxol
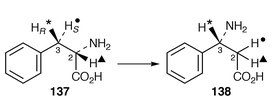
The potent antitumour properties of Taxol 136 (paclitaxel) and its scarcity continue to fuel the search for increased production of this secondary metabolite of the Pacific Yew (Taxus brevifolia). The biosynthesis158 and production of 136 in cell cultures159 has been reviewed (cf. ref. 7, p. 206; ref. 5, p. 52). The biosynthesis of the terpene framework of 136 is beyond the remit of this Report, but published work on the multiple oxygenase reactions which occur during the course of the biosynthesis of the terpene part of 136 is to be noted.160 The origin of the phenylisoserine side chain in Taxol 136 is in phenylalanine 137via
β-phenylalanine 138 (cinnamic acid is not involved) (ref. 5, p. 52; ref. 4, p. 455). It follows that an aminomutase is involved. Appropriate activity has been detected161 in cell-free extracts of T. brevifolia. The enzymic product 138 of the mutase reaction has the same absolute stereochemistry as that seen in 136. Experiments with deuteriated samples of phenylalanine gave results which show that nitrogen migration occurs with retention of configurations at C-3 in 138. The C-3 pro-R proton and the one at C-2 are retained, whereas the 3-pro-S proton migrates, and with at least some intermolecularity. The results point up several unprecedented features of this unique aminomutase from a higher plant and further results are awaited with interest.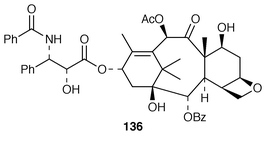
8.2 Kinamycins
The biosynthesis of the kinamycins (ref. 6, p. 370; ref. 7, p. 204) has been authoritatively reviewed.162 The genes of most of the biosynthetic steps have been cloned and expressed heterologously162 and gene expression products identified. Stealthin C 139 has been identified as an intermediate in the biosynthesis of kinamycin D 140.163,164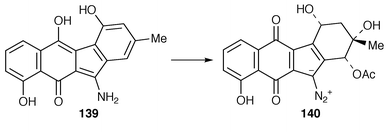
8.3 Amino sugars, blasticidin S and caffeine
The left-most ring in the glucosidase inhibitor, acarbose 141 is a C7N unit that is derived, not from the shikimate, but the pentose-phosphate, pathway165 (cf. Sections 6.1 and 6.2). The three other rings derive from maltose/maltotriose.166 Glutamate is the primary nitrogen source.167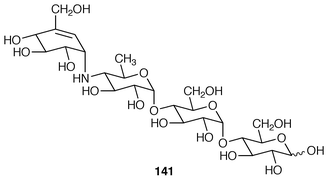
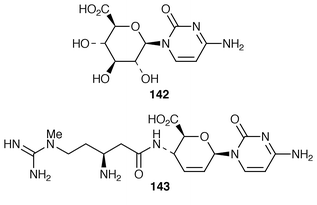
Gene fragments encoding the biosynthesis of blasticidin S 143 (ref. 5, p. 56) in Streptomyces griseochromogenes have been cloned and heterologously expressed.168 Gene expression products included 142 and other metabolites related to 143.
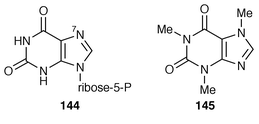
The biosynthesis and catabolism of caffeine has been reviewed.169 A sequence of N-methylations characterises the biosynthesis of caffeine 145, which was recently deduced to start with the N7-methylation of xanthosine phosphate 144 rather than xanthosine itself (ref. 7, p. 206). A xanthosine-N7-methyltransferase from Coffea arabica has been partially sequenced and this sequence has been used in a search for matching nucleic acid sequence in a C. arabica cDNA library. Matching cDNA was characterised.170 Results on the purification of the labile N-methyltransferases in C. arabica have been published.171
8.4 Coronatine
Biosynthesis of the phytotoxin coronatine 146 in Pseudomonas syringae pv. glycinea is thermoregulated at the transcriptional level.172 It has been deduced that the transcription of coronofacate ligase, putatively associated with formation of the amide bond linking the two halves of coronatine, is temperature-sensitive.173 A thioesterase involved in the biosynthesis of coronamic acid 147 has been overexpressed.174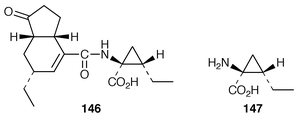
References
- R. B. Herbert, Nat. Prod. Rep., 1992, 9, 507 RSC.
