Controlled dethreading/rethreading of a scorpion-like pseudorotaxane and a related macrobicyclic self-complexing system
Vincenzo Balzani*a, Paola Ceronia, Alberto Credia, Marcos Gómez-Lópezb, Christoph Hamersb, J. Fraser Stoddart*c and Reinhard Wolfb
aDipartimento di Chimica “G. Ciamician”, Uni![[italic v]](https://www.rsc.org/images/entities/char_e0f5.gif) ersità di Bologna, ia Selmi 2, 40126, Bologna, Italy. E-mail: vbalzani@ciam.unibo.it
ersità di Bologna, ia Selmi 2, 40126, Bologna, Italy. E-mail: vbalzani@ciam.unibo.it
bSchool of Chemistry, The Uniersity of Birmingham, Edgbaston, Birmingham, UK B15 2TT
cDepartment of Chemistry and Biochemistry, Uniersity of California, 405 Hilgard Aenue, Los Angeles, California 90095-1569, USA. E-mail: stoddart@chem.ucla.edu
First published on 15th December 2000
Abstract
A scorpion-like pseudorotaxane (1·H2+), composed of a macrocyclic polyether containing a 1,5-dioxynaphthalene (1,5-DON) and a 1,3-dioxybenzene (1,3-DOB) electron-donor unit, the latter bearing a 4,4′-bipyridinium electron-acceptor tail, underwent threading/dethreading motions under control of (i) solvent polarity and (ii) solution acidity. Furthermore, the complexation of 1 ·H2+ with the trans-1,2-bis(1-benzyl-4-pyridinio)ethylene electron acceptor can be acid/base controlled. The spectroscopic properties of a related macrobicyclic compound 24+ (comprised of an electron-donor and an electron-acceptor macrocycle that share a benzene ring) have also been investigated. The results obtained for 24+ are consistent with the presence of intramolecular self-complexed species where the electron-donor macrocycle is threaded through the electron-acceptor one. Electrochemical experiments confirm the self-complexing structures of 1·H2+ and 24+. It seems likely that both compounds undergo decomplexation upon electrochemical stimulation.
Introduction
Much attention is currently being devoted to molecular-level systems in which the relative positions of the component parts can change as a result of some external stimulus.1,2 Such systems could find applications for information processing at the molecular level3 and can also be viewed as the forerunners of nanoscopic machines.1g Appealing examples of chemical compounds where controlled mechanical movements can take place are suitably designed pseudorotaxanes, rotaxanes, and catenanes.4 It has been shown that, in these systems, the relative displacements of the component parts (i) can be induced by chemical energy, electrical energy and light, and (ii) can be monitored by a variety of techniques including NMR, absorption and emission spectroscopy, and electrochemistry.1a–c,1e–g,2c–fConventional pseudorotaxanes4 are made of two covalently disconnected components, namely a macrocycle and a thread (Fig. 1a), which contain complementary recognition sites. The threading and dethreading motions can be controlled by an external stimulus capable of switching on/off one of the recognition sites.5 In the dethreaded state each of the two components, the macrocycle and the thread, are free to move independently in solution. Such a “disordered” situation can nevertheless lead to well defined properties for the supramolecular system, e.g. all the free threads are luminescent whereas the threaded ones are not.5,6 Therefore, the threading/dethreading process can exhibit binary logic behavior. It has indeed been shown that suitably designed pseudorotaxanes can perform as molecular-level logic gates.6
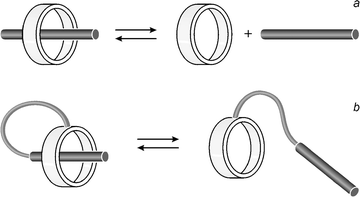 | ||
| Fig. 1 Schematic representations of (a) a conventional pseudorotaxane and (b) a self-complexing molecule in which a macrocycle is linked by a covalent tether to a tail containing a recognition unit. | ||
From the machine-like viewpoint, however, the molecular level structure and the movement of the component parts should be spatially well defined. For pseudorotaxane-type systems this goal can be reached in a molecular context by connecting7 (Fig. 1b) the macrocycle and the thread by a covalent tether. The result is a self-complementary molecule.8
Several examples of compounds consisting of a macrocyclic head and a tail that contain complementary recognition sites have recently been studied.7,9–11 In this paper we report the results of an investigation on the dethreading/rethreading motions of compound 1·H2+ and on the self-complexation of the related macrobicyclic compound 24+, which is comprised of electron-donor and electron-acceptor macrocycles sharing a benzene ring (Fig. 2).
