Ph otochemical expulsion of a Ru(phen)2 unit from a macrocyclic receptor and its thermal reco-ordination
Jean-Paul
Collin
*,
Anne-Chantal
Laemmel
and
Jean-Pierre
Sauvage
*
Laboratoire de Chimie Organo-Minérale, UMR 7513 du CNRS, Uni![[italic v]](https://www.rsc.org/images/entities/char_e0f5.gif) ersité Louis Pasteur, Faculté de Chimie, 4, rue Blaise Pascal, 67070, Strasbourg Cedex, France. E-mail: sauvage@chimie.u-strasbg.fr
ersité Louis Pasteur, Faculté de Chimie, 4, rue Blaise Pascal, 67070, Strasbourg Cedex, France. E-mail: sauvage@chimie.u-strasbg.fr
First published on 3rd November 2000
Abstract
A ruthenium complex containing two 1,10-phenanthroline ligands as well as a bipyridine fragment incorporated in a macrocycle (36 atoms) has been synthesized; photoexpulsion of the Ru(phen)2 unit from the macrocycle and its thermal reco-ordination take place efficiently and quantitatively.
Until now, most molecules or molecular assemblies considered as “machines” or “ motors”1 have been set into motion using chemical or electrochemical signals. Electrochemically driven movements are all based on the same principle: by reducing or oxidizing a given component of the compound, a new, thermodynamically unstable situation is generated which spontaneously will evolve to the thermodynamic well of the system while geometrical changes occur.2 An interesting photochemical variant of this principle has been proposed by Balzani and co-workers.3 By exciting a certain fragment of the molecule under light irradiation an excited state having a pronounced electron donor or acceptor character is formed. In the presence of a “sacrificial” agent, electron transfer from or to the excited state takes place, affording the oxidized or reduced form of the compound while the sacrificial agent is consumed.
We would now like to report another type of photochemically
induced movement based on dissociati![[italic v]](https://www.rsc.org/images/entities/i_char_e0f5.gif) e photo
excit
ed states. Ru(bipy)32+ (bipy = 2,2′-bipyridine) and its numerous
homologues have mostly been used as photochemical
electron or energy transfer agents, in relation to light energy conversion into chemical energy.4 In this respect, the photochemical stability of these compounds represents a real advantage, but a few examples of photochemical dissociation of such complexes have been reported.5 In some cases the “photoexpulsion”
process of a given ligand can be relatively efficient6
and it opens the gate to a new way of setting molecular systems
into motion. The use of a sterically hindered chelate, such as 6,6′-dimethyl-2,2′-bipyridine (dmbp), turned out to be especially promising since selective and efficient dissociation of this
particular chelate was observed under light irradiation.7
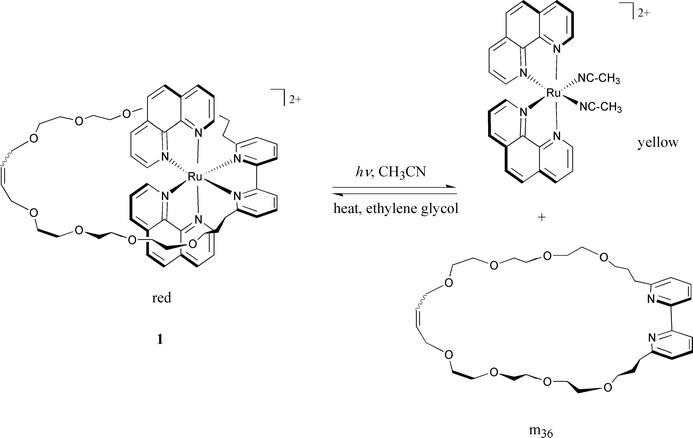
In the present system a hindered bipyridine fragment has been incorporated into a macrocycle and the ruthenium(II)-containing unit includes two 1,10-phenanthroline (phen) fragments. Ru(phen)22+ is a stable unit which will not be affected by light irradiation. The principle of the photochemically driven motion and the thermal backward reaction is shown in Scheme
1.
