Coordination and oxidative addition of octafluoronaphthalene at a nickel centre: isolation of an intermediate in C–F bond activation†
Thomas
Braun
,
Leroy
Cronin
,
Catherine L.
Higgitt
,
John E.
McGrady
,
Robin N.
Perutz
* and
Meike
Reinhold
Department of Chemistry, Uni![[italic v]](https://www.rsc.org/images/entities/char_e0f5.gif) ersity of York, York, UK YO10 5DD. E-mail: rnp1@york.ac.uk
ersity of York, York, UK YO10 5DD. E-mail: rnp1@york.ac.uk
First published on 15th November 2000
Abstract
Reaction of [Ni(COD)2] with PEt3 and octafluoronaphthalene yielded the complex [Ni(η2-1,2-C10F8)(PEt3)2] 1, which was converted thermally into the C–F activation product trans-[NiF(2-C10F7)(PEt3)2] 2. The crystal structure of 1 shows asymmetric η2 coordination with significant distortions of the naphthalene unit compared to the “ free” ligand; DFT calculations reproduce the principal features of the geometry.
Several methods have been described for the activation of carbon–fluorine bonds of fluoroaromatic compounds by reaction at transition metal centres.1–4 Although a wide variety of electron configurations and metals appear capable of C–F activation, nickel has assumed a prominent role.3–5 We have studied the oxidative addition of fluorinated aromatics and heteroaromatics such as hexafluorobenzene, pentafluoropyridine and 2,4,6-trifluoropyrimidine at a nickel(0) centre yielding trans-[NiF(C6F5)(PEt3)2], trans-[NiF(2-C5NF4)(PEt3)2] and trans-[NiF(4-C4N2F2H)(PEt3)2].3,4 We have proposed that these reactions proceed by initial coordination of the aromatic compounds through a C
![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) C bond or nitrogen followed by oxidative addition of the C–F bond, but no reaction intermediates have yet been detected.3,4
Of particular note are the different rates of these reactions: the C–F activation of the heteroaromatics is very fast, while the reaction of hexafluorobenzene is extremely slow. Mechanistic information on C–F oxidative addition
is very limited. Crespo et al. have obtained kinetic evidence for concerted intramolecular oxidative addition of fluoroaromatic substituents at platinum.6 Bach et al. have synthesized
[Ni(η2-C6F6){tBu2P(CH2)2PtBu2}] and shown that on heating
it forms [NiF(C6F5){tBu2P(CH2)2PtBu2}]. The product
was identified by 19F and 31P NMR spectroscopy, but no kinetics was reported.7 Here we show the coordination of octafluoronaphthalene to nickel in a η2 mode and its thermal
C–F bond activation at the nickel centre. The crystal structure
of the complex [Ni(η2-1,2-C10F8)(PEt3)2]
1 shows remarkable distortions when compared to structures of η2-C6F6 complexes.
C bond or nitrogen followed by oxidative addition of the C–F bond, but no reaction intermediates have yet been detected.3,4
Of particular note are the different rates of these reactions: the C–F activation of the heteroaromatics is very fast, while the reaction of hexafluorobenzene is extremely slow. Mechanistic information on C–F oxidative addition
is very limited. Crespo et al. have obtained kinetic evidence for concerted intramolecular oxidative addition of fluoroaromatic substituents at platinum.6 Bach et al. have synthesized
[Ni(η2-C6F6){tBu2P(CH2)2PtBu2}] and shown that on heating
it forms [NiF(C6F5){tBu2P(CH2)2PtBu2}]. The product
was identified by 19F and 31P NMR spectroscopy, but no kinetics was reported.7 Here we show the coordination of octafluoronaphthalene to nickel in a η2 mode and its thermal
C–F bond activation at the nickel centre. The crystal structure
of the complex [Ni(η2-1,2-C10F8)(PEt3)2]
1 shows remarkable distortions when compared to structures of η2-C6F6 complexes.
The stepwise treatment of [Ni(COD)2] with PEt3 and octafluoronaphthalene in hexane solution at room temperature results in the regioselective formation of [Ni(η2-1,2-C10F8)(PEt3)2] 1.‡ The 31P NMR spectrum displays a triplet at δ 18.0 (average JPF 18.5 Hz) for the equivalent phosphorus nuclei. The 19F NMR spectrum shows four multiplets at room temperature at δ −162.72, −160.87, −160.49 and −145.93 consistent either with a structure with mirror symmetry (e.g. η4-C10F8) or a fluxional structure (e.g. η2-C10F8). On cooling to 190 K the resonances at δ −162.72 and −160.87 broaden, but those at δ −160.49 and −145.93 stay fairly sharp, indicating the occurrence of fluxional behaviour.
