A new class of anionic phosphinooxazoline ligands in palladium and ruthenium complexes: catalytic properties for the transfer hydrogenation of acetophenone†
Pierre Braunstein*a, Frédéric Nauda and Steven J. Rettig‡b
aLaboratoire de Chimie de Coordination (CNRS UMR 7513), Uni![[italic v]](https://www.rsc.org/images/entities/char_e0f5.gif) ersité Louis Pasteur, 4 rue Blaise Pascal, F-67070, Strasbourg cedex, France. E-mail: braunst@chimie.u-strasbg.fr
ersité Louis Pasteur, 4 rue Blaise Pascal, F-67070, Strasbourg cedex, France. E-mail: braunst@chimie.u-strasbg.fr
bDepartment of Chemistry, Uniersity of British Columbia, 2036 Main Mall, Vancouer, BC V6T 1Z1, Canada
First published on 12th December 2000
Abstract
The synthesis of the cationic
Pd complex [Pd(dmba)(PCH2-oxazoline)]Cl [PCH2-oxazoline = 2-oxazoline-2-ylmethyl)diphenylphosphine] (2) has allowed for the first time the observation of hemilabile behaviour for a phosphinooxazoline ligand. This molecular dynamics is stopped upon removal of the chloride anion, by reaction with either NH4PF6, which retains the cationic nature of the complex, or ButOK. The latter reaction leads to the formation of the first example of a Pd complex bearing an anionic phosphinooxazoline ligand, [![[upper bond 1 start]](https://www.rsc.org/images/entities/char_e010.gif) Pd(dmba){Ph2PCH
Pd(dmba){Ph2PCH![[horiz bar, triple dot above]](https://www.rsc.org/images/entities/char_e0f1.gif)
![[lower bond 1 start]](https://www.rsc.org/images/entities/char_e012.gif) C(N
C(N![[upper bond 1 end]](https://www.rsc.org/images/entities/char_e011.gif) )CH2CH2O
)CH2CH2O![[lower bond 1 end]](https://www.rsc.org/images/entities/char_e013.gif) }], abreviated [Pd(dmba)(PCH-oxazoline)] (7), the anionic charge resulting from the monodeprotonation of the PCH2 group. Following this methodology, the Ru complex [RuCl(p-cymene){Ph2PCHC(N)CH2CH2O}], abreviated [RuCl(η6-p-cymene)(PCH-oxazoline)] (8) and containing this four-electron chelating anionic ligand, was prepared and shown to be more reactive for the catalytic transfer hydrogenation of acetophenone in propan-2-ol than the analogous complex bearing the neutral phosphinooxazoline ligand PCH2-oxazoline. The crystal structure of cis-[Pd{Ph2PCHC(N)CH2CH2O}2], abbreviated cis-[Pd(PCH-oxazoline)2] (6), was determined by X-ray diffraction.
}], abreviated [Pd(dmba)(PCH-oxazoline)] (7), the anionic charge resulting from the monodeprotonation of the PCH2 group. Following this methodology, the Ru complex [RuCl(p-cymene){Ph2PCHC(N)CH2CH2O}], abreviated [RuCl(η6-p-cymene)(PCH-oxazoline)] (8) and containing this four-electron chelating anionic ligand, was prepared and shown to be more reactive for the catalytic transfer hydrogenation of acetophenone in propan-2-ol than the analogous complex bearing the neutral phosphinooxazoline ligand PCH2-oxazoline. The crystal structure of cis-[Pd{Ph2PCHC(N)CH2CH2O}2], abbreviated cis-[Pd(PCH-oxazoline)2] (6), was determined by X-ray diffraction.
Oxazolines are versatile synthons for ligand design1,2 and readily available in enantiomerically pure forms, which accounts for their increasing use in asymmetric reactions catalysed by transition metal complexes.2–5 Nevertheless, reports on anionic ligands incorporating an oxazoline ring are not legion compared to their neutral analogs and such ligands may be classified according to two main families. In class A compounds, the anionic charge is carried by an external heteroatom (C−, O−, S−) and the nitrogen atom of the oxazoline remains a neutral donor. This includes ligands such as bis(oxazolinyl)phenyl,6–8 (hydroxyphenyl)oxazoline,9–15 (hydroxyferrocenyl)oxazoline16,17 and (mercaptoaryl)oxazoline,17 which upon deprotonation allowed the preparation of complexes with metals such as Ti, Zr, Hf, V, Mn, Ni, Pd, Pt or Cu. An oxazoline moiety was recently combined to a pyrrole for use in copper-catalysed enantioselective cyclopropanation reactions; the NH group is expected to be deprotonated upon in situ reaction with Cu(I) triflate.18 Some of the isolated complexes were found to be catalysts for reactions such as ethylene polymerisation,9 epoxydation,11b alkyl- and phenylzincation of aldehydes,16 and conjugate addition to ketones.17
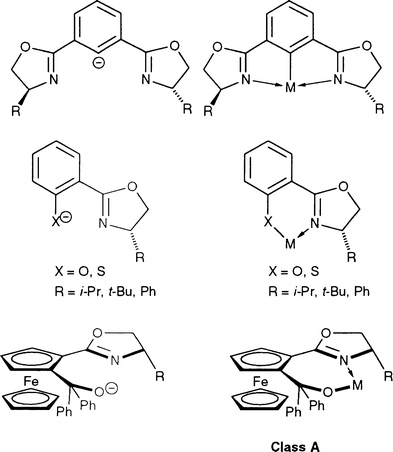
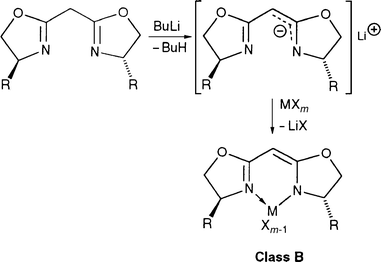
In the second class of ligands, B, the anionic charge is largely delocalised onto the nitrogen atom of the oxazoline but only one such ligand type has been reported, as illustrated in eqn. (1) . The corresponding complexes were obtained by first deprotonation of the acidic CH2 of the bis(oxazoline) ligand at low temperature and subsequent quenching with a metal salt. This strategy allowed the development of efficient Ti,19,20 Cu,21,22 Rh,23 Zn24,25 or Mg26 catalysts for several asymmetric reactions such as reduction of ketones,20 cyclopropanation of olefins,21,22 allylzincation,24,25 and hydrocyanation of aldehydes.26 The precatalysts are usually prepared in situ and not isolated (Zn, Mg, Ti) and only three crystal structures have been determined by X-ray diffraction, for the Cu,27 Rh 23 and Y 28 complexes.
