Fluorine-containing α-alkynyl amino esters and access to a new family of 3,4-dehydroproline analogues†
David Sémerila, Jérôme Le Nôtrea, Christian Bruneaua, Pierre H. Dixneuf*a, Alexey F. Kolomietsb and Sergey N. Osipov*b
aLaboratoire d'Organométalliques et Catalyse: Chimie et Electrochimie Moléculaires (CNRS UMR 6509), Uni![[italic v]](https://www.rsc.org/images/entities/char_e0f5.gif) ersité de Rennes, Campus de Beaulieu, 35042, Rennes, France. E-mail: pierre.dixneuf@univ-rennes1.fr
ersité de Rennes, Campus de Beaulieu, 35042, Rennes, France. E-mail: pierre.dixneuf@univ-rennes1.fr
bA N Nesmeyano Institute of Organoelement Compounds, Russian Academy of Sciences, Vailo str. 28, 117813, Moscow, V-334, GSP-1, Russia. E-mail: osipov@ineos.ac.ru
First published on 8th December 2000
Abstract
New α-alkynyl, α-CF3 amino esters have been prepared from electrophilic imines and used to produce 3-alkenyl-3,4-dehydroproline
derivatives, ![[italic v]](https://www.rsc.org/images/entities/i_char_e0f5.gif) ia enyne metathesis with the precatalyst [Ru
ia enyne metathesis with the precatalyst [Ru![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) CCCPh2(Cl)(PCy3)(arene)]O3SCF3.
These new conjugated fluorine-containing dienes
are active substrates for the Diels–Alder reaction and lead
to a new class of bicyclic
amino esters.
CCCPh2(Cl)(PCy3)(arene)]O3SCF3.
These new conjugated fluorine-containing dienes
are active substrates for the Diels–Alder reaction and lead
to a new class of bicyclic
amino esters.
α,α-Disubstituted α-amino acids constitute an important class of nonproteinogenic amino acids that has recently received a great deal of attention.1 In particular, the incorporation of these compounds into peptides results in conformational restrictions and increased rigidity, leading to enhanced resistance towards protease enzymes and to the stabilisation of some secondary structures.2 Moreover, such α-amino acids bearing an alkynyl or CF3 group at the α-position irreversibly inhibit the action of various pyridoxal phosphate-dependant enzymes3 such as alanine racemases4a or decarboxylases.4b On the other hand, increasing interest in new proline derivatives is connected with the fact that proline is unique among the natural amino acids for its abilities to induce β-turns and initiate peptide folding of the α-helix.5 Due to these structurally important properties, proline is often regarded as the primary contributor to the biological activity of several proteins, as well as having a key role in biological recognition processes.5 Structurally modified prolines, especially those containing multiple C–C bonds, have been described as potential enzyme inhibitors.6 For example, 3,4-dehydro-L-proline is an effective inhibitor of proline dehydrogenase.7
Simple syntheses of new α,α-disubstituted amino acids, containing α-alkynyl and α-CF3 groups, are of interest as proline derivative precursors.
As ruthenium-allenylidene complexes of the type (LnRuCCCR2)+PF6−
were recently shown to be excellent catalyst precursors for enyne metathesis in the synthesis of dihydrofuran derivatives,8 it might
be expected that α-alkynyl, α-CF3
α-amino esters could offer a direct access to unsaturated cyclic amino esters, and especially to unsaturated
proline analogs.
We now report (i) general access to a variety of α-CF3
α-alkynyl amino
esters from the readily available electrophilic imines (CF3)(CO2Me)CNR
and (ii) their use for access to 5-membered ring, fluorinated amino esters ia
enyne metathesis performed with the
ruthenium-allenylidene precatalyst
[RuCCCR2(Cl)(PCy3)(p-cymene)]+CF3SO3−
[eqn. (1)].