e photo
excit
ed states. Ru(bipy)32+ (bipy = 2,2′-bipyridine) and its numerous
homologues have mostly been used as photochemical
electron or energy transfer agents, in relation to light energy conversion into chemical energy.4 In this respect, the photochemical stability of these compounds represents a real advantage, but a few examples of photochemical dissociation of such complexes have been reported.5 In some cases the “photoexpulsion”
process of a given ligand can be relatively efficient6
and it opens the gate to a new way of setting molecular systems
into motion. The use of a sterically hindered chelate, such as 6,6′-dimethyl-2,2′-bipyridine (dmbp), turned out to be especially promising since selective and efficient dissociation of this
particular chelate was observed under light irradiation.7
In the present system a hindered bipyridine fragment has been incorporated into a macrocycle and the ruthenium(II)-containing unit includes two 1,10-phenanthroline (phen) fragments. Ru(phen)22+ is a stable unit which will not be affected by light irradiation. The principle of the photochemically driven motion and the thermal backward reaction is shown in Scheme
1.
 | ||
| Scheme 1 Principle of the photochemically driven motion and the thermal backward reaction. | ||
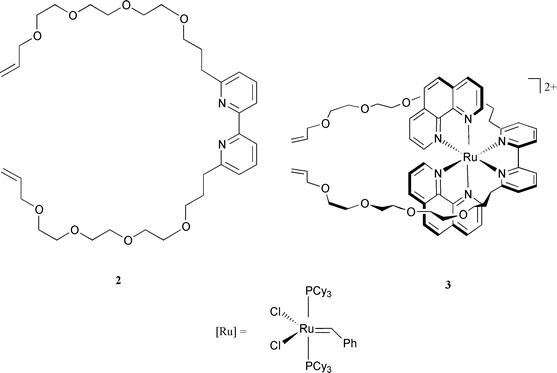
The macrocyclic complex 1 was synthesized from the ruthenium(II) precursor 3 incorporating a 2,2′-bipy ligand substituted at its 6 and 6′ positions by two long chains bearing terminal olefins (see Scheme 2). Compound 2 was prepared in 5 s teps from dmbp8 and its synthesis will be reported elsewhere. The precursor complex 3 was obtained in good yield (75%) from 2 and Ru(phen)2(CH3CN)22+ by heating a stoichiometric mixture of them in ethylene glycol at 140°C for 2 hours. 3 was isolated as its PF6− salt. It is obtained pure as a red powder after column chromatography. The 1H NMR spectrum is in accordance with its structure. The next step consists of cyclizing the two terminal functions of the co-ordinated bipy. Ring-closing metathesis of olefins (RCM), recently proposed by Grubbs and his group,9 has been used in numerous cases, most of the time with remarkable success.10 This catalytic reaction is compatible with a large variety of functions and, in particular, with transition metal complexes such as copper(I) or iron(II) polyimine compounds.11,12 In the present case the RCM reaction turned out to be equally efficient since a 67% yield of the desired complex 1 was obtained, without utilizing high dilution conditions (0.01 M dichloromethane solution of 3, room temperature, 5 days, 7% of ruthenium catalyst [Ru]). The reaction can easily be monitored by 1H NMR since the characteristic signals of the terminal olefins constitute a convenient probe. Gradually, these two sets of signals (δ 5.18 and 5.04 in acetone-d6) are replaced by those corresponding to the cyclic olefin of 1 (5.31 and 5.39), obtained as a mixture of cis and trans isomers. The mass spectrum (FAB-MS) of 1 confirms the expected structure (M − PF6, m/z 1195.3).
 | ||
| Scheme 2 The precursors 2 and 3 of the ruthenium complex 1. | ||
The photochemical behaviour of complex 1 has been examined by both UV-Visible and 1H NMR spectroscopy in acetonitrile. Under light irradiation (λ>300 nm), it undergoes an efficient and quantitative photolabilization of the macrocycle m36
as demonstrated in Fig. 1 by the different visible spectra recorded during the progress of the reaction. After 6 min of irradiation there is no change in the electronic spectrum and curve
10 is characteristic of the Ru(phen)2(CH3CN)22+ species.13
Also, two well resolved isosbestic points at 342 and 406
nm demonstrate the selectivity of the process. By analysing
the absorbance variations s. time, an apparent rate constant can be deduced (t1/2
= 75 s). The efficiency of this photochemical reaction is less than that of a previously studied system7 in which a Ru(phen)2 core is associated to dmbp itself (t1/2
= 28 s under the same conditions). Since the steric hindrance is more prononced in 1 than in Ru(phen)2dmbp2+, it seems that some deco-ordination steps of the chelate unit are probably slower due to the presence of the macrocyclic chain. The cyclic nature of the complex is also expected to favour a cis conformation for the bipy unit, making its
stepwise decomplexation slightly more difficult than with its unconstrained analogue, dmbp.