 | (1) |
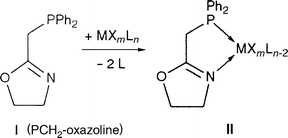
We have previously reported the synthesis of Pd and Ru complexes of type II bearing the neutral P,N-type ligand (oxazolinylmethyl)diphenylphosphine I (abreviated PCH2-oxazoline) [eqn. (2)] and studied their catalytic properties for ethylene/CO copolymerization and asymmetric transfer hydrogenation of ketones, respectively.29,30
 | (2) |
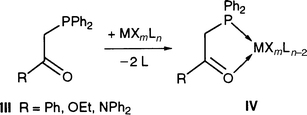
This provided an extension to work carried out by several groups, including ours, on bifunctional phosphorus ligands and more specifically on phosphorus/oxygen P,O-type ligands III [eqn. (3)].2,31–37
 | (3) |
The often encountered hemilabile character of such neutral ligands in complexes of type IV allows for the temporary pro tection and facile generation of reactive sites in the coordination sphere of their complexes and several of them have been used in homogeneous catalysis. An additional feature of these P,O-type ligands is that the acidity of their PCH2 protons allows facile deprotonation of this group to form an anionic 4-electron donor phosphino enolate ligand, with loss of the hemilabile behaviour. These strong chelates confer special reactivity to their complexes V, as found, for example, in the reversible CO2 fixation by palladium complexes,38,39 the rhodium-catalysed transfer dehydrogenation of cyclooctane40 and the highly selective nickel-catalysed conversion of ethylene into linear α-olefins.41–47
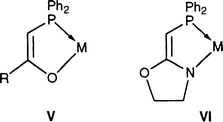
This dramatic change in reactivity observed when going from neutral to anionic P,O-type ligands triggered our interest for new anionic P,N-type ligands derived from I, in which the acidity of the PCH2 protons should also allow easy deprotonation, particularly after coordination to a metal centre. Here we report the synthesis of such complexes containing a phosphine moiety covalently linked to an oxazoline, whose nitrogen atom formally carries the partly delocalised negative charge, thus resulting in the formation of a covalent rather than dative N–M bond, as depicted in VI. The corresponding ligand, abreviated PCH-oxazoline, is the first representative of a new class of hybrid ligands, which combines some of the features of B and V type systems. In addition to the synthesis of palladium(II) complexes, we present a comparison of the catalytic activity of ruthenium(II) complexes containing PCH2-oxazoline (type II) or PCH-oxazoline (type VI) ligands for transfer hydrogenation of acetophenone in propan-2-ol. A preliminary communication on related systems where the oxazoline carries two methyl groups on the carbon α to nitrogen has appeared.48
Results and discussion
Complexes with the neutral PCH2-oxazoline ligand I
Mixing 2 equiv. of PCH2-oxazoline with 1 equiv. of [Pd(NCMe)4](BF4)2 in CH2Cl2 [eqn. (4)] led to the formation of [Pd(PCH2-oxazoline)2](BF4)2 (1) in high yield (95%).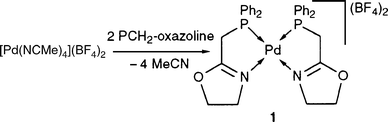 | (4) |
The spectroscopic data (Table 1) are consistent with the coordination of the ligand in a static bidentate fashion.29 The cis arrangement of the P atoms is deduced from the 31P{1H} NMR chemical shift and from the doublet observed in the 1H NMR spectrum for the PCH2 protons. Such an arrangement for two atoms of large trans influence is thermodynamically favoured.49,50 Moreover, the related Ni complex [Ni(phosphinoaryloxazoline)2]2+ has been recently shown to have a cis structure, both in solution and in the solid state.51