- R. B. Herbert, Nat. Prod., 1993, 10, 575 Search PubMed.
- R. B. Herbert, Nat. Prod. Rep, 1995, 12, 55 RSC.
- R. B. Herbert, Nat. Prod. Rep., 1995, 12, 445 RSC.
- R. B. Herbert, Nat. Prod. Rep., 1996, 13, 45 RSC.
- R. B. Herbert, Nat. Prod. Rep., 1997, 14, 359 RSC.
- R. B. Herbert, Nat. Prod. Rep., 1999, 16, 199 RSC.
- R. B. Herbert, The Biosynthesis of Secondary Metabolites, 2nd edn., Chapman and Hall, London, 1989. Search PubMed.
- R. A. Creelman and J. E. Mullet, Annu. Rev. Plant Physiol. Plant Mol. Biol., 1997, 48, 355 Search PubMed.
- C. Wasternack, O. Miersch, R. Kramell, B. Hause, J. Ward, M. Beale, W. Boland, B. Parthier and I. Feussner, Fett/Lipid, 1998, 100, 139 Search PubMed.
- M. J. Mueller, Physiol. Plant, 1997, 100, 653 CrossRef CAS.
- H. G. Floss, Nat. Prod. Rep., 1997, 14, 433 RSC.
- M. F. Roberts, in Alkaloids: Biochemistry, Ecology and Medicinal Applications, eds. M. F. Roberts and M. Wink, Plenum, New York, 1988, p. 109. Search PubMed.
- W.-M. Chou and T. M. Kutchan, Plant J., 1998, 15, 289 CrossRef CAS.
- J. V. Shanks and R. Bhadra, in Hairy Roots, ed. P. M. Doran, Harwood, Amsterdam, 1997, p. 51; Search PubMed; D. Hallard, A. Geerlings, R. van der Heijden, M. Ines Lopez Cardoso, J. H. C. Hoge and R. Verpoorte, in Hairy Roots, ed. P. M. Doran, Harwood, Amsterdam, 1997, p. 43. Search PubMed.
- D. O’Hagan and R. J. Robins, Chem. Soc. Rev., 1998, 27, 207 RSC.
- R. J. Robins, in Alkaloids: Biochemistry, Ecology and Medicinal Applications, eds. M. F. Roberts and M. Wink, Plenum, New York, 1988, p. 199. Search PubMed.
- R. J. Robins, T. W. Abraham, A. J. Parr, J. Eagles and N. J. Walton, J. Am. Chem. Soc., 1997, 119, 10929 CrossRef CAS.
- C. H. Chang, M. D. Ballinger, G. H. Reed and P. A. Frey, Biochemistry, 1996, 35, 11081 CrossRef CAS and references cited therein; P. A. Frey, G. H. Reed, M. L. Moss, R. M. Petrovitch, M. D. Ballinger, K. W. Lieder, W. Wu, C. H. Chang, V. Bandarian, F. J. Ruzicka, R. LoBrutto and H. Beinert, in Vitamin B12 and Vitamin B12-Proteins, ed. B. Kräutler, D. Arigoni and B. T. Golding, Wiley-VCH, Weinheim, Chichester, 1998, p. 435. Search PubMed.
- P. Baldet, C. Alban and R. Douce, FEBS Lett., 1997, 419, 206 CrossRef CAS; N. M. Shaw, O. M. Birch, A. Tinschert, V. Venetz, R. Dietrich and L.-A. Savoy, Biochem. J., 1998, 330, 1079 CAS and references cited therein..
- S. Ollagnier, E. Kervio and J. Rétey, FEBS Lett., 1998, 437, 309 CrossRef CAS.
- C. W. Wong, J. T. G. Hamilton, D. O’Hagan and R. J. Robins, Chem. Commun., 1998, 1045 RSC.
- I. Zabetakis, R. Edwards, J. Hamilton and D. O’Hagan, Plant Cell Rep., 1998, 18, 341 CrossRef CAS.
- I. Zabetakis, R. Edwards and D. O’Hagan, Phytochemistry, 1998, 50, 53 CrossRef CAS.
- M. Botte, F. Mabon, M. Le Mouillour and R. J. Robins, Phytochemistry, 1997, 46, 117 CrossRef CAS.