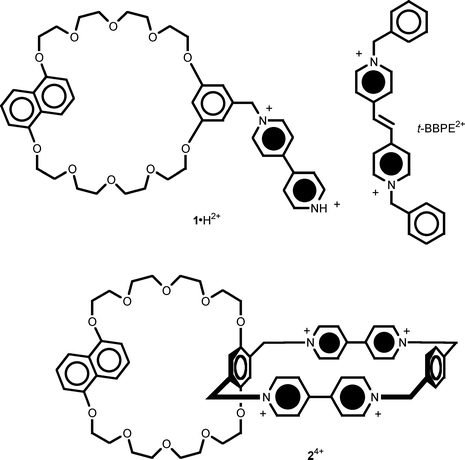 | ||
| Fig. 2 Structural formulae of the investigated compounds 1·H2+ and 24+, as well as of the t-BBPE2+ species that has been used in the experiment described in Fig. 8. | ||
Experimental
Materials
The synthesis and characterization of compounds 1 ·H2+9b and 24+9a (Fig. 2) as their PF6− salts have been reported previously. The trans-1,2-bis(1-benzyl-4-pyridinio)ethylene dication12 (t-BBPE2+, Fig. 2), and model compounds 1,5-dimethoxynaphthalene (1,5-DMN),133·2PF6,144,125·4PF614 and 6·4PF613 (Fig. 3) were all available from previous investigations. 2,6-Lutidine (2,6-dimethylpyridine), tributylamine (n-Bu3N) and trifluoroacetic acid were of the highest commercial purity used without further purification.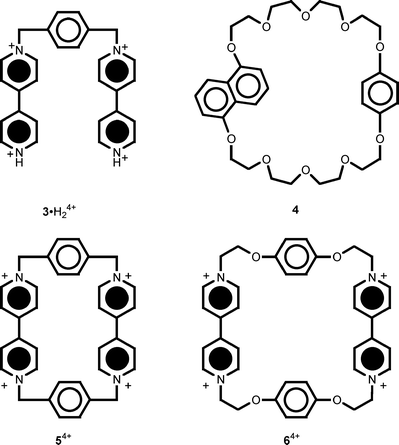 | ||
| Fig. 3 Structural formulas of the model compounds 3 ·H24+, 4, 54+ and 64+ for the electroactive units of compounds 1·H2+ and 24+. | ||
Spectroscopic measurements
Absorption and luminescence measurements were carried out at room temperature in MeCN or CH2Cl2 (Merck UvasolTM) solutions, or in mixtures of such solvents, with concentrations ranging from 2 × 10−3 to 3 × 10−5 mol L−1. UV-Vis absorption spectra were recorded with a Perkin-Elmer λ16 spectrophotometer, using quartz cells with pathlength 0.2, 0.5, 1.0 or 5.0 cm. Luminescence spectra were obtained with a Perkin-Elmer LS-50 spectrofluorimeter, equipped with a Hamamatsu R928 phototube, on air-equilibrated solutions. Right-angle excitation with 1 cm × 1 cm square quartz cuvettes and front-face excitation with triangular quartz cells were employed for luminescence experiments carried out on dilute and concentrated solutions, respectively. 1,5-DMN (Φ = 0.38, τ = 7.5 ns in aerated MeCN)13 was used as a luminescence standard in relative quenching experiments. In these experiments excitation was performed at an isosbestic wavelength for the sample and the standard; when this approach was not possible, corrections for differences in the excitation conditions were applied.15 Luminescence spectra at 77 K were recorded on a CH2Cl2–MeOH 1:1 v/v rigid matrix contained in a quartz tube which, in turn, was immersed in a quartz Dewar, filled with liquid nitrogen. Luminescence lifetimes were measured by time-correlated single-photon counting with Edinburgh Instruments DS199 equipment (D2 lamp, λex = 300 nm). Experimental errors: wavelengths, ±1 nm; molar absorption coefficients, and luminescence intensities and lifetimes, 15%.Electrochemical experiments
Conventional electrochemical experiments were carried out in argon-purged CH2Cl2 or MeCN (Romil Hi-DryTM) solutions at room temperature or at 228 K (by employing a liquid N2/EtOH low-temperature bath) with an EcoChemie Autolab 30 multipurpose instrument interfaced to a personal computer. In the cyclic voltammetry (CV) and differential pulse voltammetry (DPV) experiments the working electrode was a glassy carbon electrode (0.08 cm2, Amel); its surface was routinely polished with a 0.05 μm alumina–water slurry on a felt surface, immediately prior to use. In all cases the counter electrode was a platinum spiral, separated from the bulk solution with a fine glass frit, and a silver wire was employed as a quasi-reference electrode (QRE). The potentials reported are referred to the SCE by measuring the AgQRE potential with respect to ferrocene. The concentration of the compounds examined was of the order of 5 × 10−4 mol L−1; 0.1 mol L−1 tetraethylammonium hexafluorophosphate (TEAPF6) and tetrabutylammonium hexafluorophosphate (TBAPF6) were added to MeCN and CH2Cl2 solutions, respectively, as supporting electrolytes. Cyclic voltammograms were obtained with scan rates in the range 0.05–10 V s−1; DPV experiments were performed with a scan rate of 0.02 V s−1, a pulse height of 75 mV, and a duration of 40 ms. The number of electrons exchanged in each process was estimated by comparing the current intensity of the corresponding CV wave with that observed for the monoelectronic oxidation of ferrocene, after correction for differences in the diffusion coefficients.16 The experimental error on the potential values was estimated to be ±10 mV.Results and discussion
Solvent-driven dethreading/rethreading of compound 1·H2+
Compound 1·H2+ (Fig. 2) is composed of a macrocyclic polyether containing a 1,5-dioxynaphthalene (1,5-DON) and a 1,3-dioxybenzene (1,3-DOB) electron-rich unit, the latter bearing a covalently linked 4,4′-bipyridinium electron-deficient tail. As its PF6− salt, 1 ·H2+ is highly soluble in MeCN, moderately soluble in CH2Cl2 (at least up to 2 × 10−3 mol L−1), and is thermally stable in both solvents.In very dilute solutions (3 × 10−5 mol L−1) the absorption spectrum of compound 1 ·H2+ in MeCN at 298 K (Fig. 4, curve a) shows only a very weak tail (ε<70 L mol−1 cm−1) in the visible region and it is very similar to the sum of the spectra of its chromophoric units, namely 1,5-DON, 1,3-DOB, and 4,4′-bipyridinium. On increasing concentration, however, the structure of the 1,5-DON band is lost, the intensity of the tail in the visible spectral region increases and the solution becomes colored. For a 2 × 10−3 mol L−1 solution a band with maximum at 480 nm (ε = 150 L mol−1 cm−1) can be observed (Fig. 4, curve b).