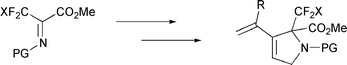 | (1) |
Electrophilic α-CF3, α-CO2Me imines constitute excellent starting materials for the formation of bifunctional α-CF3
α-amino esters.9 The starting electrophilic imines 2a and 2b were prepared in quantitative yields (90–95%) from the fluorinated keto ester CF3COCO2Me, 1,10 upon addition of H2NTs, H2NSO2Ph or H2NCbz, followed by dehydration11 (Scheme 1). The addition of lithium acetylide to an imine 2, followed by hydrolysis,12
led to the new α-CF3, α-alkynyl amino esters 3. The treatment
of the amino esters 3 with NaH, followed by allyl bromide addition
in DMF, afforded α-CF3 amino esters 4 with the 1,6-enyne structure (Scheme 1). Thus, the derivatives 4a
(R = H), 4b (R = Bun), 4c
(R = CH2OMe), 4d (R = cyclopropyl) and 4e
[R = C(Me)CH2] were obtained in overall yields of 40, 42, 60, 50 and 28%,
respectively, directly from the imines 2a–c without purification of the intermediates 3. Similarly, the α-CF2Cl-containing
imine CF2Cl(CO2Me)CN-Boc, 2d,13 led to the new α-alkynyl-N-allyl amino ester 5
[eqn. (2)].
![Reagents and conditions: (i) H2N-PG [H2NTs or H2NSO2Ph (without solvent) or H2NBoc or H2NCbz (both in dichloromethane)]; (ii) SOCl2 in excess (>5 equiv.) with a catalytic amount of pyridine for PG = Ts or SO2Ph; (iii) 1 equiv. of trifluoroacetic anhydride (TFAA), 2 equiv. of pyridine for PG = Cbz or Boc; (iv) 1 equiv. of RCCLi in THF and then hydrolysis; (v) 2 equiv. of NaH and then allyl bromide (3 equiv.) in DMF.](/image/article/2001/NJ/b007396m/b007396m-s1.gif) | ||
Scheme 1 Reagents and conditions: (i) H2N-PG [H2NTs or H2NSO2Ph (without solvent) or H2NBoc or H2NCbz (both in dichloromethane)]; (ii) SOCl2 in excess (>5 equiv.) with a catalytic amount of pyridine for PG = Ts or SO2Ph; (iii) 1 equiv. of trifluoroacetic anhydride (TFAA), 2 equiv. of pyridine for PG = Cbz or Boc; (iv) 1 equiv. of RC![[triple bond, length half m-dash]](https://www.rsc.org/images/entities/char_e007.gif) CLi in THF and then hydrolysis; (v) 2 equiv. of NaH and then allyl bromide (3 equiv.) in DMF. CLi in THF and then hydrolysis; (v) 2 equiv. of NaH and then allyl bromide (3 equiv.) in DMF. | ||
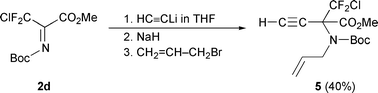 | (2) |
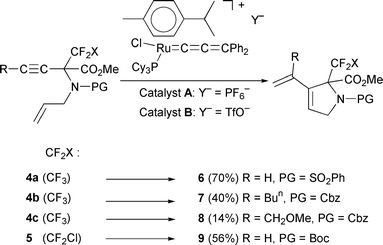
The catalytic enyne metathesis on the fluorinated enynes 4
was then attempted. A solution of the α-CF3 amino ester 4a (0.2 mmol) in toluene (5 mL) with 10 mol.% of catalyst [RuCCCPh2(Cl)(PCy3)(p-cymene)]+PF6−,
A,14,15 was irradiated at 300 nm for 0.5 h at room temperature and then the mixture was heated at 80°C
for 69 h in order to reach 90% conversion of 4a. The 5-membered cyclic amino ester 6
resulting from enyne metathesis was isolated in 70% yield (Scheme 2). Thus, the catalyst A appears to be less active towards the bulky enynes 4 than in the enyne metathesis of smaller mixed propargyl
allyl ethers to give dihydrofurans.8 The reaction of 4a was then performed with 5 mol.% of the
salt catalyst, but with a different counter anion, [RuCCCPh2(Cl)(PCy3)(p-cymene)]+CF3SO3−,
B.15,16 After irradiation for 0.5 h at room temperature,
only 24 h of heating in toluene at 80°C led to 95% conversion of 4a and to the isolation of 6 in 58% yield. Thus, the triflate
catalyst B appeared to tolerate the CF3 group as well, brought
the best compromise for this enyne metathesis and was selected
for further use.