- Z.-P. Zhang, T. Krumm and I. T. Baldwin, J. Chem. Ecol., 1997, 23, 2777 Search PubMed; S. Imanishi, K. Hashizume, M. Nakakita, H. Kojima, Y. Matsubayashi, T. Hashimoto, Y. Sakagami, Y. Yamada and K. Nakamura, Plant Mol. Biol., 1998, 38, 1101 CrossRef CAS.
- P. Duspara, S. I. Jenkins, D. W. Hughes and P. H. M. Harrison, Chem. Commun., 1998, 2643 RSC; P. H. M. Harrison, D. W. Hughes and R. W. Riddoch, Chem. Commun., 1998, 273 RSC.
- D. J. Robins, Chem. Soc. Rev., 1989, 18, 375 RSC; D. J. Robins in The Alkaloids, Academic Press, NY, 1995, vol. 46, p. 1. Search PubMed.
- I. R. Stirling, I. K. A. Freer and D. J. Robins, J. Chem. Soc., Perkin Trans. 1, 1997, 677 RSC.
- G. Graser and T. Hartmann, Phytochemistry, 1997, 45, 1591 CrossRef CAS.
- T. Hartmann and B. Dietrich, Planta, 1998, 208, 443 CrossRef.
- S. Sagner, Z.-W. Shen and M. H. Zenk, Tetrahedron Lett., 1997, 38, 2443 CrossRef CAS; S. Sagner, Z.-W. Shen, B. Deus-Neumann and M. H. Zenk, Phytochemistry, 1998, 47, 375 CrossRef CAS.
- G. Bringmann, M. Rueckert, M. Wenzel, C. Guenther, K. Wolf, J. Holenz and J. Schlauer, Pharm. Pharmacol. Lett., 1998, 8, 5 Search PubMed.
- R. Wilhelm and M. H. Zenk, Phytochemistry, 1997, 46, 701 CrossRef CAS.
- R. B. Herbert, S. Pos and H. Venter, Nat. Prod. Rep., 2000, 17, 317 RSC.
- P. J. Facchini and D. A. Bird, In Vitro Cell Dev. Biol.: Plant, 1998, 34, 69 Search PubMed.
- P. J. Facchini, Phytochemistry, 1998, 42, 481 CrossRef.
- P. J. Facchini, C. Penzes-Yost, N. Samanami and B. Kowalchuk, Plant Physiol., 1998, 118, 69 CrossRef CAS.
- H. H. Pauli and T. M. Kutchan, Plant J., 1998, 13, 793 CrossRef CAS.
- K. Hauschild, H. H. Pauli and T. M. Kutchan, Plant Mol. Biol., 1998, 36, 473 CrossRef CAS.
- W. Roos, S. Evers, M. Hieke, M. Tschope and B. Schumann, Plant Physiol., 1998, 118, 349 CrossRef CAS.
- G. Haider, T. Kislinger and T. M. Kutchan, Biochem. Biophys. Res. Commun., 1997, 24, 606 CrossRef.
- G. B. Mahady, C. Liu and C. W. W. Beecher, Phytochemistry, 1998, 48, 93 CrossRef CAS.
- Y. Sugimoto, S. Uchida, S. Inanga and A. Isogai, J. Plant Physiol., 1997, 150, 376 Search PubMed.
- U. H. Maier and M. H. Zenk, Chem. Commun., 1997, 2313 RSC.
- N. Nagakura, G. Höfle, D. Coggiola and M. H. Zenk, Planta Med., 1978, 34, 381 CAS.
- W. De-Eknamkul, A. Ounaroon, T. Tanahashi, T. M. Kutchan and M. H. Zenk, Phytochemistry, 1997, 45, 477 CrossRef CAS.
- R. Robinson, The Structural Relations of Natural Products, Oxford University Press, Oxford, 1955. Search PubMed.
- A. R. Battersby, R. B. Herbert, E. McDonald, R. Ramage and J. H. Clements, J. Chem. Soc., Perkin Trans. 1, 1972, 1741 RSC.
- R. B. Herbert, A. E. Kattah and E. Knagg, Tetrahedron, 1990, 46, 7119 CrossRef CAS.
- A. Nasreen, H. Gundlach and M. H. Zenk, Phytochemistry, 1997, 46, 107 CrossRef CAS.
- E. McDonald, R. Ramage, R. N. Woodhouse, E. W. Underhill, L. R. Wetter and A. R. Battersby, J. Chem. Soc., Perkin Trans. 1, 1998, 2979 RSC.