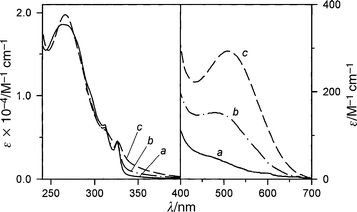 | ||
| Fig. 4 Absorption spectrum at 298 K of compound 1 ·H2+ in: (a) 3 × 10−5 mol L−1 MeCN solution; (b) 2 × 10−3 mol L−1 MeCN solution; (c) 3 × 10−5 mol L−1 CH2Cl2 solution. | ||
The absorption spectrum of compound 1·H2+ in CH2Cl2 solution is similar, but not identical, to that observed in concentrated MeCN solution (Fig. 4, curve c). In this case, however, the spectrum does not depend on the concentration. The most important feature is the presence of an absorption band in the visible region (λmax = 510 nm, ε = 300 L mol−1 cm−1), responsible for the pink color of the solution.
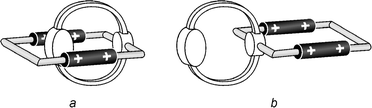
The different results obtained for MeCN and CH2Cl2 solutions can be accounted for as follows. The absorption bands observed in the visible region are typical5c,7a,12 of the charge-transfer (CT) interaction between bipyridinium dications and 1,5-DON. In CH2Cl2 the dicationic bipyridinium unit is self-complexed with the macrocyclic part of the molecule, whose oxygen atoms offer a much more favorable environment than the non-polar solvent molecules (1,1′-dimethyl-4,4′-bipyridinium is highly insoluble in CH2Cl2). Although intermolecular complexation is possible, evidence for intramolecular complexation is given by the fact that the absorption spectrum does not change with concentration.17 Inspection of CPK (Corey-Pauling-Koltun) space-filling molecular models shows that the bipyridinium tail can easily be accommodated inside the macrocycle. A threaded structure (Fig. 5a) seems therefore more likely than a side-on one (Fig. 5b) because the former facilitates formation of additional [C–H···O] hydrogen bonds between the α bipyridinium hydrogen atoms and some of the polyether oxygen atoms.18
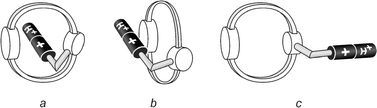 | ||
| Fig. 5 Cartoons representing the threaded (a), side-on (b) and dethreaded (c) conformations of compound 1·H2+. | ||
In MeCN the dicationic bipyridinium moiety is well solvated by the polar solvent molecules, so that, in dilute solutions, the more stable isomer of compound 1·H2+ is the decomplexed one (Fig. 5c). This suggestion is supported by the low values of association constants between aromatic crown ethers and 1,1′-dimethyl-4,4′-bipyridinium in MeCN solution.19 The weak absorption tail in the visible region observed under such conditions (Fig. 4, curve a) can be assigned to a through-bond CT interaction between the 1,3-DOB moiety of the macrocycle and the appended bipyridinium unit. As the concentration increases, however, intermolecular complexation takes place and a CT band arising from the interaction between the 1,5-DON and bipyridinium units appears.
Evidence for the presence of decomplexed species in dilute MeCN and self-complexed ones in CH2Cl2 is also provided by the fluorescence measurements. Although the intense fluorescence band characteristic of the 1,5-DON unit (λmax = 328 nm, τ = 7.5 ns in aerated MeCN)13 is strongly quenched,20,21 in both solvents, in CH2Cl2 solution the fluorescence intensity quenching is ten times larger than that observed in dilute MeCN solution. This observation suggests that, in CH2Cl2 solution, compound 1·H2+ adopts a conformation that maximizes the quenching of the potentially luminescent 1,5-DON unit, as would be the case when the bipyridinium tail is threaded (Fig. 5a) in the crown ether ring.22 Addition of MeCN to a 5 × 10−5 mol L−1 CH2Cl2 solution of 1·H2+ causes the disappearance of the visible absorption band and an increase in the fluorescence intensity (Fig. 6), showing that the larger fluorescence quenching observed in CH2Cl2 solution is indeed related to the conformation which is responsible for the presence of the CT absorption band in the visible region. Concentrated (1 × 10−3 mol L−1) MeCN solutions of 1·H2+ show a much stronger fluorescence quenching20 than dilute (2 × 10−5 mol L−1) ones, as expected because of the intermolecular complexation (see above).
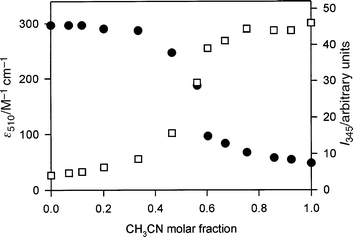 | ||
| Fig. 6 Changes in the absorbance at 510 nm (●) and in the fluorescence intensity at 345 nm (λex = 295 nm) (□) upon addition of MeCN to a 5 × 10−5 mol L−1 CH2Cl2 solution of compound 1·H2+ at 298 K. | ||
In conclusion, the conformation of compound 1 ·H2+ is solvent dependent:23 the dethreaded conformation is the more abundant one in very dilute solutions of the polar solvent MeCN and the self-threaded18 one is preferred in the non-polar solvent CH2Cl2. Since the conformational change is accompanied by a visible color change and by a change in the fluorescence intensity, 1·H2+ can be viewed as an optical sensor for solvent polarity.