 | ||
| Scheme 2 | ||
Catalyst B is easily prepared in situ, just before use for the
transformation of 0.2 mmol of enyne, from 14 mg (2 × 10−2 mmol) of the very stable salt [RuCl(PCy3)(p-cymene)]+TfO−, C,16 and 4 mg of HCC–CPh2OH (1 equiv.) in 2 mL of toluene. After 30 min of stirring at room temperature, the enyne (0.2 mmol) was then introduced and the enyne metathesis performed as described above. The stable salt C simply results from
the quantitative one-pot transformation of the commercial precursor [RuCl2(p-cymene)]2 on addition of 1 equiv. of PCy3, to produce RuCl2(PCy3)(p-cymene),16
followed by the reaction with 1 equiv. of AgOTf. The metathesis of enynes 4b and 4c, containing a disubstituted CC bond, required more forcing conditions. Enyne 4b (0.2 mmol) with 10 mol.% of catalyst B in
toluene, after 0.5 h UV irradiation followed by 4 days at 80°C, led to 86% conversion and 40% of isolated derivative 7. Analogously, after 3 days at 80°C, 4c led to 48% conversion into 8
isolated in 14% yield (Scheme 2).
In contrast, the novel cyclopropyl (4d) and isopropenyl (4e)
derivatives were not transformed under these conditions. This
is likely due to the bulkiness of the cyclopropyl and isopropenyl
groups, since the CC bond is expected to initially interact with
the catalyst.17 The N-Boc-protected α-CF2Cl enyne 5 containing the
α-HCC group is transformed under similar conditions as for 4a (5
mol.% of B, 0.5 h irradiation, and 25 h at 80°C) to yield the cyclic
amino ester 9 (56%) (Scheme 2). The amino esters 6–9
contrast well with the cyclic amino esters obtained ia alkene
metathesis18 as they contain a diene moiety and are potentially
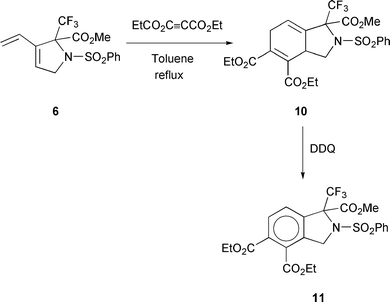
suitable precursors for Diels–Alder reactions. Thus, the diene
6 was refluxed with 3 equiv. of EtO2CCCCO2Et in toluene
for 18 h and the Diels–Alder adduct 10 was isolated in 77%
yield, showing the presence of two diastereoisomers in a 7:1
ratio. Compound 10 was aromatized on treatment with dichlorodicyanobenzoquinone
(DDQ; 5 equiv.). After 24 h of
reflux in toluene only 50% of 10 was converted, however, the
new fluorine-containing bicyclic amino ester 11 was isolated
in 37% yield (Scheme 3).
 | ||
| Scheme 3 | ||
The above reactions show that the ruthenium-allenylidene complex B is a useful catalyst for access to bicyclic amino esters.
Experimental
General procedure for the preparation of 3
The imine 2 (5 mmol) in dry THF (15 ml) was added dropwise to a stirred solution of 6 mmol of lithium acetylide at − 78°C. After 1 h at − 78°C the reaction mixture was allowed to warm up to room temperature and stirred for 5 h. The reaction was quenched with a saturated solution of NH4Cl and extracted with diethyl ether (2 × 20 ml). The organic layer was washed with brine (25 ml), dried over MgSO4 and filtered. The solvent was removed under reduced pressure and the crude product, characterized by 1H NMR, was used directly without purification.General procedure for the preparation of 4 and 5
A solution of 3 (3 mmol) in dry DMF (9 ml) was added to a suspension of NaH (6 mmol) in dry DMF (15 ml) at 0°C. The mixture was stirred at room temperature for 0.5 h and then 9 mmol of allyl bromide (in solution in 6 ml of DMF) were added. After stirring for an additional 5 h, the mixture was hydrolyzed with water (20 ml) and extracted with diethyl ether (2 × 20 ml). The organic layer was washed with water (4 × 20 ml), dried over MgSO4 and filtered. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography (diethyl ether–pentane). Compounds 4a–e and 5 were characterized by 1H, 13C and 19F NMR and gave satisfactory elemental analyses.CH), 3.97 (s, 3 H, OCH3), 4.00–4.27 (m, 2 H, CH2N), 4.87 (dm, 1
H, 3J
= 10.1, cis-CH2CH), 4.94 (dm, 1 H, 3J
= 16.9 Hz, trans-CH2
CH), 5.52–5.74 (m, 1 H, CH2CH), 7.44–7.65 (m, 3 H, Ph), 7.88–7.97 (m, 2 H, Ph). 19F NMR (CDCl3, 188.292 MHz): δ
− 71.9 (s, 3 F,
CF3). 13C NMR (CDCl3, 50.329 MHz): δ
50.60 (NCH2), 54.35 (OCH3), 72.59 (quat. C), 80.15 (CCH), 97.95 (CCH), 117.44 (CH2CH), 122.39 (q, 1JCF
= 288.7 Hz, CF3), 128.65, 128.99, 133.60 (aromatic CH), 133.48 (CH2CH), 139.22 (ipso C), 163.294 (CO). Anal.