- U. H. Maier and M. H. Zenk, Tetrahedron Lett., 1997, 38, 7357 CrossRef CAS.
- A. C. Barker, D. R. Julian, R. Ramage, R. N. Woodhouse, G. Hardy, E. McDonald and A. R. Battersby, J. Chem. Soc., Perkin Trans. 1, 1998, 2989 RSC.
- R. N. Woodhouse, E. McDonald, R. Ramage and A. R. Battersby, J. Chem. Soc., Perkin Trans. 1, 1998, 2995 RSC.
- P. W. Sheldrake, K. E. Suckling, R. N. Woodhouse, A. J. Murtagh, R. B. Herbert, A. C. Barker, J. Staunton and A. R. Battersby, J. Chem. Soc., Perkin Trans. 1, 1998, 3003 RSC.
- M. Rueffer and M. H. Zenk, FEBS Lett., 1998, 438, 111 CrossRef CAS.
- J. Eichhorn, T. Takada, Y. Kita and M. H. Zenk, Phytochemistry, 1998, 49, 1037 CrossRef CAS.
- R. Verpoorte, R. Van der Heijden and P. R. H. Moreno, in The Alkaloids, ed. G. A. Cordell, Academic Press, NY, 1997, vol. 49, p. 221. Search PubMed.
- V. De Luca, B. St-Pierre, F. V. Vazquez and P. Laflamme, NATO ASI Ser., Ser. H, 1998, 104, 171 Search PubMed.
- D. Hallard, R. Van der Heijden, A. Contin, E. M. T. Jiminez, W. Snoeijer, R. Verpoorte, S. R. Jenson, M. I. L. Cardoso, G. Pasquali, J. Memelink and J. H. C. Hoge, Phytochem. Anal., 1998, 9, 162 CrossRef CAS.
- R. Verpoorte, Acta Hortic., 1998, 457, 403 Search PubMed.
- C. Canel, M. I. Lopes-Cardoso, S. Whitmer, L. Van der Fits, G. Pasquale, R. Van der Heijden, J. H. C. Hoge and R. Verpoorte, Planta, 1998, 205, 414 CrossRef CAS.
- M.-P. Bonzom, C. Andary, A. Gueiffier, J.-L. Roussel and A. Gargadennec, Phytochemistry, 1997, 46, 235 CrossRef CAS.
- M. I. Lopes-Cardoso, A. H. Meijer, S. Rueb, J. Q. Machado, J. Memelink and J. H. C. Hoge, Mol. Gen. Genet., 1997, 256, 674 Search PubMed.
- M. J. Leech, K. May, D. Hallard, R. Verpoorte, V. De Luca and P. Christou, Plant Mol. Biol., 1998, 38, 765 CrossRef CAS.
- S. Whitmer, C. Canel, D. Hallard, C. Goncalves and R. Verpoorte, Plant Physiol., 1998, 116, 853 CrossRef CAS.
- S. Rijhwani and J. V. Shanks, Biotechnol. Prog., 1998, 14, 442 CrossRef CAS.
- F. A. Vazquez-Flota and V. De Luca, Phytochemistry, 1998, 49, 395 CrossRef CAS.
- S. Whitmer, R. Verpoorte and C. Canel, Plant Cell, Tissue Organ Cult., 1998, 53, 135 Search PubMed.
- B. St-Pierre, P. Laflamme, A.-M. Alarco and V. De Luca, Plant. J., 1998, 14, 1703 CrossRef CAS.
- F. Vazquez-Flota, E. De Carolis, A.-M. Alarco and V. De Luca, Plant Mol. Biol., 1997, 34, 935 CrossRef CAS.
- F. A. Vazquez-Flota and V. De Luca, Plant Physiol., 1998, 117, 1351 CrossRef CAS.
- M. Lopez-Meyer and C. L. Nessler, Plant J., 1997, 11, 1167 CrossRef CAS.
- S.-W. Yang and G. A. Cordell, J. Nat. Prod., 1997, 60, 788 CrossRef CAS.
- S.-W. Yang and G. A. Cordell, J. Nat. Prod., 1997, 60, 230 CrossRef CAS.
- S. Weidner, M. Kittelmann, K. Goeke, O. Ghisalba and H. Zahner, J. Antibiot., 1998, 51, 679 Search PubMed.
- T. Hoshino and M. Yamamoto, Biosci. Biotech. Biochem., 1997, 61, 2134 Search PubMed.