Base/acid-driven dethreading/rethreading of compound 1·H2+
We have shown above that, in CH2Cl2 solution, 1·H2+ is present in a self-complexed (threaded)18 conformation (Fig. 5a). On addition of tributylamine (1 equivalent) or 2,6-lutidine (ca. 4 equivalents) the CT band at 510 nm completely disappears and the structured bands at 311 and 326 nm, characteristic of the uncomplexed 1,5-DON unit, are recovered. Furthermore, the fluorescence intensity of the 1,5-DON unit partially revives (ca. 2.5 times enhancement). Identical titration curves are obtained by plotting the decrease in absorbance of the CT band and the increase in the fluorescence intensity of the 1,5-DON unit![[italic v]](https://www.rsc.org/images/entities/i_char_e0f5.gif) s. amine equivalents. This process
can be reversed quantitatively by adding an excess of trifluoroacetic acid (ca. 6 equivalents with respect to the added amine).
s. amine equivalents. This process
can be reversed quantitatively by adding an excess of trifluoroacetic acid (ca. 6 equivalents with respect to the added amine).These results can be accounted for as shown in Fig. 7. Upon addition of a base the terminal pyridinium ring of compound 1·H2+ undergoes deprotonation, so that the tail becomes a monocationic 4-pyridyl(4-pyridinium) unit. Since a pyridyl unit is not a good electron acceptor, while a pyridinium unit is, this tail is a much poorer electron acceptor than the protonated tail, so that the original CT interaction with the electron donor macrocycle is disabled. At the same time deprotonation makes the tail much more soluble in the non-polar CH2Cl2 solvent. As a consequence, deprotonation causes dethreading, while addition of acid directs the system back to the threaded conformation.18,24
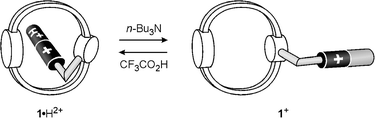 | ||
| Fig. 7 Base/acid-driven dethreading/rethreading of compound 1·H2+. | ||
Base/acid control of the complexation of an external guest by compound 1·H2+
The base/acid driven dethreading/rethreading cycle (see above) has been used to trigger the complexation of an external guest, namely the trans-1,2-bis(1-benzyl-4-pyridinio)ethylene electron acceptor (t-BBPE2+, Fig. 2), into the macrocyclic cavity of compound 1+. In CH2Cl2 solution t-BBPE2+ forms a stable (Kstabca. 6.5 × 104 L mol−1, as determined from fluorescence titrations) CT adduct with the crown ether 1,5-dinaphtho-38-crown-10 (1,5DN38C10), which contains the same 1,5-DON donor unit that is present in 1·H2+. By analogy with many other similar systems,4a, 5c,6a,12,19 this adduct has most likely a pseudorotaxane-type structure. The formation of the adduct can be evidenced by (i) the modification of the absorption spectrum with respect to the sum of the spectra of the com ponents, and (ii) the quenching of the fluorescence characteristic of 1,5DN38C10.The absorption spectrum of a CH2Cl2 solution containing 3.2 × 10−4 mol L−1 compound 1·H2+ and 9 × 10−6 mol L−1 t-BBPE2+ (whose solubility in CH2Cl2 is very low) matches exactly the sum of the spectra of the isolated components in the same concentration. After addition of 6.4 × 10−4 mol L−1 tributylamine the absorption spectrum becomes different from the sum of the spectra of the components; in particular, a decrease in the intensity of the absorption bands characteristic of the 1,5-DON unit (311 and 326 nm) and of t-BBPE2+ (340–350 nm), and the appearance of an absorption tail in the 350–380 nm region, are observed. These changes are the same as those observed in the absorption spectrum of the solution containing 1,5DN38C10 and t-BBPE2+. Owing to the lack of suitable luminescence signals,25 it is difficult to assess the amount of t-BBPE2+ threaded through 1+. A rough calculation done on the basis of the absorption tail intensity at 370 nm, compared with that of the 1,5DN38C10/t-BBPE2+ system, suggests that t-BBPE2+ is quantitatively engaged by 1+. Evidence of strong complexation comes also from a comparison of low-temperature emission spectra. At 77 K, t-BBPE2+ shows an intense and structured fluorescence band that can no longer be seen in frozen solutions which also contain, besides 9 × 10−6 mol L−1 t-BBPE2+, 3.2 × 10−4 mol L−11·H2+ and 6.4 × 10−4 mol L−1 tributylamine. This behavior can be explained by the fact that the fluorescence is quenched by low-lying CT levels when t-BBPE2+ is encapsulated within the macrocyclic crown ether. Although it is difficult to push this comparison to a fully quantitative level, there is evidence that a large amount of t-BBPE2+ is complexed by 1+.
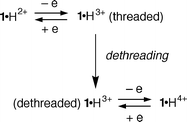
The results can be rationalized as schematized in Fig. 8. t-BBPE2+ is a poorer electron acceptor than the 4,4′-bipyridinium unit;12 moreover, the latter is entropically advantaged, as it is covalently linked to the host crown ether. Thus, t-BBPE2+ cannot dethread compound 1·H2+. When tributylamine is added 1·H2+ undergoes deprotonation and then dethreading (see above) so that the external guest can enter the open macrocyclic cavity. This process can be reversed by addition of 2.5 × 10−3 mol L−1 CF3CO2H: the terminal pyridine unit of 1+ is protonated and the 1 ·H2+ species undergoes self-threading, pushing t-BBPE2+ out of the macrocycle cavity. In conclusion, amines can open and protons can close the ‘door’ of the macrocyclic cavity of 1·H2+, thereby allowing or preventing the threading of an external guest.