| NMRa | IR/cm−1 (CH2Cl2) | ||||
|---|---|---|---|---|---|
| Complex | 1Hb | 13C{1H}c (1JPC/Hz) | 31P{1H}d | ν(C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) N) N) | ν(CCN) |
| a In CD2Cl2, unless otherwise stated.b 300.13 MHz.c 75.4 MHz.d 121.5 MHz.e In CDCl3. | |||||
| cis-[Pd(PCH2-oxazoline)2](BF4)2, 1 | 3.75 (d) PCH2e | — | 29.7e | 1633 | — |
| [Pd(dmba)Cl(PCH2-oxazoline)], 2 | 3.65 (d) PCH2e | 31.5 (25.2) | 32.6e | 1660, 1631 | — |
| Pd(dmba)(PCH2-oxazoline)](PF6), 3 | 3.45 (d) PCH2e | — | 32.8e | 1635 | — |
| [RuCl(η6-p-cymene)(PCH2-oxazoline)]Cl, 4 | 3.30 (dd), 3.40 (dd), PCH2 | 30.5 (34.0) | 50.5 | 1641 | — |
| cis-[Pd(PCH-oxazoline)2], 6 | 2.60 (s) PCH | 41.2 (78.5) | 20.5 | — | 1538 |
| [Pd(dmba)(PCH-oxazoline)], 7 | 2.82 (s) PCH | 40.7 (82.5) | 24.4 | — | 1537 |
| [RuCl(η6-p-cymene)(PCH-oxazoline)], 8 | 3.10 (s) PCH | 37.0 (78.8) | 40.5 | — | 1546 |
 | (5) |
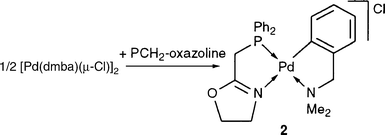
Reaction of PCH2-oxazoline with [Pd(dmba)(μ-Cl)]2 afforded
2![[italic v]](https://www.rsc.org/images/entities/i_char_e0f5.gif) ia opening of the chloride bridges [eqn. (5)].
ia opening of the chloride bridges [eqn. (5)].
The IR spectrum of 2 in CH2Cl2 shows two bands at 1660
and 1631 cm−1, assigned to the CN vibrations of the uncoordinated and coordinated oxazolines, respectively, by analogy with the values for the free ligand (1660 cm−1)29 and for 1. For comparison, we prepared the PF6 analogue of 2 by carrying out the reaction in the presence of NH4PF6 (see Experimental). As expected, its IR spectrum in CH2Cl2 exhibits only one band at 1635 cm−1. Therefore, the existence of coordinated and uncoordinated oxazolines in solutions of 1 suggests
competition between the oxazoline nitrogen and the chloride for coordination to palladium. Comparison of the 31P{1H} and 1H NMR spectra with those of the free ligand confirms that PCH2-oxazoline forms a P,N-chelate, but the broad resonances of the oxazoline protons are again consistent with a
hemilabile behaviour of the ligand through reversible oxazoline nitrogen (de)coordination. For the first time, this ligand is shown to display a hemilabile behaviour.2
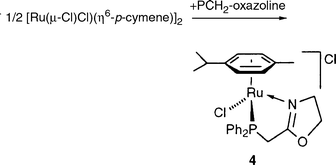
Reaction of 2 equiv. of PCH2-oxazoline with 1 equiv. of [Ru(μ-Cl)Cl(η6-p-cymene)]2 in refluxing EtOH [eqn. (6)] led to the isolation of [RuCl(η6-p-cymene)(PCH2-oxazoline)]Cl (4) in good yield (89%).
 | (6) |
In contrast to 2, which also has Cl− as a counter ion, the IR spectrum of 4 exhibits only one CN vibration band at 1641 cm−1, which corresponds to a coordinated oxazoline. The 1H NMR spectrum shows that the protons of the PCH2-oxazoline ligand appear at six distinct resonances and are thus diastereotopic. This is consistent with the ruthenium centre becoming a stereogenic centre upon chelation of PCH2-oxazoline. The PCH2 protons exhibit an ABX spin system with two
different 2JPH coupling constants (8.5 and 13.0 Hz).
The analogous complex, [RuCl(η6-C6H6)(PCH2-oxazoline)](O3SCF3) (5),
was prepared
for catalytic purposes in order to allow comparison
with related complexes containing a tridentate NPN ligand (see
below). Its spectroscopic properties are very similar to
those of 4 (see Experimental).
These cationic PCH2-oxazoline complexes were mainly used as precursors for the synthesis of complexes containing the anionic PCH-oxazoline ligand (see below).