- A. Z. M. R. Momen, T. Mizuoka and T. Hoshino, J. Chem. Soc., Perkin Trans. 1, 1998, 3087 RSC.
- E. M. Stocking, J. F. Sanz-Cervera, R. M. Williams and C. J. Unkefer, J. Am. Chem. Soc., 1997, 119, 9588 CrossRef CAS.
- M. Zook, Plant Physiol., 1998, 118, 1389 CrossRef CAS.
- M. Rohmer, Nat. Prod. Rep., 1999, 16, 565 RSC.
- H. Seto, H. Watanabe and K. Furihata, Tetrahedron Lett., 1996, 37, 7979 CrossRef CAS.
- M. Kaneda, K. Kitahara, K. Yamasaki and S. Nakamura, J. Antibiot., 1990, 43, 1623 Search PubMed.
- N. Orihara, K. Furihata and H. Seto, J. Antibiot., 1997, 50, 979 Search PubMed.
- H. S. Vageeshbabu and V. L. Chopra, J. Plant Biochem. Biotechnol., 1997, 6, 53 Search PubMed.
- R. Verkerk, M. Dekker and W. M. F. Jongen, Natural Toxicants in Food, ed. D. Watson, Sheffield Academic Press, Sheffield, 1998, p. 29. Search PubMed.
- R. F. Mithen, H. Quiroz and H. Campos-de, Acta Hortic., 1998, 459, 373 Search PubMed.
- J. E. Rodman, P. S. Soltis, D. E. Soltis, K. J. Sytsma and K. G. Karol, Am. J. Bot., 1998, 85, 997 Search PubMed.
- R. N. Bennett, G. Kiddle and R. M. Wallsgrove, Plant Physiol., 1997, 114, 1283 CAS.
- S. Bak, H. L. Nielsen and B. A. Halkier, Plant Mol. Biol., 1998, 38, 725 CrossRef CAS.
- L. Du and B. A. Halkier, Phytochemistry, 1998, 48, 1145 CrossRef CAS.
- R. A. Kahn, S. Bak, I. Svendsen, B. A. Halkier and B. L. Møller, Plant Physiol., 1997, 115, 1661 CrossRef CAS.
- S. Bak, R. A. Kahn, H. L. Nielsen, B. L. Møller and B. A. Halkier, Plant Mol. Biol., 1998, 36, 393 CrossRef CAS.
- A. R. Knaggs, Nat. Prod. Rep., 2000, 17, 269 RSC.
- D. H. Williams and B. Bardsley, Angew. Chem., Int. Ed., 1999, 38, 1173 CAS.
- D. J. Wilson, S. Patton, G. Florova, V. Hale and K. A. Reynolds, J. Ind. Microbiol. Biotechnol., 1998, 20, 299 CrossRef.
- I. Sattler, R. Thiericke and A. Zeeck, Nat. Prod. Rep., 1998, 15, 221 RSC.
- Y. Hu, C. R. Melville, S. J. Gould and H. G. Floss, J. Am. Chem. Soc., 1997, 119, 4301 CrossRef CAS.
- M. Müller, R. Müller, T.-W. Yu and H. G. Floss, J. Org. Chem., 1998, 63, 9753 CrossRef.
- C.-G. Kim, T.-W. Yu, C. B. Fryhle, S. Handa and H. G. Floss, J. Biol. Chem., 1998, 273, 6030 CrossRef CAS.
- P. R. August, L. Tan, Y. J. Yoon, S. Ning, R. Müller, T.-W. Yu, M. Taylor, D. Hoffmann, C.-G. Kim, X. Zhang, C. R. Hutchinson and H. G. Floss, Chem. Biol., 1998, 5, 69 CrossRef CAS.
- T. Schupp, C. Toupet, N. Engel and S. Goff, FEMS Microbiol. Lett., 1998, 159, 201 CrossRef CAS.
- L. Tan, Y. J. Yoon, C.-Y. Choi and C. R. Hutchinson, Gene, 1998, 216, 255 CrossRef.
- D. Hunziker, T.-W. Yu, C. R. Hutchinson, H. G. Floss and C. Khosla, J. Am. Chem. Soc., 1998, 120, 1092 CrossRef CAS.
- M. G. Anderson, D. Monypenny, R. W. Rickards and J. M. Rothschild, J. Chem. Soc., Chem. Commun., 1989, 311 RSC.