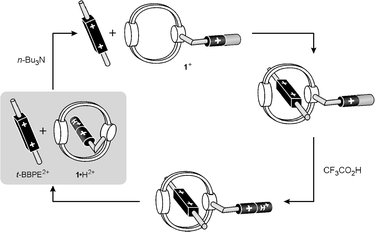 | ||
| Fig. 8 Base/acid control of the complexation of t-BBPE2+ by compound 1+. | ||
Electrochemical behavior of compound 1·H2+
The electrochemical behavior of compound 1·H2+ was investigated by cyclic and differential pulse voltammetry in MeCN and CH2Cl2. The potentials of the various processes are gath ered in Table 1, where the values of some reference compounds (Fig. 3) are also reported for comparison purposes.| Compound | Solvent | E/V s. SCEa | |||
|---|---|---|---|---|---|
| a Halfwave potential values (E1/2), unless otherwise noted.b Chemically irreversible process; Ep value at scan rate of 0.2 V s−1.c Two-electron transfer process.d After addition of tributylamine in a stoichiometric amount with respect to the number of protonated 4-pyridyl groups.e After addition of tributylamine and subsequent treatment with trifluoroacetic acid in a stoichiometric amount with respect to the previously added amine.f At scan rates higher than 1 V s−1 this process gains reversibility (E1/2 = +1.20 V).g Four-electron transfer process. | |||||
| 1·H2+ | CH2Cl2 | +1.45bc | −0.40 | −0.82 | |
| 1+d | CH2Cl2 | +1.28bc | −0.67b | −0.82 | |
| 1·H2+e | CH2Cl2 | +1.51bc | −0.42 | −0.72 | |
| 1·H2+ | MeCN | +1.31cf | −0.42 | −0.80 | |
| 1+d | MeCN | +1.28cf | −0.86c | ||
| 1·H2+e | MeCN | +1.30cf | −0.44 | −0.78 | |
| 3·H24+ | MeCN | −0.43c | −0.79c | ||
| 32+d | MeCN | −0.82g | |||
| 3·H24+e | MeCN | −0.47c | −0.80c | ||
| 4 | CH2Cl2 | +1.48b | +1.28b | ||
| 4 | MeCN | +1.32b | +1.16b | ||
Under the experimental conditions used (6.0 × 10−4 mol L−1), the 1 ·H2+ species adopts a self-threaded conformation in CH2Cl2 , while in MeCN most of the molecules are involved in intermolecular complexes, as evidenced by the spectroscopic investigation at different 1·H2+ concentrations (see above). In both solvents the experimental cyclic voltammetries show two reversible one-electron reduction processes, and a chemically irreversible two-electron oxidation process which becomes reversible only at scan rates higher than 1 V s−1 in MeCN solution.
By comparison with model compound 3·H24+ (Fig. 3, Table 1), which is soluble only in MeCN, we can assign the two reduction processes of 1·H2+ to the two successive reductions of the bipyridinium unit. After addition of a stoichiometric amount of tributylamine, the reduction potential is shifted toward more negative potentials, as expected for the pyridyl(pyridinium) unit of the deprotonated form 1+ (Table 1). It should be noted that, in MeCN solution, after the addition of the amine, both 1+ and the model compound 32+ show only one peak, which corresponds to the transfer of two electrons. This behavior can be explained tentatively by an ECE (electrochemical–chemical–electrochemical) mechanism, as illustrated in eqn. (1) for 1+. The monoreduced pyridyl(pyridinium) unit of 1 is protonated by the tributylammonium cation, leading to a 1·H+ species with a standard potential less negative than that of 1+. Addition of trifluoroacetic acid restores the initial electrochemical pattern, confirming the reversibility of the deprotonation process and the resistance of the system to base/acid stimulation, as observed in the spectroscopic studies.
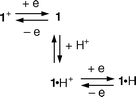 | (1) |
The two-electron oxidation process of compound 1·H2+ is likely to involve both the 1,5-DON and 1,3-DOB units. Crown ether 4 (Fig. 3, Table 1) is a satisfactory model for the 1,5-DON, but not for the 1,3-DOB, unit of 1·H2+ since (i) 1,3-dimethoxybenzene is oxidized at more positive potentials than 1,4-dimethoxybenzene,26 and (ii) the 1,3-DOB unit of 1·H2+ bears a 4,4′-bipyridinium substituent. The results show that the oxidation of the 1,5-DON unit of 1 ·H2+ occurs at more positive potentials than in the case of compound 4. This observation is fully consistent with the engagement of the 1,5-DON unit in CT interactions with the 4,4′-bipyridinium unit, and is consistent with the spectroscopic results (see above).
In CH2Cl2 solution it might be expected (eqn. 2) that the one-electron oxidized form 1·H3+ undergoes dethreading because of electronic repulsions and the disruption of CT interactions. Oxidation of the 1,3-DOB unit should therefore take place in the dethreaded structure, but the lack of suitable model compounds does not allow us to discuss this possibility.
 | (2) |
Spectroscopic properties of compound 24+
Compound 24+ (Fig. 2) is composed of two macrocycles sharing a benzene ring. As expected, this tetracationic species exhibits a very poor solubility in CH2Cl2 (<5 × 10−5 mol L−1). The spectroscopic properties are the same in CH2Cl2 and MeCN solutions.Inspection of CPK space-filling molecular models show that compound 24+ can undergo self-complexation, as indicated in Fig. 9. In the solid state, the 1,5-DON unit is indeed sandwiched between the two bipyridinium units, a structure which is stabilized by π–π stacking interactions and by [C–H···O] hydrogen bonds.9a NMR Spectroscopic analysis indicated that such a self-complexed structure is maintained in CD3CN solution at 233 K, and that a high temperature (309 K) rotation can occur about the –CH2–C6H2R2–CH2– axis of the disubstituted p-xylyl group.9a The absorption spectrum of 24+ in MeCN solution is shown in Fig. 10, together with the sum of the spectra of its chromophoric component units, taken as 1,1′-dibenzyl-4,4′-bipyridinium, 1,4-dimethoxybenzene and 1,5-dimethoxynaphthalene. The rather intense tail in the 320–400 nm region (ε = 2500 L mol−1 cm−1 at 350 nm) can be assigned to a through-bond CT interaction between the 1,4-DOB and 4,4′-bipyridinium units, whereas the much weaker and broad absorption band with a maximum at 560 nm compares well with the CT band found for pseudorotaxanes comprised of a 1,5-DON unit threaded through a tetracationic macrocycle containing two 4,4′-bipyridinium units.5c,7a These results confirm that the stable conformation of compound 24+ is the self-complexed one (Fig. 9a).