A suitable single crystal of complex 1 was obtained from hexane at 253 K and its structure determined by X-ray crystallography (Fig. 1).§8 The structure shows an η2-C10F8 ligand coordinated to a nickel centre with approximately trigonal planar geometry. The Ni–C(1)–C(2) plane forms an angle of 108° with the naphthalene ligand, and an angle of 13.0° with the Ni–P(1)–P(2) plane. The bond C(1)–C(2) is significantly lengthened to 1.438(6) Å on coordination, but the C(1)–C(10) separation is also extremely long [1.472(6) Å], while the C(3)–C(4) bond length is shortened to 1.341(6) Å. No reliable structural data for free C10F8 are available for comparison, but the bond lengths of the uncoordinated ring in 1 may be used as a reference; they range from 1.356(8) to 1.412(7) Å (the C–C bond length of free C6F6 is 1.394(7) Å).9 A similar disruption of the aromaticity in the bound naphthalene ring is found in [Ru(η5-C5Me5)(η2-C10H8)(NO)].10
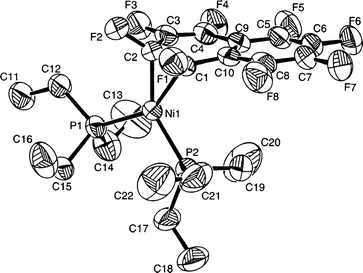 | ||
| Fig. 1 An ORTEP8 diagram of 1. Ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) and angles (°): Ni–C(1) 1.959(4), Ni–C(2) 1.899(4), C(1)–F(1) 1.390(5), C(2)–F(2) 1.397(5), C(3)–F(3) 1.350(6), C(4)–F(4) 1.353(6), C(1)–C(2) 1.438(6), C(2)–C(3) 1.429(6), C(3)–C(4) 1.341(6), C(4)–C(9) 1.435(7), C(9)–C(10) 1.412(7), C(1)–C(10) 1.472(6); C(1)–Ni–C(2) 43.7(2), Ni–C(1)–F(1) 111.2(3), Ni–C(2)–F(2) 119.6(3), Ni–C(2)–C(3) 112.9(3), Ni–C(1)–C(10) 123.4(3), C(1)–Ni–P(1) 146.19(14), C(2)–Ni–P(2) 146.82(14). | ||
The benzenoid ring that is bound to nickel in 1 is very distorted: C(1) lies 0.197(6) Å above the plane defined by C(3) to C(10). The buckling effect is also apparent in the torsion angles (Table 1). As found for the fluorine atoms attached to the η2-bound carbons in [Ni(η2-C6F6){tBu2P(CH2)2PtBu2}]7 and other η2-C6F6 complexes,11 the atoms F(1) and F(2) in 1 lie out of the octafluoronaphthalene plane with F(2) further out than F(1) (Fig. 1). In line with this observation, the Ni–C(2) bond [1.899(4) Å] is shorter than the Ni–C(1) bond [1.959(4) Å]. For comparison, the Ni–C bond length in a related σ bond in trans-[NiF(C6F5)(PEt3)2] is 1.878(7) Å.3 The C(1)–F(1) and C(2)–F(2) bonds of 1 are extended to 1.390(5) and 1.397(5) Å compared to C(3)–F(3) to C(8)–F(8) which range from 1.341(6) to 1.356(6) Å.