- A. M. Hooper and R. W. Rickards, J. Antibiot., 1998, 51, 958 Search PubMed.
- A. M. Hooper and R. W. Rickards, J. Antibiot., 1998, 51, 845 Search PubMed.
- D. Melanson, M.-D. Chiltern, D. Masters-Moore and W. S. Chilton, Proc. Natl. Acad. Sci. USA, 1997, 94, 13345 CrossRef CAS.
- E. Glawischnig, W. Eisenreich, A. Bacher, M. Frey and A. Gierl, Phytochemistry, 1997, 45, 715 CrossRef CAS.
- D. V. Mavrodi, B. M. Chatuev, K. N. Ksenzenko, L. S. Tomashov and A. M. Boronin, Dokl. Akad. Nauk, 1997, 352, 117 Search PubMed; D. V. Mavrodi, V. N. Ksenzenko, B. M. Chatuev, L. S. Thomashov and A. M. Boronin, Mol. Biol. (Engl. Transl.), 1997, 31, 62 Search PubMed.
- D. W. Wood, F. Gong, M. M. Daykin, P. Williams and L. S. Pierson III, J. Bacteriol, 1997, 179, 7663 CAS.
- D. Mavrodi, V. N. Ksenzenko, R. F. Bonsall, R. J. Cook, A. M. Boronin and L. S. Thomashov, J. Bacteriol, 1998, 180, 2541 CAS.
- C. Lin, S. Ashish and L. C. Vining, Microbiology, 1998, 144, 1971 Search PubMed.
- K. T. Junghanns, R. E. Kneusel, D. Gröger and U. Matern, Phytochemistry, 1998, 49, 403 CrossRef CAS.
- U. G. Hinz, J. Fivaz, P.-A. Girod and J. P. Zryd, Mol. Gen. Genet., 1997, 256, 1 Search PubMed.
- L. A. Mueller, A. Hinz and J.-P. Zryd, Phytochemistry, 1997, 44, 567 CrossRef CAS.
- L. A. Mueller, U. Hinz, M. Uzé, C. Sautter and J.-P. Zryd, Planta, 1997, 203, 260 CrossRef CAS.
- A. L. Demain and J. Zhang, Crit. Rev. Biotechnol., 1998, 18, 283 Search PubMed.
- P. L. Skatrud, T. Schwecke, H. Van Liempt, M. B. Tobin, in Biotechnology, 2nd edn., eds. H. Kleinkauf and H. Von Doehren, VCH, Weinheim, Germany, 1997. Search PubMed.
- A. S. Paradkar, S. E. Jensen and R. H. Mosher, Drugs Pharm. Sci., 1997, 82, 241 Search PubMed.
- A. A. Brakhage, Microbiol. Mol. Biol. Rev., 1998, 62, 547 Search PubMed.
- J. E. Thirkettle, C. J. Schofield and M. W. Walter, Amino Acids, Pept., Proteins, 1997, 28, 281 Search PubMed.
- A. A. Brakhage, FEMS Microbiol. Lett., 1997, 148, 1 CrossRef CAS.
- K. H. Baggaley, A. G. Brown and C. J. Schofield, Nat. Prod. Rep., 1997, 14, 309 RSC.
- C. J. Schofield, J. E. Baldwin, M. F. Byford, I. Clifton, J. Hajdu, C. Hensgens and P. Roach, Curr. Opin. Struct. Biol., 1997, 7, 857 CrossRef CAS.
- H. Kleinkauf and H. von Döhren, Acta Biochim. Pol., 1997, 44, 839 Search PubMed.
- N. Rius and A. L. Demain, J. Microbiol. Biotechnol., 1997, 7, 95 Search PubMed.
- J. Romero , J. F. Martín, P. Liras, A. L. Demain and N. Rius, J. Ind. Microbiol. Biotechnol., 1997, 18, 241 CrossRef CAS.
- O. Litzka, P. Papagiannopolous, M. A. Davis, M. J. Hynes and A. A. Brakhage, Eur. J. Biochem., 1998, 251, 758 CrossRef CAS; K. T. Bergh and A. A. Brakhage, Appl. Environ. Microbiol., 1998, 64, 843 CAS.
- W. Kallow, H. von Döhren and H. Kleinkauf, Biochemistry, 1998, 37, 5947 CrossRef CAS.
- W. Kallow, T. Neuhof, B. Arezi, P. Jungblut and H. von Döhren, FEBS Lett., 1997, 414, 74 CrossRef CAS.