Complexes with the anionic PCH-oxazoline ligand
The deprotonation of the PCH2 group in complexes 1, 2 and 4 in THF was achieved in 78–86% yields, by reaction with ButOK (Scheme 1).48 This simple and efficient methodology has been reported before with β-ketophosphines of the type R21PCH2C(O)R2 coordinated to Re,52 Fe,53 Ru,54–56 Rh,40,57 Pd,58 or Pt,59 using various bases.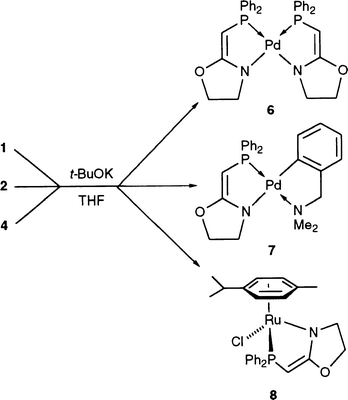 | ||
| Scheme 1 | ||
In all cases, the typical ν(CN) IR absorption band of the coordinated oxazoline in the precursor complex is replaced by a strong
absorption in the range 1537–1546 cm−1 (see Table 1). This is consistent with the IR data of a Cu complex described by Lehn et al., which shows an absorption band at 1530 cm−1.27
The shift towards lower energy by about 100 cm−1 on going from PCH2-oxazoline to anionic PCH-oxazoline is consistent with previous observations made with P,O-ligands when going from IV to V. The PCH carbon appears in the 13C{1H} NMR spectrum as a doublet downfield shifted (6.5–9 ppm) compared to the corresponding PCH2 and exhibits a larger 1JPC coupling constant, 78.5–82.5 Hz s. 25.2–34.0 Hz, respectively. This increase is attributed to the change in hybridisation of the carbon atom α to P from sp3 in the neutral chelate
to sp2 in the anionic one. While the PCH2 protons of complexes 1–5
exhibit in the 1H NMR spectrum a doublet, the PCH proton
of 6–8 exhibits only a singlet. Complexes 6–8 are air-stable and, in contrast to their precursors, they react with CDCl3
to
give uncharacterised side products. An X-ray diffraction study on single crystals of 6 confirmed that the two phosphorus atoms occupy cis positions (Fig. 1 and Table 2). The
coordination around the metal centre approximates a square planar geometry with a slight displacement of the Pd–N(2) vector from the plane defined by the two phosphorus, two nitrogen
and palladium atoms [0.150(3) Å]. The non-ideal geometry
is also reflected by the angles P(1)–Pd–P(2) and N(1)–Pd(1)–N(2) of 102.16(4)° and
94.40(1)°, respectively. The two planes defining the backbone of the P,N-chelates, Pd(1), P(1), N(1), C(1), C(2) and Pd(1), P(2), N(2), C(17), C(18), form a dihedral angle of 1.9°. However, the angle between the two planes O(1), N(1), C(2), C(3), C(4) and O(2), N(2), C(18), C(19), C(20), corresponding to the two oxazolines, form a dihedral angle of
28.8°, which results in a staggered arrangement for the NCH2
protons. It is interesting to compare the distances and angles
of PCHox in 6 with those of the neutral PCH2-oxazoline-Me2
ligand in [PdCl2(PCH2-oxazoline-Me2)] where PCH2-oxazoline-Me2
is similar to PCH2-oxazoline but has two methyl substituents on the carbon α to the nitrogen atom.29 Of special interest are the bond lengths and angles within the five-membered ring chelates. The C–C bond distance decreases on going from the neutral to the anionic ligand [1.480(4) to 1.370(5) Å, respectively]
and this latter value is close to the average length of a CC (sp2–sp2) bond (1.34 Å). At the same time, the C–N bond length increases from 1.276(4) to 1.335(4) Å, which is in favour
of a single C–N bond [average d(Csp2
–N) = 1.36 Å]. Even though it is
likely that electron delocalisation occurs within the chelate ring, we prefer to describe the bonding within the five-membered P,N-chelate as involving a pronounced CC double bond character. The X-ray crystal structure of 6 is significant since there are only three reported X-ray crystal structures of
an anionic oxazoline in a class B
complex (M = Cu,27 Rh,23 Y28)
although this bonding mode is often invoked, and the only example with a PCH-oxazoline-type ligand.48
![ORTEP view of the crystal structure of cis-[Pd(PCH-oxazoline)2] (6).](/image/article/2001/NJ/b004786o/b004786o-f1.gif) | ||
| Fig. 1 ORTEP view of the crystal structure of cis-[Pd(PCH-oxazoline)2] (6). | ||
| Pd(1)–P(1) | 2.286(1) | N(1)–C(2) | 1.335(4) |
| Pd(1)–P(2) | 2.293(1) | C(2)–C(1) | 1.370(5) |
| Pd(1)–N(1) | 2.074(3) | C(1)–P(1) | 1.737(4) |
| Pd(1)–N(2) | 2.073(3) | C(2)–O(1) | 1.380(4) |
| N(1)–Pd(1)–N(2) | 94.40(11) | N(1)–C(2)–C(1) | 125.6(3) |
| N(2)–Pd(1)–P(2) | 81.42(8) | C(2)–C(1)–P(1) | 112.1(3) |
| P(2)–Pd(1)–P(1) | 102.16(4) | C(1)–P(1)–Pd(1) | 102.0(1) |
| P(1)–Pd(1)–N(1) | 82.20(8) | O(1)–C(2)–C(1) | 121.3(3) |
| Pd(1)–N(1)–C(2) | 116.2(2) | O(1)–C(2)–N(1) | 113.1(3) |
Catalytic study
As part of our interest in the properties of multidentate ligands that contain both phosphine and oxazoline in a chelating array,2 we have tested their ruthenium complexes in the catalytic transfer hydrogenation of ketones by propan-2-ol [eqn. (7)].30,48,60,61 There is considerable current research activity around this reaction in order to both develop new classes of catalysts and to understand the different steps involved in the overall catalytic process.62–67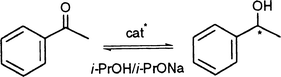 | (7) |
We recently described a precursor catalyst for this reaction, [RuCl(η6-C6H6)(N,P-NPN)](O3SCF3) (9) where NPN is a bis(oxazoline)phosphine that forms a bidentate P,N-chelate.60 It was shown that the dangling oxazoline arm of this ligand tends to have a detrimental effect on the catalytic activity, possibly owing to the formation of a bis-chelating system of type 10.60 This is now further supported by the observation that complex 5 is more active than 9 and 10 (see Table 3).