| X-ray | L = PEt3a | L = PEt3b | |
|---|---|---|---|
| a Optimisation starting from X-ray coordinates. b Optimisation with Ni–C bond lengths constrained equal at 1.985 Å. | |||
| Ni–C(1) | 1.959(4) | 2.017 | 1.985 |
| Ni–C(2) | 1.899(4) | 1.959 | 1.985 |
| C(1)–C(2) | 1.438(6) | 1.448 | 1.449 |
| C(9)–C(10) | 1.412(7) | 1.437 | 1.438 |
| C(1)–F(1) | 1.390(5) | 1.388 | 1.391 |
| C(2)–F(2) | 1.397(5) | 1.393 | 1.389 |
| C(3)–F(3) | 1.350(6) | 1.358 | 1.357 |
| C(2)–C(3)–C(4)–C(10) | 7.1 | 6 | 7 |
| C(1)–C(9)–C(8)–C(7) | −175.3 | −177 | −178 |
| C(1)–C(9)–C(10)–C(4) | −6.1 | −5 | −5 |
| Δr[Ni–C(1) −Ni–C(2)] | 0.060(6) | 0.058 | 0 |
The pattern of crystallographic bond lengths in 1 is suggestive of an incipient transition state for concerted C–F activation at C(2).12 We examined the origin of the unusual geometry of [Ni(η2-1,2-C10F8)(PEt3)2] using density functional theory.¶ Optimised structural parameters are summarised in Table 1, together with the corresponding crystallographically determined values. The calculations correctly reproduce the principal features of the molecular structure, most noticeably the difference between Ni–C(1) and Ni–C(2) (calc.: 0.058, X-ray 0.060(6) Å). (Calculations on the PH3 analogue of 1 gave similar results.) However, an examination of the potential energy surface in the region of the minimum reveals that small (0.05 Å) variations in the two Ni–C bond lengths have an almost negligible influence on the calculated total energy. In fact, when Ni–C(1) and Ni–C(2) are constrained to be equal, the resulting optimised structure lies less than 3 kJ mol−1 above the global minimum. Thus DFT calculations suggest that the Ni(η2-1,2-C10F8) interaction exhibits a soft potential for distortion of the coordination geometry. Since the distortion energies are so small we cannot describe the structure of 1 as an incipient transition state with confidence.
The solution NMR spectra are consistent with the crystallographic data if the Ni(η2-C10F8) unit undergoes a suprafacial [1,3] shift from the 1,2-η2-coordinated form to the 3,4-η2-isomer (Scheme 1). A similar low energy process, involving an exchange of coordinated and non-coordinated CC bonds, has been observed for [Ni(η2-C10H8){iPr2P(CH2)2PiPr2}] and [Ni(η2-C10H8){iPr2P(CH2)3PiPr2}] as well as for the hexafluorobenzene complex [Ni(η2-C6F6){tBu2P(CH2)2PtBu2}].7,17
An η4-coordinated arene species is proposed as an intermediate for these dynamic processes. However, ring-whizzing ![[italic v]](https://www.rsc.org/images/entities/i_char_e0f5.gif) ia an η3-bound transition state as predicted in extended Hückel calculations for [Ni{η2-C6(CF3)6}(CO)2] is also conceivable.18
ia an η3-bound transition state as predicted in extended Hückel calculations for [Ni{η2-C6(CF3)6}(CO)2] is also conceivable.18
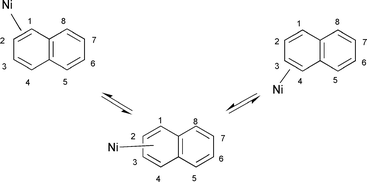 | ||
| Scheme 1 Fluxional behaviour of 1. | ||
On isolating 1, redissolving in toluene and heating to 348 K for 24 h, the C–F activation product trans-[NiF(2-C10F7)(PEt3)2] 2 is obtained.|| The 19F NMR spectrum of 2 shows a triplet of virtual triplets at δ −387.25 (JPF 47, ∣JFF + JFF′ ∣ 12 Hz) characteristic of the metal fluoride and seven further signals indicating the presence of a heptafluoronaphthyl group. The assignment of 2 as trans-[NiF(2-C10F7)(PEt3)2] is based on the values of JFF measured through selective 19F–19F NMR decoupling experiments and the presence of two low field 19F resonances (Fig. 2).** The 31P NMR spectrum displays a doublet resonance at δ 13.3 (JPF 47.6 Hz) for the two equivalent phosphorus nuclei coupled to the metal-bound fluoride.
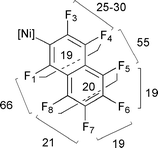 | ||
| Fig. 2 Coupling constants JFF Hz in 2. | ||
The rates of loss of 1 and formation of 2 were monitored by 19F NMR spectroscopy at 348 K with an integration standard contained in a capillary. They are both first order and the same within the experimental error (ca. 10−5 s−1). There is appreciable variation (10–15%) in the rates from one run to the next due to decomposition reactions. However, the kinetic results are compatible with an intramolecular reaction of 1 forming the nickel(II) C–F activation product 2 (Scheme 2).