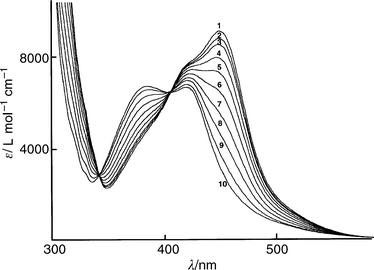 | ||
| Fig. 1 Electronic spectra of acetonitrile solution of complex 1 after visible light irradiation: 1(0); 2(6); 3(12); 4(30); 5(40); 6(60); 7(90); 8(130); 9(192); 10(370 s). | ||
Similar results could be obtained using 1H NMR spectroscopy with direct light irradiation of the NMR tube. The characteristic spectrum of complex 1 is gradually replaced by the sum of the spectra of Ru(phen)2(CD3CN)22+ and free macrocyclic m36 .
The thermal back reaction was also proved to be quantitative. An equimolecular mixture of Ru(phen)2(CH3CN)22+ and m36 leads back to the starting complex 1 in quantitative yield (refluxing ethylene glycol; two hours).
These first results are promising for the construction of photomechanical devices in which the macrocycle m36 and the Ru(phen)2 unit will be respectively the ring and the core of the string of a rotaxane.
Experimental
Characterization of 1, 2 and 3
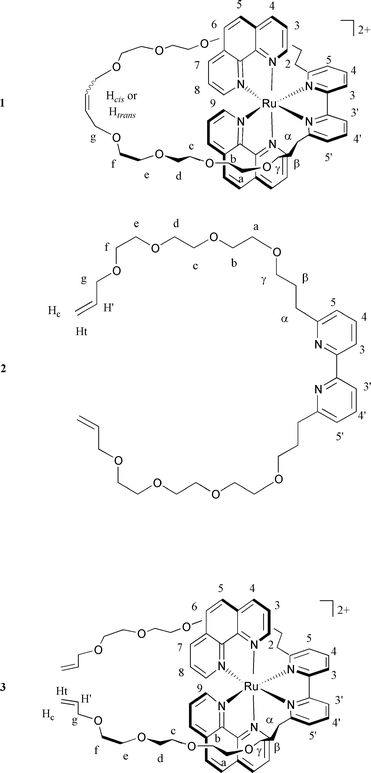
1: 1H NMR (acetone-d6 , 200 MHz) δ 8.94 (dd, 2 H, H4, J = 8.36 and 1.24) 8.76 (dd, 2 H2, J = 5.41 and 1.23), 8.68 (dd, 2 H, H7, J = 8.37 and 1.23), 8.62 (d, 2 H, H3,3′ , J = 7.38), 8.47 (d, 2 H, H5, J = 8.86), 8.38 (d, 2 H, H6, J = 8.86), 8.09 (dd, 2 H, H4,4′ , J = 8.12 and 7.14), 8.09 (dd, 2 H, H3), 7.92 (dd, 2 H, H9, J = 5.29 and 1.11), 7.67 (dd, 2 H, H8, J = 5.42 and 8.12), 7.43 (d, 2 H, H5,5′, J = 7.88 Hz), 5.39 (m, 2 H, Hcis, 75%), 5.31 (m, 2 H, Htra ns, 25%), 3.90–1.50 (m, 40 H, Hg, Ha,b,c,d,e,f, Hα,β,γ); FAB-MS: m/z = 1195.3, [M − PF6]+, calc. 1195.35, 8%; 1049.3, [M − 2 PF6 + e−]+, calc. 1050.38, 11%; 462.0 [M − 2 PF6-m36 + e−]+, calc. 462.04, 32%. UV-Vis (CH3CN) λ 450 nm (ε 9200 L mol−1 cm−1).