 | ||
| Fig. 9 Cartoon representation of the two possible conformations of compound 24+. | ||
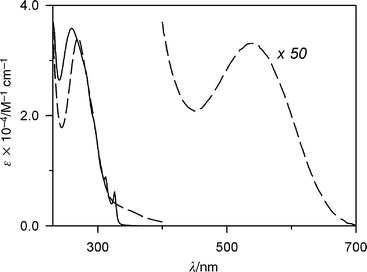 | ||
| Fig. 10 Absorption spectrum of compound 24+ (dashed line), and sum of the spectra of its chromophoric units, 4,4′-bipyridinium, 1,4-DOB and 1,5-DON (full line) in MeCN at 298 K. | ||
In compound 24+ the fluorescence of the 1,5-DON unit is strongly quenched (>200 times), with a residual emission that, by evaluation of its lifetime, can be assigned to small amounts of 1,5-dimethoxynaphthalene-type impurities.
Electrochemical control of the self-complexation process of compound 24+
The electrochemical behavior of compound 24+ could only be investigated by cyclic and differential pulse voltammetry in MeCN since this compound is not sufficiently soluble in CH2Cl2. By comparison with the behavior of compounds 54+ and 64+ (Fig. 3, Table 2), the two reversible reduction peaks of 24+ can be ascribed to successive reduction of the bipyridinium units. Each reduction involves the transfer of two electrons, thus evidencing that the two bipyridinium units of 24+ are electrochemically equivalent. The negative shift of the reduction processes of 24+, with respect to 54+, demonstrates the involvement of the bipyridinium units in CT interactions with the electron-donating 1,5-DON and 1,4-DOB groups, as evidenced by the spectroscopic investigations. The occurrence of these kinds of interactions between the bipyridinium and 1,4-DOB units of 24+ is also suggested by comparison with the behavior13 of macrocycle 64+. Although the lack of a good model compound for the tetracationic part of the molecule prevents us from entering into a detailed discussion of the results, it is reasonable6b,7a to assume that the injection of two electrons into 24+ weakens the CT interaction responsible for self-complexation, thus leading to escape of the 1,5-DON unit from the macrocyclic cavity. Therefore, the successive two-electron reduction should occur on the uncomplexed species (Fig. 11a). An investigation of the cathodic region at 228 K reveals splitting of the first reduction peak. This result is consistent with the presence of two redox couples (complexed and decomplexed 24+/2+) that can be interconverted according to a square scheme.27| Compound | E/V s. SCEa | |||
|---|---|---|---|---|
| a Halfwave potential values (E1/2), unless otherwise noted.b Chemically irreversible process; Epa value at scan rate of 0.2 V s−1.c Two-electron transfer process.d At 228 K a splitting of this process is observed.e Data from ref. 12.f Data from ref. 13. | ||||
| 24+ | +1.62b | +1.50b | −0.40cd | −0.80c |
| 4 | +1.32b | +1.16b | ||
| 54+e | −0.29c | −0.71c | ||
| 64+f | +1.51bc | −0.35c | −0.80c | |
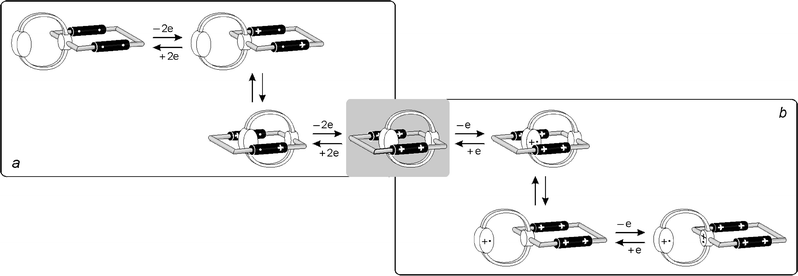 | ||
| Fig. 11 Scheme of the electrochemically controlled mechanical movements that can take place in compound 24+. | ||
In the anodic region compound 24+ shows two distinct one-electron chemically irreversible oxidation processes that have to involve the 1,5-DON and 1,4-DOB units. Although 4 is a good model for the 1,5-DON unit of 24+, it is not so good for the 1,4-DOB unit, which carries, in 24+, strong electron-acceptor substituents.28 The results show (Table 2) that oxidation of the 1,5-DON unit of 24+ occurs at much more positive potentials than in the case of compound 4. This observation is fully consistent with a self-complexed conformation (Fig. 9a), as suggested by the spectroscopic results (see above). The second oxidation process of 24+ can be assigned to its 1,4-DOB unit. The lack of good model compounds does not allow us to establish whether such an oxidation occurs in the original self-complexed conformation or in the decomplexed one. Clearly, in the one-electron oxidized form 25+, the presence of electrostatic repulsion and the disappearance of the CT interaction that stabilizes the self-complexed conformation of 24+ are expected to cause escape of the positively charged 1,5-DON unit from the cavity of the tetracationic macrocycle (Fig. 11b).