| Catalyst | Time/h | Yieldb (%) | Turnover/h−1 |
|---|---|---|---|
| a The reactions were carried out in refluxing propan-2-ol using a 0.1 M substrate concentration and a 0.1 M solution of PriONa as a base and a 1: 200:5 Ru–ketone–base ratio.b Chemical yields determined by GC analysis of the crude reaction mixture at the reported time.c Reaction carried out in propan-2-ol at 30°C.d Taken from ref. 60 with a 1:200:24 Ru–ketone–base ratio. | |||
| 4 | 0.25 | 5 | 40 |
| 1 | 34 | 68 | |
| 3 | 75 | 50 | |
| 6 | 94 | 32 | |
| 5 | 0.25 | 23 | 184 |
| 1 | 61 | 122 | |
| 8 | 0.25 | 25 | 200 |
| 1 | 49 | 98 | |
| 3 | 86 | 58 | |
| 6 | 94 | 32 | |
| 8c | 8 | 16 | 4 |
| 9d | 0.25 | 13.5 | 108 |
| 1 | 54 | 108 | |
| 10d | 0.25 | 11 | 88 |
| 1 | 45 | 90 | |
In light of our synthetic route to prepare complexes bearing the anionic ligand, it is likely that under catalytic conditions, the PriONa base, which is present in a 5-fold excess to Ru, reacts with the precursor catalyst 4 to form compound 8. To test this hypothesis we used 8 as the catalyst precursor. Whereas the conversion obtained after 6 h at reflux temperature in propan-2-ol was identical to that observed with 4 (94% yield), the turnover frequency (TOF) was higher with [RuCl(η6-p-cymene)(PCH-oxazoline)] than with [RuCl(η6-p-cymene)(PCH2-oxazoline]Cl during the first hour (Table 3). This may correspond to an induction period to form the catalyst 8. Although it appears that 4 gives 8 under catalytic conditions, we cannot rule out that both 4 and 8 may be independently active catalysts for transfer hydrogenation. Noyori et al. have recently published67 the structure of a preformed catalyst precursor 11, which upon in situ deprotonation of the NH group generates the true catalyst 12 which is to date among the best catalysts for transfer hydrogenation of ketones.68,69 These results emphasise the spectacular conse quences that ligand deprotonation may have on the catalytic properties of the metal complex.
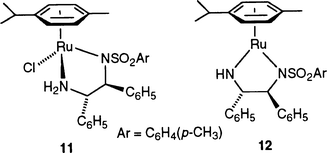
Compound [RuCl(η6-p-cymene)(PCH-oxazoline)] (8) is very similar to 11 with the difference that the PCHox ligand in 8 cannot be further deprotonated to form species analogous to 12.67a This could account for the higher reactivity of 11 compared to 8. Nevertheless, our results are encouraging and complexes analogous to [RuCl(η6-p-cymene)(PCH-oxazoline)] with a chiral version of PCH2-oxazoline should be synthesised in the future. Furthermore, Noyori et al. have shown that the arene ligand has a strong influence on the reactivity and enantioselectivity of the catalysts, suggesting that complexes containing other arene ligands such as benzene or 1,3,5-trimethylbenzene should also be evaluated.70
Conclusion
The versatility of the PCH2-oxazoline ligand allows its coordination as hemilabile or static neutral P,N- or static anionic P,N-type ligand. It was shown that in the latter case its structural and electronic properties induce higher reactivity in the catalytic transfer hydrogenation of acetophenone in propan-2-ol than its neutral analogue. This finding should lead to further investigations with chiral versions of PCH-oxazoline in order to study their influence on the enantioselectivity of this catalytic reaction. Also as an extension to this work, it would be interesting to evaluate our Ru-phosphinooxazoline complexes in Diels–Alder reactions, since a [Ru(H2O)(η6-p-cymene)-phosphinoaryloxazoline]2+ complex has been recently reported to be a good catalyst for such reactions.71Experimental
General procedures