![Reactivity of [Ni(COD)2] towards octafluoronaphthalene.](/image/article/2001/NJ/b006368l/b006368l-s2.gif) | ||
| Scheme 2 Reactivity of [Ni(COD)2] towards octafluoronaphthalene. | ||
When the solution of initial reagents is heated directly
without isolating complex 1, product 2 is formed together
with other fluorine containing compounds. The principal C–F activation products are 2 and another compound 3 containing a (Et3P)2Ni–F unit [δ(19F) −385.94 (t, average JPF 49.6 Hz)]. The structure of 3 is not yet proven but the spectra are consistent with trans-[NiF(1-C10F7)(PEt3)2], isomeric to 2. However, we have not observed conversion of 2 into 3 or ice ersa. There is also a loss of selectivity for 2 when a solution of pure compound 1 is heated in the presence of free PEt3 (10 equivalents); the products include 3. These observations provide evidence for a second mechanism of C–F activation.2,6,20,21
The results described lead to the conclusion that one
mechanism of C–F activation of octafluoronaphthalene by [Ni(COD)(PEt3)2]
proceeds ia coordination at the nickel centre and subsequent intramolecular oxidative addition. Another
mechanism may operate when the initial reagents are heated directly. The rate determining step is intramolecular insertion of nickel in the C–F bond in the 2 position. The unusual structure of [Ni(η2-1,2-C10F8)(PEt3)2] 1 has been simulated by theory, but the energetic stabilisation induced by distortion is
very slight. Intermediates similar to 1 with the fluorinated molecules coordinated to the metal centre ia a CC bond or a nitrogen atom are probable in the C–F activation of hexafluorobenzene,
pentafluoropyridine and 2,4,6-trifluoropyrimidine at Ni(PEt3)2.3–5,7,22
Experimental
A suspension of [Ni(COD)2] (82.2 mg, 0.30 mmol) in hexane (5 mL) was treated with PEt3 (111 μL, 0.75 mmol). The solution was stirred for 5 min at room temperature until it changed from red-purple to yellow. After adding octafluoronaphthalene (128 mg, 0.47 mmol) the solution was stirred for 2 h and the volatiles were removed under vacuum. The orange residue was dissolved in hexane (10 mL). Orange crystals were obtained at −20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C
overnight. The NMR spectra of 1 always revealed the presence
of free octafluoronaphthalene (ratio 1: C10F8>4:1) preventing
us from determining yields and analytical data. Heating
a sample of 1 in toluene at 348 K for 24 h led to complex 2.
After removing the solvent under vacuum, the orange solid
was recrystallised from hexane.
°C
overnight. The NMR spectra of 1 always revealed the presence
of free octafluoronaphthalene (ratio 1: C10F8>4:1) preventing
us from determining yields and analytical data. Heating
a sample of 1 in toluene at 348 K for 24 h led to complex 2.
After removing the solvent under vacuum, the orange solid
was recrystallised from hexane.
Acknowledgements
We would like to acknowledge Dr. S. B. Duckett and J. K. Mitchell for experimental assistance, and the EPSRC, DSM Research and the European Commission for financial support.Notes and references
- J. Burdeniuc, B. Jedlicka and R. H. Crabtree, Chem. Ber./Recl., 1997, 130, 145, and references therein Search PubMed; J. L. Kiplinger, T. G. Richmond and C. E. Osterberg, Chem. Re., 1994, 94, 373, and references therein Search PubMed; E. F. Murphy, R. Murugavel and H. W. Roesky, Chem. Re., 1997, 97, 3425, and references therein Search PubMed.
- (a) B. L. Edelbach and W. D. Jones, J. Am. Chem. Soc., 1997, 119, 7734 CrossRef CAS; (b) B. L. Edelbach, A. K. F. Rahman, R. J. Lachicotte and W. D. Jones, Organometallics, 1999, 18, 3170 CrossRef CAS; (c) B. L. Edelbach, B. M. Kraft and W. D. Jones, J. Am. Chem. Soc., 1999, 121, 10327 CrossRef CAS; (d) J. L. Kiplinger and T. G. Richmond, Chem. Commum., 1996, 1115 RSC; (e) F. Godoy, C. L. Higgitt, A. H. Klahn, B. Oelckers, S. Parsons and R. N. Perutz, J. Chem. Soc., Dalton Trans., 1999, 2039 RSC.