Conclusion
We have investigated the spectroscopic and electrochemical properties of the scorpion-like compound 1 ·H2+ and of the related macrobicyclic compound 24+, which is composed of two macrocycles sharing a benzene ring. Both compounds contain electron-donor and electron-acceptor units that promote CT interactions capable of stabilizing self-complexed structures (Figs. 5 and 9). We have shown that 1·H2+ can be dethreaded (i) by increasing the solvent polarity and (ii) by addition of a base. Both 1·H2+ and 24+ are also likely to undergo decomplexation/recomplexation upon electrochemical stimulation.Acknowledgements
We thank Professor Margherita Venturi for stimulating discussions. This research was supported in Bologna by MURST (Supramolecular Devices Project) and the University of Bologna (Funds for Selected Research Topics), and in Birmingham by the Engineering and Physical Sciences Research Council.References and notes
- (a) For reviews, see: V. Balzani, M. Gómez-López and J. F. Stoddart, Acc. Chem. Res., 1998, 31, 405 CrossRef CAS; (b) J.-P. Sauvage, Acc. Chem. Res., 1998, 31, 611 CrossRef CAS; (c) A. E. Kaifer, Acc. Chem. Res., 1999, 32, 62 CrossRef CAS; (d) L. Fabbrizzi, M. Licchelli and P. Pallavicini, Acc. Chem. Res., 1999, 32, 846 CrossRef CAS; (e) D. A. Leigh and A. Murphy, Chem. Ind., 1999, 178 Search PubMed; (f) M. D. Ward, Chem. Ind., 2000, 22 Search PubMed; (g) V. Balzani, A. Credi, F. M. Raymo and J. F. Stoddart, Angew. Chem., Int. Ed., 2000, 39, 3349 CrossRef.
- (a) For recent and representative examples, see: N. Koumura, R. W. J. Zijlstra, R. A. van Delden, N. Harada and B. L. Feringa, Nature (London), 1999, 401, 152 CrossRef CAS; (b) T. R. Kelly, H. De Silva and R. A. Silva, Nature (London), 1999, 401, 150 CrossRef CAS; (c) L. Raehm, J.-M. Kern and J.-P. Sauvage, Chem. Eur. J., 1999, 5, 3310 CrossRef CAS; (d) V. Balzani, A. Credi, S. J. Langford, R. M. Raymo, J. F. Stoddart and M. Venturi, J. Am. Chem. Soc., 2000, 122, 3542 CrossRef CAS; (e) V. Bermudez, N. Capron, T. Gase, F. G. Gatti, F. Kajzar, D. A. Leigh, F. Zerbetto and S. Zhang, Nature (London), 2000, 406, 608 CrossRef CAS; (f) P. R. Ashton, R. Ballardini, V. Balzani, A. Credi, R. Dress, E. Ishow, C. J. Kleverlaan, O. Kocian, J. A. Preece, N. Spencer, J. F. Stoddart, M. Venturi and S. Wenger, Chem. Eur. J., 2000, 6, 3558 CrossRef.
- D. H. Rouvray, Chem. Br., 1998, 34, 26 Search PubMed; C. P. Collier, E. W. Wong, M. Belohradsky, F. M. Raymo, J. F. Stoddart, P. J. Kuekes, R. S. Williams and J. R. Heath, Science, 1999, 285, 391 CrossRef CAS; V. Balzani, A. Credi and M. Venturi, in Stimulating Concepts in Chemistry, eds. M. Shibasaki, J. F. Stoddart and F. Vögtle, Wiley-VCH, Weinheim, 2000, p. 255 Search PubMed; C. P. Collier, G. Mattersteig, E. W. Wong, Y. Luo, K. Beverly, J. Sampaio, F. M. Raymo, J. F. Stoddart and J. R. Heath, Science, 2000, 289, 1172 Search PubMed; J. M. Tour, Acc. Chem. Res., 2000, 33, in press CrossRef CAS.
-
(a) D. B. Amabilino and J. F. Stoddart, Chem. Re., 1995, 95, 2725 Search PubMed;
(b) Molecular Catenanes, Rotaxanes and Knots, eds. J.-P. Sauvage
and C. Dietrich-Buchecker, Wiley-VCH, Weinheim, 1999. Search PubMed.
- (a) See, e.g.: P. R. Ashton, R. Ballardini, V. Balzani, E. C. Constable, A. Credi, O. Kocian, S. J. Langford, J. A. Preece, L. Prodi, E. R. Schofield, N. Spencer, J. F. Stoddart and S. Wenger, Chem. Eur. J., 1998, 4, 2413 CrossRef CAS; (b) P. R. Ashton, V. Balzani, O. Kacian, L. Prodi, N. Spencer and J. F. Stoddart, J. Am. Chem. Soc., 1998, 120, 11190 CrossRef CAS; (c) A. Credi, M. Montalti, V. Balzani, S. J. Langford, F. M. Raymo and J. F. Stoddart, New J. Chem., 1998, 22, 1061 RSC; (d) E. Ishow, A. Credi, V. Balzani, F. Spadola and L. Mandolini, Chem. Eur. J., 1999, 5, 984 CrossRef CAS; (e) V. Balzani, J. Becher, A. Credi, M. B. Nielsen, F. M. Raymo, J. F. Stoddart, A. M. Talarico and M. Venturi, J. Org. Chem., 2000, 65, 1947 CrossRef CAS.
- (a) A. Credi, V. Balzani, S. J. Langford and J. F. Stoddart, J. Am. Chem. Soc., 1997, 119, 2679 CrossRef CAS; (b) M. Asakawa, P. R. Ashton, V. Balzani, A. Credi, G. Mattersteig, O. A. Matthews, M. Montalti, N. Spencer, J. F. Stoddart and M. Venturi, Chem. Eur. J., 1997, 3, 1992 CrossRef CAS.
- (a) P. R. Ashton, R. Ballardini, V. Balzani, S. E. Boyd, A. Credi, M. T. Gandolfi, M. Gómez-López, S. Iqbal, D. Philp, J. A. Preece, L. Prodi, H. G. Ricketts, J. F. Stoddart, M. S. Tolley, M. Venturi, A. J. P. White and D. J. Williams, Chem. Eur. J., 1997, 3, 152 CrossRef CAS; (b) P. R. Ashton, M. Gómez-López, S. Iqbal, J. A. Preece and J. F. Stoddart, Tetrahedron Lett., 1997, 38, 3635 CrossRef CAS.