All reactions were performed under purified nitrogen. Solvents were purified and dried under nitrogen by conventional methods. The 1H, 13C and 31P{1H} NMR spectra were recorded at 300.13, 75.4 and 121.5 MHz, respectively, on an FT Bruker AC300 instrument, 1H{31P} NMR at 500.13 MHz on an FT Bruker ARX500 instrument, chemical shifts are positive downfield from external TMS for 1H and 13C, and from H3PO4 (85% in H2O) for 31P. IR spectra were recorded in the 4000–400 cm−1 range on a Bruker IFS66 FT spectrometer.Syntheses
The compounds [Pd(dmba)(μ-Cl)]2 ,72 [Ru(μ-Cl)Cl(η6-p-cymene)]2,73 and [Pd(NCMe)4](BF4)274 were prepared according to literature procedures.acuo. The product was obtained as a white solid (1.035 g, yield 95%). 1H NMR (CDCl3
, 300.13 MHz): δ 3.75 (d, 4 H, 2JPH
= 11.9 Hz, PCH2), 4.40 (t, 4 H, 3JHH
= 9.5 Hz, NCH2), 4.80 (t, 4 H, 3JHH
= 9.5 Hz, OCH2), 7.30–7.65 (m, 20 H, aryl). 31P{1H} NMR (CDCl3, 121.5
MHz): δ 29.7. Anal. calc. for C32H32B2F8N2O2P2Pd:
C, 46.95; H, 3.94; Found: C, 47.22; H, 3.68%.N). 31P{1H} NMR (CDCl3
, 121.5 MHz): δ 32.6. Anal. calc. for C25H28ClN2OPPd: C, 55.06; H, 5.18; N, 5.14. Found: C, 55.20; H, 5.17; N, 5.04%.acuo. The product was obtained as a pale yellow solid
(0.438 g, 91%). 1H NMR (CDCl3, 300.13 MHz): δ 2.90 [s, 6 H, N(CH3)2], 3.45 (d, 2 H, 2JPH
= 10.5 Hz, PCH2), 4.00 [s, 2 H, (CH3)2NCH2],
4.05 (t, 2 H, 3JHH
= 11.1 Hz, oxazoline NCH2), 4.60 (t, 2 H, 3JHH
= 11.1 Hz, OCH2), 6.40–7.00 (m, 4 H, aryl from dmba), 7.40–7.90 (m, 10 H, aryl). 31P{1H} NMR (CDCl3, 121.5 MHz): δ 32.8. Anal. calc. for C25H28N2OP2Pd: C, 45.85; H, 4.31. Found: C, 45.61; H, 4.22%.N). 31P{1H} NMR (CD2Cl2, 121.5 MHz): δ 50.5. Anal. calc. for C26H30NOPCl2Ru: C, 54.20; H, 5.21; N, 2.43. Found: C, 54.17; H, 5.31; N, 2.27%.acuo. The product was obtained as an orange powder (0.616 g, yield 78%). 1H NMR (CD2Cl2, 300.13 MHz): δ 2.60 (s, 1 H, PCH), 3.90 (t, 2 H, 3JHH
= 7.9 Hz, NCH2),
4.35 (t, 2 H, 3JHH
= 7.9 Hz, OCH2), 7.00–7.40 (m, 20 H, aryl). 13C{1H} NMR (CD2Cl2
, 75.4 MHz): δ 41.2 (d, 1JPC
= 78.5 Hz, PCH), 52.7 (s, NCH2), 70.1 (s, OCH2), 126.5–135.0 (m, aryl),
180.5 (d, CN). 31P{1H} NMR (CD2Cl2, 121.5 MHz): δ 20.5. Anal. calc. for C32H30N2O2P2Pd: C, 59.78; H, 4.70; N, 4.36. Found: C,
60.04; H, 4.62; N, 4.18%.acuo. The product was obtained as a yellow powder (0.800 g, yield 84%). 1H NMR (CD2Cl2, 300.13 MHz): δ 2.80 [s, 6 H, N(CH3)2], 2.82 (s, 1 H, PCH), 3.70 (t,
2 H, 3JHH
= 7.9 Hz, oxazoline NCH2), 3.95 [s, 2 H, (CH3)2NCH2], 4.30 (t, 2 H, 3JHH
= 7.9 Hz, OCH2), 6.55–7.00 (m, 4 H, aryl
from dmba), 7.40–7.70 (m, 10 H, aryl). 13C{1H} (CD2Cl2, 75.4 MHz): δ 40.7 (d, 1JPC
= 82.5 Hz, PCH), 50.9 [s, N(CH3)2], 52.2 (s, oxazoline NCH2), 69.3 [s, (CH3)2NCH2], 73.3 (s, OCH2), 122.8–149.5 (m, aryl). 31P{1H} NMR (CDCl3, 121.5 MHz): δ 24.4. Anal. calc. for C25H27N2OPPd: C, 59.01; H, 5.35; N, 5.50. Found: C, 58.79; H, 5.22; N, 5.46%.acuo. The product was isolated as a brown–orange powder (0.355 g, yield: 86%). 1H
NMR (CD2Cl2, 500.13 MHz): δ 0.95 [d, 3 H, 2JHH
= 6.9 Hz, CH(CH3)(CH3)],
1.10 [d, 3 H, 2JHH
= 6.9 Hz, CH(CH3)(CH3)], 2.10 (s, 3 H, p-CH3), 2.20 [sept, 1 H, 2JHH
= 6.9 Hz, CH(CH3)2], 3.10 (s, 1 H, PCH), 3.70 (m with appearance of t of d, 2JHH
= 9.0 Hz, 3JHHcis
= 9.0 Hz, 3JHHtrans
= 3.6 Hz, OCHH), 3.85 (m with appearance of overlapping dt, 1 H, JHH
= 10.2 Hz, JHH
= 9.0 Hz, NCHH),
4.25 (m with appearance of overlapping dt, 1 H, JHH
= 10.2
Hz, JHH
= 7.8 Hz, OCHH), 4.25 (overlapping m, 1 H, JHH
= 1.2
Hz, p-cymene), 4.35 (m with appearance of t of d of
d, 2JHH
= 7.8 Hz, 3JHHcis
= 7.8 Hz, 3JHHtrans
= 3.6 Hz, 5JHH
= 1.2
Hz, NCHH), 5.20 (d, 1 H, 3JHH
= 6.2 Hz, p-cymene), 5.40 (d, 1 H, 3JHH
= 5.7 Hz, p-cymene), 5.70 (d, 1 H, 3JHH