- L. Cronin, C. L. Higgitt, R. Karch and R. N. Perutz, Organometallics, 1997, 16, 4920 CrossRef CAS; T. Braun, S. Parsons, R. N. Perutz and M. Voith, Organometallics, 1999, 18, 1710 CrossRef CAS; S. J. Archibald, T. Braun, J. F. Gaunt, J. E. Hobson and R. N. Perutz, J. Chem. Soc., Dalton Trans., 2000, 2013 RSC.
- T. Braun, S. P. Foxon, R. N. Perutz and P. H. Walton, Angew. Chem., Int. Ed., 1999, 38, 3326 CrossRef CAS.
- T. Yamamoto and M. Abia, J. Organomet. Chem., 1997, 535, 209 CrossRef CAS; N. Y. Adonin and V. F. Starichenko, Mendelee Commun., 2000, 60 Search PubMed.
- M. Crespo, M. Martinez and E. de Pablo, J. Chem. Soc., Dalton Trans., 1997, 1231 RSC; M. Crespo, M. Martinez and J. Sales, J. Chem. Soc., Chem Commun., 1992, 823 RSC.
- I. Bach, K.-R. Pörschke, R. Goddard, C. Kopiske, C. Krüger, A. Rufinska and K. Seevogel, Organometallics, 1996, 15, 4959 CrossRef CAS.
- C. K. Johnson, ORTEP, Report ORNL-5138, Oak Ridge National Laboratory, Oak Ridge, TN, 1976; G. M. Sheldrick, SHELXS 86, program for crystal structure determination, University of Göttingen, 1986; G. M. Sheldrick, SHELXL 93, program for crystal structure refinement, University of Göttingen, 1993..
- A. Almenningen, O. Bastiansen, R. Seip and H. M. Seip, Acta Chem. Scand., 1964, 18, 2115 Search PubMed.
- C. D. Tagge and R. G. Bergman, J. Am. Chem. Soc., 1996, 118, 6908 CrossRef CAS.
- C. L. Higgitt, A. H. Klahn, M. H. Moore, B. Oelckers, M. G. Partridge and R. N. Perutz, J. Chem. Soc., Dalton Trans., 1997, 1269 RSC.
- R. Bosque, E. Clot, S. Fantacci, F. Maseras, O. Eisenstein, R. N. Perutz, K. B. Renkema and K. G. Caulton, J. Am. Chem. Soc., 1998, 120, 12634 CrossRef CAS.
- ADF 1999.02, E. J. Baerends, D. E. Ellis and P. Ros, Chem. Phys., 1973, 2, 41 CrossRef CAS; L. Verluis and T. Ziegler, J. Chem. Phys., 1988, 88, 322 CrossRef CAS; G. te Velde and E. J. Baerends, J. Comput. Phys., 1992, 99, 84 CrossRef CAS; C. Fonseca Guerra, J. G. Snijders, G. te Velde and E. J. Baerends, Theor. Chim. Acta, 1998, 99, 391 Search PubMed.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200 CrossRef CAS.
- A. D. Becke, Phys. Re. A, 1988, 38, 3098 Search PubMed.
- J. P. Perdew, Phys. Re. B, 1986, 33, 8822 Search PubMed.
- R. Benn, R. Mynott, I. Topalovic and F. Scott, Organometallics, 1989, 8, 2299 CrossRef CAS.
- J. Silvestre and T. A. Albright, J. Am. Chem. Soc., 1985, 107, 6829 CrossRef CAS.
- M. Carravetta, F. Castiglione, G. De Luca, M. Edgar, J. W. Emsley, R. D. Farrant, E. K. Foord, J. C. Lindon, M. Longeri, W. E. Palke and D. L. Turner, J. Magn. Reson., 1998, 135, 298 CrossRef CAS; L. Cassidei, O. Sciacovelli and L. Folani, Spectrochim. Acta Part A, 1982, 38, 755 CrossRef; J. W. Emsley, L. Phillips and V. Wray, Prog. Nucl. Magn. Reson. Spectrosc., 1976, 10, 83.