- For early examples of self-complexing macrocycles, see: Y. Nakatsuji, H. Kobayashi and M. Okahara, J. Chem. Soc., Chem. Commun., 1983, 800 RSC; S. Shinkai, M. Ishihara, K. Ueda and O. Manabe, J. Chem. Soc., Perkin Trans. 2, 1985, 511 RSC.
- (a) R. Wolf, M. Asakawa, P. R. Ashton, M. Gómez-López, C. Hamers, S. Menzer, I. W. Parsons, N. Spencer, J. F. Stoddart, M. S. Tolley and D. J. Williams, Angew. Chem., Int. Ed., 1998, 37, 975 CrossRef CAS; (b) P. R. Ashton, I. W. Parsons, F. M. Raymo, J. F. Stoddart, A. J. P. White, D. J. Williams and R. Wolf, Angew. Chem., Int. Ed., 1998, 37, 1913 CrossRef CAS; (c) P. R. Ashton, I. Baxter, S. J. Cantrill, M. C. T. Fyfe, P. T. Glink, J. F. Stoddart, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 1998, 37, 1294 CrossRef CAS; (d) N. Yamaguchi, D. Nagvekar and H. W. Gibson, Angew. Chem., Int. Ed., 1998, 37, 2361 CrossRef CAS; (e) M. B. Nielsen, S. B. Nielsen and J. Becher, Chem. Commun., 1998, 475 RSC; (f) A. Mirzoian and A. E. Kaifer, Chem. Commun., 1999, 1603 RSC.
- Besides self-complexation, compounds of this type can give rise to dimers and polymeric arrays (see, e.g., refs. 9b–d, f)..
- The interesting case of a ‘tailed’ macrocycle which exhibit a hermaphroditic character, based on the template effect of copper(I) ions, has recently been reported (See: M. C. Jiménez, C. Dietrich-Buchecker, J.-P. Sauvage and A. De Cian, Angew. Chem., Int. Ed., 2000, 39, 1295). This compound, however, is not able to undergo intramolecular complexation since the tail is not sufficiently flexible; rather, it gives rise to doubly threaded dimers that have been used to construct an interlocked multicomponent system capable of contracting or stretching under chemical stimulation (See: M. C. Jiménez, C. Dietrich-Buchecker and J.-P. Sauvage, Angew. Chem., Int. Ed., 2000, 39, 3284). Search PubMed.
- P. R. Ashton, R. Ballardini, V. Balzani, A. Credi, M. T. Gandolfi, S. Menzer, L. Pérez-García, L. Prodi, J. F. Stoddart, M. Venturi, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 1995, 117, 11171 CrossRef.
- P. R. Ashton, V. Baldoni, V. Balzani, A. Credi, C. G. Claessens, H. D. A. Hoffmann, F. M. Raymo, J. F. Stoddart, M. Venturi, A. J. P. White and D. J. Williams, Eur. J. Org. Chem., 2000, 1121 CrossRef CAS.
- P.-L. Anelli, P. R. Ashton, R. Ballardini, V. Balzani, M. Delgado, M. T. Gandolfi, T. T. Goodnow, A. E. Kaifer, D. Philp, M. Pietraszkiewicz, L. Prodi, M. V. Reddington, A. M. Z. Slawin, N. Spencer, J. F. Stoddart, C. Vicent and D. J. Williams, J. Am. Chem. Soc., 1992, 114, 193 CrossRef CAS.
- A. Credi and L. Prodi, Spectrochim. Acta, Part A, 1998, 54, 159 Search PubMed.
- J. B. Flanagan, S. Margel, A. J. Bard and F. C. Anson, J. Am. Chem. Soc., 1978, 100, 4248 CrossRef CAS.
- The alternative explanation according to which compound 1·H2+ undergoes complete intermolecular complexation (e.g. formation of dimers) in CH2Cl2 solution under the conditions (down to 2 × 10−5 mol L−1) employed is very unlikely since it would imply an unusually large value for the association constant..
- Although a threaded self-complexation seems more likely than a side-on self-complexation, there is no easy way to distinguish between the two conformations. It should be noted, however, that the conclusions about the occurrence of a switching process are not affected in the least by the actual conformation that is adopted by the self-complexed species..
- R. E. Gillard, F. M. Raymo and J. F. Stoddart, Chem. Eur. J., 1997, 3, 1933 CrossRef CAS.
- Quenching may occur by electron transfer from the excited state of the 1,5-DON unit to the bipyridinium tail or by energy transfer to the lower energy CT excited states originating from the interaction between the 1,3-DOB and 1,5-DON units and the appended bipyridinium tail..
- Under these conditions the occurrence of bimolecular quenching phenomena can be neglected because of the short excited-state lifetime of the 1,5-DON unit..
- The low-energy excited state(s) arising from the CT interaction between the bipyridinium tail and the 1,5-DON unit can offer an additional radiationless deactivation pathway to the fluorescent excited state of the latter unit..
- C. Reichardt, Solents and Solent Effects in Organic Chemistry,
Wiley-VCH, Weinheim, 1990. Search PubMed.
- When trifluoroacetic acid is replaced by the stronger triflic acid (CF3SO3H) at the end of the experiment the CT band is blue shifted (10 nm). This observation could arise from conformational changes induced by protonation of the weakly basic oxygen atoms in the polyether chain or by ion pairing effects..
- t-BBPE2+ shows a very weak fluorescence band (λmax = 370 nm) at room temperature that cannot be used for monitoring the threading process since it is covered by the residual emission of compound 12+..
- M. Goez and G. Eckert, Ber. Bungenses. Phys. Chem., 1991, 95, 1179 Search PubMed.
- A. E. Kaifer and M. Gómez-Kaifer, Supramolecular Electrochemistry, Wiley-VCH, Weinheim, 1999. Search PubMed.
- A more realistic model for the 1,4-DOB unit of compound 24+ might be 64+..
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2001 |