= 6.2 Hz, p-cymene), 7.30–7.40 (m, 6 H, aryl), 7.50–7.70 (m, 4 H, aryl H). 13C{1H} NMR (CD2Cl2
, 75.4 MHz): δ 18.4 (s, CH3), 22.0 [s, CH(CH3)(CH3)], 23.0 [s, CH(CH3)(CH3)], 31.4 [s, CH(CH3)2], 37.0 (d, 1JPC
= 78.8 Hz, PCH), 55.4 (s, NCH2), 69.3 (s, OCH2), 83.5 (s, Pri–C···CH from p-cymene), 84.5 (s, Me–C···CH
from p-cymene), 88.6 (d, JPC
= 5.9 Hz, Pri–C···CH from p-cymene),
93.3 (d, JPC
= 5.9 Hz, Me–C···CH from p-cymene), 97.4 (s, quat
C p-cymene), 103.1 (s, quat C p-cymene), 126.8–142.2
(m, aryl), 179.3 (d, 2JPC
= 43.4 Hz, CN). 31P{1H} NMR (CD2Cl2
, 121.5 MHz): δ 40.5. Anal. calc. for C26H29NOPRuCl: C, 57.88;
H, 5.38; N, 2.59. Found: C, 57.65; H, 5.29; N, 2.56%.X-Ray structural analysis75
The intensity data were collected on a Rigaku/ADSC CCD diffractometer at 180(1) K in 0.50° oscillations with 35.0 s exposures. The structure was solved by heavy-atom Patterson methods and expanded using Fourier techniques. The non-hydrogen atoms were refined anisotropically. Hydrogen atoms were fixed in calculated positions with C–H = 0.98 Å. Selected data are given in Table 4.| a R = Σhkl(‖Fobs∣ − ∣Fcalc‖)/Σhkl∣Fobs ∣; Rw = [Σhklw(∣Fobs∣ − ∣Fcalc∣)2/ΣhklwFobs2]1/2, w = 1/σ2(Fobs). | |
|---|---|
| Formula | C32H30N2O2P2Pd |
| FW | 642.95 |
| Crystal system | Monoclinic |
| Space group | P21/n (no. 14) |
| T/K | 180(1) |
| a/Å | 17.293(2) |
| b/Å | 9.7528(13) |
| c/Å | 17.6784(6) |
| β/° | 115.2840 |
| U/Å3 | 2695.9(4) |
| Z | 4 |
| μ/mm−1 | 8.42 |
| No. of reflections collected | 24477 |
| No. of unique reflections | 6934 |
| No. of variable parameters | 352 |
| No. of reflections [I>3σ(I)] | 4082 |
| Residuals (R, Rw)a | 0.037, 0.031 |
CCDC reference number 440/211. See http://www.rsc.org/suppdata/nj/b0/b004786o/ for crystallographic files in cif format.
Acknowledgements
We are grateful to Prof. M. D. Fryzuk (Vancouver) for his hospitality toward F.N. and to the Centre National de la Recherche Scientifique (Paris) and the Ministère de l'Enseignement Supérieur de la Recherche et de la Technologie (Paris) for financial support and a PhD grant to F.N.References
- T. G. Gant and A. I. Meyers, Tetrahedron, 1994, 50, 2297 CrossRef CAS.
- P. Braunstein and F. Naud, Angew. Chem., Int. Ed., 2001, 40, in press.
- C. Bolm, Angew. Chem., Int. Ed. Engl., 1991, 30, 542 CrossRef.
- A. K. Ghosh, P. Mathivanan and J. Cappiello, Tetrahedron: Asymmetry, 1998, 9, 1 CrossRef CAS.
- A. Pfaltz, Synlett, 1999, 835 CAS.
- S. E. Denmark, R. A. Stavenger, A.-M. Faucher and J. P. Edwards, J. Org. Chem., 1997, 62, 3375 CrossRef CAS.
- Y. Motoyama, N. Makihara, Y. Mikami, K. Aoki and H. Nishiyama, Chem. Lett., 1997, 951 CrossRef CAS.
- Y. Motoyama, Y. Mikami, H. Kawakami, K. Aoki and H. Nishiyama, Organometallics, 1999, 18, 3584 CrossRef CAS.
- P. G. Cozzi, E. Gallo, C. Floriani, A. Chiesi–Villa and C. Rizzoli, Organometallics, 1995, 14, 4994 CrossRef CAS.
- P. G. Cozzi, C. Floriani, A. Chiesi–Villa and C. Rizzoli, Inorg. Chem., 1995, 34, 2921 CrossRef CAS.
-
(a) C. Bolm, K. Weickhardt, M. Zehnder and D. Glasmacher, Hel. Chim. Acta, 1991, 74, 717 Search PubMed;
(b) C. Bolm, G. Schlingloff and K. Weickhardt, Angew. Chem.,
Int. Ed. Engl., 1994, 33, 1848 CrossRef;
(c) C. Bolm and G. Schlingloff, J. Chem. Soc., Chem. Commun., 1995, 1247 RSC;
(d) C. Bolm, T. K. K. Luong and K. Harms, Chem. Ber.
Recueil, 1997, 130, 887 Search PubMed.
- (a) H. Brunner and J. Berghofer, J. Organomet. Chem., 1995, 501, 161 CrossRef CAS; (b) H. Brunner and B. Hassler, Z. Naturforsch. B, 1998, 53, 126 Search PubMed.
- H. R. Hoveyda, V. Karunaratne, S. J. Rettig and C. Orvig, Inorg. Chem., 1992, 31, 5408 CrossRef CAS.