2: 1H NMR (CDCl3, 200 MHz) δ 8.24 (d, 2 H, H3,3′, J = 7.86), 7.68 (t, 2 H, H4,4′, J = 7.75), 7.14 (dd, 2 H, H5,5′, J = 7.62 and 0.98), 5.91 (ddt, 2 H, H′, J = 17.22, 10.32 and 5.66), 5.26 (dtd, 2 H, Ht, J = 17.10, 3.44 and 1.60), 5.16 (m, 2 Hc, J = 10.32, 1.72 and 1), 4.01 (dt, 4 H, Hg, J = 5.66 and 1.36), 3.67–3.50 (m, 28 H, Ha,b,c,d,e,f, 4 Hγ), 2.92 (t, 4 H, Hα, J = 7.50), 2.11 (tt, 4 Hβ, J = 6.52 and 6.88 Hz); FAB-MS: m/z = 617.5, [M + H]+, calc. 617.38, 100%.
3: 1H NMR (acetone-d6 , 200 MHz) δ 8.94 (dd, 2 H, H4, J = 8.15 and 1.10), 8.74 (dd, 2 H, H2, J = 5.17 and 1.22), 8.67 (dd, 2 H, H7, J = 8.35 and 1.22), 8.63 (d, 2 H, H3,3, J = 8.86), 8.47 (d, 2 H, H5, J = 8.86), 8.38 (d, 2 H, H6, J = 8.86), 8.10 (dd, 2 H, H4,4′, J = 7.86 and 7.88), 8.10 (dd, 2 H, H3, J = 5.23 and 8.24), 7.97 (dd, 2 H, H9, J = 5.40 and 1.23), 7.62 (dd, 2 H, H8, J = 5.28 and 8.24), 7.44 (dd, 2 H, H5,5′ , J = 7.87 and 0.99), 5.82 ( ddt, 2 H, H′, J = 17.34, 10.44 and 5.23), 5.18 (m, 2 H, Ht), 5.04 (m, 2 H, Hc), 3.88 (dt, 4 H, Hg, J = 5.42 and 1.54 Hz), 3.60–3.20 (m, 24 H, Ha,b,c,d,e,f), 3.20–1.00 (12 H, Hα,β,γ).
Photoirradiation was performed in a quartz UV cell (P = 1.0 cm) or in a NMR tube (diameter = 5.0 mm) by the use of a Hanimex slide projector (150 W halogen lamp).
Acknowledgements
This work was supported by the French CNRS . We thank European Commission COST programme D11/0004/98 and the French Ministry of Education, Research and Technology for a fellowship to A-CL.Notes and references
- V. Balzani, M. Gomez-Lopez and J. F. Stoddart, Acc. Chem. Res., 1998, 31, 405 CrossRef CAS; J.-P. Sauvage, Acc. Chem. Res., 1998, 31, 611 CrossRef CAS; L. Fabbrizzi, M. Licchelli and P. Pallavicini, Acc. Chem. Res., 1999, 32, 846 CrossRef CAS; T. R. Kelly, H. De Silva and R. A. Silva, Nature (London), 1999, 401, 150 CrossRef CAS; for a photochemical organic system see: N. Koumura, R. W. J. Zijlstra, R. A. van Delden, N. Harada and B. L. Feringa, Nature (London), 1999, 401, 152. Search PubMed.
- R. A. Bissel, E. Còrdova, A. E. Kaifer and J. F. Stoddart, Nature (London), 1994, 369, 133 CrossRef; A. Livoreil, C. O. Dietrich-Bucheker and J.-P. Sauvage, J. Am. Chem. Soc., 1994, 116, 9399 CrossRef CAS; L. Zelikovich, J. Libman and A. Shanzer, Nature (London), 1995, 374, 790 CrossRef CAS.