- J. L. de la Fuente, A. Rumbero, J. F. Martín and P. Liras, Biochem. J., 1997, 327, 59 CAS; F. J. Perez-Llarena, A. Rodriguez-Garcia, F. J. Enguita, J. F. Martín and P. Liras, J. Bacteriol., 1998, 180, 4753 CAS.
- D. C. Alexander and S. E. Jensen, J. Bacteriol., 1998, 180, 4068 CAS.
- F. J. Perez-Llarena, P. Liras, A. Rodriguez-Garcia and J. F. Martín, J. Bacteriol., 1997, 179, 2053 CAS.
- P. L. Roach, I. J. Clifton, C. M. H. Hensgens, N. Shibata, C. J. Schofield, J. Hajdu and J. E. Baldwin, Nature, 1997, 387, 827 CrossRef CAS.
- S. Lange and L. Que, Curr. Opin. Chem. Biol., 1998, 2, 159 CrossRef CAS.
- Z. Zhang, J. N. Barlow, J. E. Baldwin and C. J. Schofield, Biochemistry, 1997, 36, 15999 CrossRef CAS.
- P. Loke and T.-S. Sim, Biochem. Biophys. Res. Commun., 1998, 252, 472 CrossRef CAS; P. Loke and T.-S. Sim, ibid., 1998, 248, 559 Search PubMed; O. Landman, I. Bororok, Y. Aharonowitz and G. Cohen, FEBS Lett., 1997, 405, 172 CrossRef CAS; M. Sami, T. J. N. Toby, P. L. Roach, C. J. Schofield and J. E. Baldwin, FEBS Lett., 1997, 405, 191 CrossRef CAS.
- P. Loke and T.-S. Sim, FEMS Microbiol. Lett., 1998, 165, 353 CrossRef CAS.
- P. Loke and T.-S. Sim, FEMS Microbiol. Lett., 1998, 164, 107 CrossRef CAS.
- P. Loke, J. Sim and T.-S. Sim, FEMS Microbiol. Lett., 1997, 157, 137 CrossRef CAS.
- D. S. H. Tan, S. G. Ang and T.-S. Sim, Biochem. Mol. Biol. Int., 1998, 44, 333 Search PubMed.
- J. K. Ryu and D. H. Nam, J. Microbiol. Biotechnol., 1997, 7, 373 Search PubMed.
- C. J. Rowe, C. P. Shorrock, T. D. W. Claridge and J. D. Sutherland, Chem. Biol., 1998, 5, 229 CrossRef CAS; S. Petursson and J. E. Baldwin, Tetrahedron, 1998, 54, 6001 CrossRef CAS; F. Cavalier, A. T. Russell, C. J. Schofield and J. E. Baldwin, J. Chem. Res. Synop, 1997, 200 RSC; J. E. Baldwin, R. M. Adlington, N. P. Crouch and I. A. Pereira, J. Labelled Compd. Radiopharm., 1998, 41, 1145 CrossRef CAS.
- K. H. Baggaley, A. Brown and C. J. Schofield, Nat. Prod. Rep., 1997, 14, 309 RSC.
- J. E. Thirkettle, J. E. Baldwin, J. Edwards, J. P. Griffin and C. J. Schofield, Chem. Commun., 1997, 1025 RSC.
- L. A. Egan, R. W. Busby, D. Iwata-Reuyl and C. A. Townsend, J. Am. Chem. Soc., 1997, 119, 2348 CrossRef CAS.
- E. G. Pavel, J. Zhou, R. W. Busby, M. Gunsior, C. A. Townsend and E. I. Solomon, J. Am. Chem. Soc., 1998, 120, 743 CrossRef CAS.
- J. Zhou, M. Gunsior, B. O. Bachmann, C. A. Townsend and E. I. Solomon, J. Am. Chem. Soc., 1998, 120, 13539 CrossRef CAS.
- H. J. McNaughton, J. E. Thirkettle, Z. Zhang, C. J. Schofield, S. E. Jensen, B. Barton and P. Greaves, Chem. Commun., 1998, 2325 RSC.
- B. O. Bachmann, R. Li and C. A. Townsend, Proc. Natl. Acad. Sci. USA, 1998, 95, 9082 CrossRef CAS.