- T. T. Tsou and J. K. Kochi, J. Am. Chem. Soc., 1976, 101, 6319.
- R. Chukwu, A. D. Hunter and B. D. Santarsiero, Organometallics, 1991, 10, 2141 CrossRef CAS.
- M. W. Holtcamp, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 1997, 119, 848 CrossRef CAS.
Footnotes |
| † Supplementary data available: rotatable 3-D crystal structure diagram in CHIME format. See http://www.rsc.org/suppdata/nj/b0/b006368l/ |
| ‡ NMR spectroscopic data for 1 ([2H8]toluene, 300 K, referenced to [2H7]toluene at δ 2.10): 1H (500 MHz) δ 0.8 (m, br, 3H), 1.2 (m, br, 2H); 31P (202.4 MHz, referenced to external H3PO4 at δ 0) δ 18.0 (t, average JPF 18.5 Hz); 19F (470.4 MHz referenced to external CFCl3 at δ 0) δ −162.72 (m, 2 F), −160.87 (m, 2 F), −160.49 (m, 2 F), −145.93 (m, 2 F). |
§
Crystal data for 1: C22H30F8NiP2, M = 567.11, triclinic, space group, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a
= 11.344(10), b
= 12.614(6), c
= 9.846(13) Å, α
= 112.43(10), β
= 96.62(10), γ
= 98.50(9)°, U
= 1265(3) Å3, T
= 293 K, Z
= 2, μ(MoKα) = 0.960 mm−1, 4701 data, 4457 unique data, Rint
= 0.0158. Structure solution by direct methods (SHELXS), refined against F2 using 4455 data (SHELXL 93). Final R1, wR2 values on all data 0.0857, 0.1334; [Io>2σ(Io)] data 0.0442, 0.1114.CCDC reference number 440/216. See http://www.rsc.org/suppdata/nj/b0/b006368l/ for crystallographic files in .cif format. , a
= 11.344(10), b
= 12.614(6), c
= 9.846(13) Å, α
= 112.43(10), β
= 96.62(10), γ
= 98.50(9)°, U
= 1265(3) Å3, T
= 293 K, Z
= 2, μ(MoKα) = 0.960 mm−1, 4701 data, 4457 unique data, Rint
= 0.0158. Structure solution by direct methods (SHELXS), refined against F2 using 4455 data (SHELXL 93). Final R1, wR2 values on all data 0.0857, 0.1334; [Io>2σ(Io)] data 0.0442, 0.1114.CCDC reference number 440/216. See http://www.rsc.org/suppdata/nj/b0/b006368l/ for crystallographic files in .cif format.
|
| ¶ All calculations were performed using the Amsterdam Density Functional package (ADF 1999.02).13 In all cases, the local density approximation14 was used in conjunction with the gradient corrections of Becke15 and Perdew.16 Triple-ζ and double-ζ + polarisation basis sets were used to describe nickel and main group atoms, respectively. |
| || NMR spectroscopic data for 2 (300 K): 1H (500 MHz, C6D6 referenced to C6D5H at δ 7.15) δ 1.03 (m, 3H), 1.16 (m, 2H); 31P (202.4 MHz, C6D6) δ 13.3 (d, JPF 47.6 Hz); 19F (376.3 MHz, [2H8]toluene) δ −387.25 (tvt, JPF 47, ∣JFF + JFF′∣ 12, 1 F, NiF), −160.61 (dd, JF5F6 19, JF6F7 19, 1F, F6), −159.07 (dd, JF7F8 21, JF6F7 19, 1F, F7), −153.56 (ddd, JF4F5 55, JF3F4 25–30, JF1F4 19, 1F, F4), −148.19 (ddd, JF4F5 55, JF5F6 19, JF5F8 20, 1F, F5), −147.52 (ddd, JF1F8 66, JF7F8 21, JF5F8 20, 1F, F8), −109.08 (d, br, JF3F4 25–30 Hz, 1F, F3), −95.15 (dm, JF1F8 66, JF1F4 19 Hz, 1F, F1).** |
| ** The assignment of the 19F NMR spectrum was assisted by the coupling constants JFF in free octafluoronaphthalene, 1,5-C10H6F2, 1,8-C10H8F2 and rhenium fluoroaryl complexes.2e,19 |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2001 |