found: C, 49.29; H, 3.68%. Calc. for C15H14F3NO4S: C, 49.86; H, 3.90%.CH), 3.83 (s, 3 H, OCH3), 4.10–4.25 (m, 2 H, CH2N), 5.13 (dm, 1 H, 3J
= 10.2, cis-CH2CH), 5.25 (dm, 1 H,
3J
= 17.2 Hz, trans-CH2
CH), 5.76–6.04 (m, 1 H, CH2
CH). 13C NMR (CDCl3,
50.329 MHz): δ 28.00 (3 × CH3 Boc), 50.22 (NCH2), 53.45 (OCH3), 73.93 (quat. CCF2Cl), 79.14 (CCH), 82.45 [C(CH3)3], 97.86 (CCH), 116.19 (CH2CH), 128.06 (t, 1JCF
= 288.6 Hz, CF2Cl),
134.25 (CH2CH), 152.58 (CO Boc), 163.23 (CO).General procedure for the preparation of 6–9
A mixture of enyne 4 or 5 (1 mmol) and catalyst A or B (5 or 10 mol.%) in toluene was irradiated at room temperature for 0.5 h and then heated at 80°C. The solvent was removed in vaccum and the crude product was purified by flash chromatography (diethyl ether–pentane). Compounds 6–9 were characterized by 1H, 13C and 19F NMR and gave satisfactory elemental analyses.CH), 5.47 (d, 1 H, 3J
= 17.6, trans-CH2CH), 6.12 (dd, 1 H, 3J
= 11.4, 3J
= 17.7 Hz, CH2CH), 6.19–6.27 (m, 1 H, CH2CHC), 7.43–7.65 (m, 3 H, Ph), 7.82–7.93 (m, 2 H, Ph).
19F NMR (CDCl3, 188.292 MHz): δ
− 71.9 (s, 3 F, CF3). 13C NMR (CDCl3, 50.329 MHz): δ
53.53 (OCH3), 55.02 (CH2N), 68.02 (CCF3), 119.16 (CH2CH), 123.40 (q, 1JCF
= 287.4 Hz, CF3),
126.71 (CH2CH), 128.58 (CH2CH), 127.40, 129.07,
133.22 (aromatic CH), 135.40 (quat. C), 139.51 (ipso C), 165.65 (CO). Anal found: C, 49.71; H, 4.17%. Calc. for C15H14F3NO4S: C, 49.86; H, 3.90%.Characterization of 10 and 11
CHCH2), 3.50–3.64 (m, 1 H, CHCH2), 3.84 (m, 3 H, CH2N and CHCH2N), 5.88 (m, 1 H, CH2CH), 7.40–7.62 (m, 3 H, Ph), 7.79–7.91 (m, 2 H, Ph). 19F NMR (CDCl3,
282.408 MHz): δ
− 71.62 (s, 3 F, CF3). 13C NMR (CDCl3, 50.329 MHz): δ 13.83 (CH3CH2), 14.00 (CH3CH2), 29.71 (CH2CH), 38.35 (CHCH2N), 52.48 (CH2N), 53.43 (OCH3), 61.78 (CH3CH2), 62.71 (CH3CH2), 72.79 (q, 2JCF
= 29.7, CCF3), 122.21 (CH2CHC), 128.43 (q, 1JCF
= 285.5 Hz, CF3), 127.26, 129.15, 133.33 (aromatic CH), 133.83 [CH2C(CO)C], 135.60 (ipso C), 139.85 [CHC(CO)C], 165.70 (quat. CHC), 164.87, 166.39,
167.21 (CO).O), 132.7 (q, 1JCF
= 283.0 Hz, CF3), 128.60, 136.60, 138.53, 139.22 (quat. aromatic C), 165.44, 165.72, 166.61 (CO).Acknowledgements
The authors wish to thank the European Union INTAS programme 97-1874 for support, the COST programme D12/0025/99 and the Region Bretagne for a grant to J. LN.References
- For a review see: H. Heimgartner, Angew. Chem., Int. Ed. Engl., 1991, 30, 238 CrossRef.