- R. Ballardini, V. Balzani, M. T. Gandolfi, L. Prodi, M. Venturi, D. Philp, H. G. Ricketts and J. F. Stoddart, Angew. Chem., Int. Ed. Engl., 1993, 32, 1301 CrossRef; A. Livoreil, J.-P. Sauvage, N. Armaroli, V. Balzani, L. Flamigni and B. Ventura, J. Am. Chem. Soc., 1997, 119, 12114 CrossRef CAS; N. Armaroli, V. Balzani, J.-P. Collin, P. Gaviña, J.-P. Sauvage and B. Ventura, J. Am. Chem. Soc., 1999, 121, 4397 CrossRef CAS.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. Von Zelewsky, Coord. Chem. Re., 1988, 84, 85 Search PubMed;
V. Balzani and F. Scandola, Supramolecular Photochemistry, Ellis Horwood, Chichester, 1991;
Search PubMed; J.-P. Sauvage, J.-P. Collin, J.-C. Chambron, S. Guillerez, C. Coudret, V. Balzani, F. Barigelletti, L. De Cola and L. Flamigni, Chem. Re., 1994, 94, 993 Search PubMed.
- P. E. Hoggaed and G. B. Porter, J. Am. Chem. Soc., 1978, 100, 1457 CrossRef CAS; B. Durham, J. V. Caspar, J. K. Nagle and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4803 CrossRef CAS; B. Durham, J. L. Walsh, C. L. Carter and T. J. Meyer, Inorg. Chem., 1980, 19, 860 CrossRef CAS; P. J. Steel, F. Lahousse, D. Lerner and C. Marzin, Inorg. Chem., 1983, 22, 1488 CrossRef CAS; B. E. Buchanan, P. Degn, J. M. Pavon Velasco, H. Hughes, B. S. Creaven, C. Long, J. G. Vos, R. A. Howie, R. Hage, J. H. van Diemen, J. G. Haasnoot and J. Reedijk, J. Chem. Soc., Dalton Trans., 1992, 1177 RSC; B. E. Buchanan, H. Hughes, J. H. van Diemen, R. Hage, J. G. Haasnoot, J. Reedijk and J. G. Vos, J. Chem. Soc., Chem. Commun., 1991, 300 RSC.
- A. von Zelewsky and G. Gremaud, Hel. Chim. Acta, 1988, 71, 1108 Search PubMed; H. Hichida, S. Tachiyashiki and Y. Sasaki, Chem. Lett., 1989, 1579.
- A.-C. Laemmel, J.-P. Collin and J.-P. Sauvage, Eur. J. Inorg. Chem., 1999, 383 CrossRef CAS.
- G. R. Newkome, D. C. Pantaleo, W. E. Puckett, P. L. Ziefle and W. A. Deutsch, J. Inorg. Nucl. Chem., 1981, 43, 1529 Search PubMed.
- (a) P. Schwab, M. B. France, J. W. Ziller and R. H. Grubbs, Angew. Chem., Int. Ed. Engl., 1995, 34, 2039 CrossRef CAS; (b) P. Schwab, R. H. Grubbs and J. W. Ziller, J. Am. Chem. Soc., 1996, 118, 100 CrossRef CAS.
- A. Fürstner and N. Kindler, Tetrahedron Lett., 1996, 37, 7005 CrossRef; B. König and C. Horn, Synlett, 1996, 1013 CrossRef; M. J. Marsella, H. D. Maynard and R. H. Grubbs, Angew. Chem., Int. Ed. Engl., 1997, 36, 1101 CrossRef CAS; Z. Yang, Y. He, D. Vourloumis, M. Vallberg and K. C. Nicolaou, Angew. Chem., Int. Ed. Engl., 1997, 36, 166 CrossRef CAS.
- M. Weck, B. Mohr, J.-P. Sauvage and R. H. Grubbs, J. Org. Chem., 1999, 64, 5463 CrossRef.
- C. O. Dietrich-Buchecker, G. Rapenne and J.-P. Sauvage, Chem. Commun., 1997, 2053 RSC; G. Rapenne, C. O. Dietrich-Buchecker and J.-P. Sauvage, J. Am. Chem. Soc., 1999, 121, 994 CrossRef CAS.
- P. Bonneson, J. L. Walsh, W. T. Pennington, A. W. Cordes and B. Durham, Inorg. Chem., 1983, 22, 1761 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2001 |