- A. S. Paradkar, K. A. Aidoo and S. E. Jensen, Mol. Microbiol., 1998, 27, 831 CrossRef CAS; R. Perez-Redondo, A. Rodriguez-Garcia, J. Martín and P. Liras, Gene, 1998, 211, 311 CrossRef CAS.
- H. J. Kwon and S.-U. Kim, Appl. Microbiol. Biotechnol., 1998, 49, 77 CrossRef CAS.
- L. Liu and P. D. Shaw, J. Bacteriol., 1997, 179, 507 CAS; L. Liu and P. D. Shaw, J. Bacteriol., 1997, 179, 5922 CAS; S. J. McGowan, M. Sebaihia, S. O’Leary, K. R. Hardie, P. Williams, G. S. A. B. Stewart, B. W. Bycroft and G. P. C. Salmond, Mol. Microbiol., 1997, 26, 545 CrossRef CAS.
- A. M. Reeve, S. D. Breazeale and C. A. Townsend, J. Biol. Chem., 1998, 273, 30695 CrossRef CAS; A. M. Reeve and C. A. Townsend, Tetrahedron, 1998, 54, 15959 CrossRef CAS.
- T. M. Smith, Y.-F. Jiang and H. G. Floss, Drugs Pharm. Sci., 1997, 82, 393 Search PubMed.
- J. Rohr, Angew. Chem., Int. Ed. Engl., 1997, 36, 2190 CrossRef CAS; M. Hezari and R. Croteau, Planta Med., 1997, 63, 291 CAS; H. Sohn and M. R. Okos, J. Microbiol. Biotechnol., 1998, 8, 427 Search PubMed.
- R. E. B. Ketchum and R. B. Croteau, Int. Congr. Ser., 1998, 1157, 339 Search PubMed; V. Ciddi and C. Kokarte, Indian Drugs, 1997, 34, 354 Search PubMed.
- W. Eisenreich, B. Meinhard, M. S. Lee, M. H. Zenk and A. Bacher, J. Am. Chem. Soc., 1998, 120, 9694 CrossRef CAS.
- K. D. Walker and H. G. Floss, J. Am. Chem. Soc., 1998, 120, 5333 CrossRef CAS.
- S. J. Gould, Chem. Rev., 1997, 97, 2499 CrossRef CAS.
- S. J. Gould, S.-T. Hong and J. R. Carney, J. Antibiot., 1998, 51, 50 Search PubMed.
- S. J. Gould, C. R. Melville, M. C. Cone, J. Chen and J. R. Carney, J. Org. Chem., 1997, 62, 320 CrossRef CAS.
- U. Degwert, R. Van Hülst, H. Pape, R. E. Herrold, J. M. Beale, P. J. Keller, J. P. Lee and H. G. Floss, J. Antibiot., 1987, 40, 855 Search PubMed.
- S. Lee, B. Sauerbrei, J. Niggemann and E. Egelkrout, J. Antibiot., 1997, 50, 954 Search PubMed.
- S. Lee and E. Egelkraut, J. Antibiot., 1998, 51, 225 Search PubMed.
- M. C. Cone, A. K. Petrich, S. J. Gould and T. M. Zabriskie, J. Antibiot., 1998, 51, 570 Search PubMed.
- A. Crozier, T. W. Baumann, H. Ashihara, T. Suzuki and G. R. Waller, Colloq. Sci. Int. Cafe, 1997, 17th, 106 Search PubMed.
- S. Moisyadi, K. R. Neupane and J. I. Stiles, Acta Hortic., 1998, 461, 367 Search PubMed.
- S. S. M. Waldhauser, F. M. Gillies, A. Crozier and T. W. Baumann, Phytochemistry, 1997, 45, 1407 CrossRef CAS.
- M. Ullrich, A. Penaloza-Vazquez, A. M. Bailey and C. L. Bender, Dev. Plant Pathol., 1997, 9, 230 Search PubMed; D. A. Palmer, C. L. Bender and S. B. Shashi, Can. J. Microbiol., 1997, 43, 517 CAS; B. H. Rhode, B. Pohlack and M. S. Ullrich, J. Basic Microbiol., 1998, 38, 41 CrossRef CAS.
- V. Rangaswamy, M. Jones, W. Jones, R. Mitchell, R. J. Parry, P. Reynolds and C. L. Bender, FEMS Microbiol. Lett., 1997, 154, 65 CrossRef CAS.
- J. Patel, J. C. Hoyt and R. J. Parry, Tetrahedron, 1998, 54, 15927 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2001 |