- C. Tomiolo, M. Crisma, S. Pegorato, E. L. Becker, S. Polinelli, W. H. J. Boesten, H. E. Schoemaker, E. M. Meijer, J. Kamphuis and R. Freer, Peptide Res., 1991, 4, 66 Search PubMed.
- C. Walsh, Tetrahedron, 1982, 38, 871 and references cited therein CrossRef.
- (a) Y. Kuroda, M. Okuhara, T. Goto, E. Iguchi, M. Kohsaka, H. Aoki and H. Imanaka, J. Antibiot., 1980, 33, 125 Search PubMed; (b) D. Schirlin, J. B. Ducep, S. Baltzer, P. Bey, F. Piriou, J. Wagner, J. M. Hornsperger, J. G. Heydt, M. J. Jung, C. Danzig, R. Weiss, J. Fischer, A. Mitschler and A. De Cian, J. Chem. Soc., Perkin Trans. 1, 1992, 1053 RSC.
- (a) D. J. Barlow and J. M. Thornton, J. Mol. Biol., 1988, 201, 601 CAS; (b) A. M. Koskinen and H. Rapoport, J. Org. Chem., 1989, 54, 1859 CrossRef CAS; (c) H. H. Ibrahim and W. D. Lubell, J. Org. Chem., 1993, 58, 6438 CrossRef CAS.
- (a) D. O. Gray and L. Fowden, Nature (London), 1962, 193, 1285 CAS; (b) D. O. Gray and L. Fowden, Phytochemistry, 1972, 11, 745 CrossRef CAS.
- F. Manfre, J.-M. Kern and J.-F. Biellmann, J. Org. Chem., 1992, 57, 2060 CrossRef CAS.
- M. Picquet, C. Bruneau and P. H. Dixneuf, Chem. Commun., 1998, 2249 RSC.
- N. Sewald and K. Burger, in Fluorine-containing Amino Acids: Synthesis and Properties, ed. V. P. Kukhar and V. A. Soloshonok, Wiley, Chichester, UK, 1995, p. 139, and references cited therein. Search PubMed.
- I. L. Knunyants, V. V. Shokina and V. V. Tyuleneva, Dokl. Akad. Nauk SSSR, 1966, 169, 594 Search PubMed; I. L. Knunyants, V. V. Shokina and V. V. Tyuleneva, Chem. Abs., 1966, 65, 15218e Search PubMed.
-
(a) S. N. Osipov, N. D. Chkanikov, A. F. Kolomiets and A. V. Fokin, Bull. Acad.
Sci. USSR, Di. Chem. Sci. (Engl. Transl.), 1986, 1256 Search PubMed;
(b) K. Burger, E. Hoess, K. Gaa, N. Sewald and C. Schierlinger, Z. Naturforsch. B, 1991, 46, 361 Search PubMed;
(c) S. N. Osipov and K. Burger, Tetrahedron Lett., 2000, 41, 5659 CrossRef CAS.
- The protocol described for analogous compounds in K. Burger and N. Sewald, Synthesis, 1990, 115 was used. Search PubMed.
- S. N. Osipov, A. S. Golubev, N. Sewald, T. Michel, A. F. Kolomiets, A. V. Folkin and K. Burger, J. Org. Chem., 1996, 61, 7521 CrossRef CAS.
- A. Fürstner, M. Picquet, C. Bruneau and P. H. Dixneuf, Chem. Commun., 1998, 1315 RSC.
- A. Fürstner, M. Liebl, C. W. Lehmann, M. Picquet, R. Kunz, C. Bruneau, D. Touchard and P. H. Dixneuf, Chem. Eur. J., 2000, 6, 1847 CrossRef CAS.
- M. Picquet, D. Touchard, C. Bruneau and P. H. Dixneuf, New J. Chem., 1999, 23, 141 RSC.
- (a) T. J. Katz and T. M. Sivavec, J. Am. Chem. Soc., 1985, 107, 737 CrossRef CAS; (b) T. M. Sivavec, T. J. Katz, M. Y. Chiang and G. X.-Q. Yang, Organometallics, 1989, 8, 1620 CrossRef CAS.
- S. Osipov, C. Bruneau, M. Picquet, A. F. Kolomiets and P. H. Dixneuf, Chem. Commun., 1998, 2053 RSC.
Footnote |
| † Dedicated to the memory of Professor Olivier Kahn |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2001 |