- H. Yang, M. A. Khan and K. M. Nicholas, Organometallics, 1993, 12, 3485 CrossRef CAS.
- M. Gómez–Simón, S. Jansat, G. Muller, D. Panyella, M. Font-Bardía and X. Solans, J. Chem. Soc., Dalton Trans., 1997, 3755 RSC.
- (a) C. Bolm, K. Muñiz–Fernández, A. Seger, G. Raabe and K. Günther, J. Org. Chem., 1998, 63, 7860 CrossRef CAS; (b) C. Bolm and K. Muñiz, Chem. Commun., 1999, 1295 RSC.
- Q.-L. Zhou and A. Pfaltz, Tetrahedron, 1994, 50, 4467 CrossRef CAS.
- H. Brunner and B. Hassler, Z. Naturforsch. B, 1998, 53, 476 Search PubMed.
- R. P. Singh, Synth. React. Inorg. Met.-Org. Chem., 1997, 27, 155 CAS.
- M. Bandini, P. G. Cozzi, L. Negro and A. Umani–Ronchi, Chem. Commun., 1999, 39 RSC.
- R. E. Lowenthal, A. Abiko and S. Masamune, Tetrahedron Lett., 1990, 31, 6005 CrossRef CAS.
- D. A. Evans, K. A. Woerpel, M. M. Hinman and M. M. Faul, J. Am. Chem. Soc., 1991, 113, 726 CrossRef CAS.
- J. M. Brown, P. J. Guiry, D. W. Price, M. B. Hursthouse and S. Karalulov, Tetrahedron: Asymmetry, 1994, 5, 561 CrossRef CAS.
- M. Nakamura, M. Arai and E. Nakamura, J. Am. Chem. Soc., 1995, 117, 1179 CrossRef CAS.
- M. Nakamura, A. Hirai and E. Nakamura, J. Am. Chem. Soc., 1996, 118, 8489 CrossRef CAS.
- E. J. Corey and Z. Wang, Tetrahedron Lett., 1993, 34, 4001 CrossRef CAS.
- J. Hall, J.-M. Lehn, A. DeCian and J. Fischer, Hel. Chim. Acta, 1991, 74, 1 Search PubMed.
- H. W. Görlitzer, M. Spiegler and R. Anwander, J. Chem. Soc., Dalton Trans., 1999, 4287 RSC.
- P. Braunstein, M. D. Fryzuk, M. Le Dall, F. Naud, S. J. Rettig and F. Speiser, J. Chem. Soc., Dalton Trans., 2000, 1067 RSC.
- P. Braunstein, C. Graiff, F. Naud, A. Pfaltz and A. Tiripicchio, Inorg. Chem., 2000, 39, 4468 CrossRef CAS.
- A. Bader and E. Lindner, Coord. Chem. Re., 1991, 108, 27 Search PubMed.
- S.-E. Bouaoud, P. Braunstein, D. Grandjean, D. Matt and D. Nobel, Inorg. Chem., 1986, 25, 3765 CrossRef CAS.
- J. Andrieu, P. Braunstein and A. D. Burrows, J. Chem. Res., Synop., 1993, 380 Search PubMed.
- W. Keim, H. Maas and S. Mecking, Z. Naturforsch. B, 1995, 50, 430 Search PubMed.
- G. J. P. Britovsek, W. Keim, S. Mecking, D. Sainz and T. Wagner, J. Chem. Soc., Chem. Commun., 1993, 1632 RSC.
- C. S. Slone, D. A. Weinberger and C. A. Mirkin, Prog. Inorg. Chem., 1999, 48, 233 Search PubMed.
- J. Andrieu, P. Braunstein, F. Naud and R. D. Adams, J. Organomet. Chem., 2000, 601, 43 CrossRef CAS.
- P. Braunstein, D. Matt, Y. Dusausoy, J. Fischer, A. Mitschler and L. Ricard, J. Am. Chem. Soc., 1981, 103, 5115 CrossRef CAS.
- P. Braunstein, D. Matt and D. Nobel, J. Am. Chem. Soc., 1988, 110, 3207 CrossRef CAS.
- P. Braunstein, Y. Chauvin, J. Nähring, A. DeCian, J. Fischer, A. Tiripicchio and F. Ugozzoli, Organometallics, 1996, 15, 5551 CrossRef CAS.
- W. Keim, Angew. Chem., Int. Ed. Engl., 1990, 29, 235 CrossRef.
- W. Keim, New J. Chem., 1994, 18, 93 Search PubMed.
- U. Klabunde, T. H. Tulip, D. C. Roe and S. D. Ittel, J. Organomet. Chem., 1987, 334, 141 CrossRef CAS.
- P. Braunstein, Y. Chauvin, S. Mercier, L. Saussine, A. DeCian and J. Fischer, J. Chem. Soc., Chem. Commun., 1994, 2203 RSC.
- D. Matt, M. Huhn, M. Bonnet, I. Tkatchenko, U. Englert and W. Kläui, Inorg. Chem., 1995, 34, 1288 CrossRef CAS.
- X. Gao, M. D. Fryzuk and S. J. Rettig, Can. J. Chem., 1995, 73, 1175 CAS.
- J. Pietsch, P. Braunstein and Y. Chauvin, New J. Chem., 1998, 221, 467 RSC.
- P. Braunstein, C. Graif, F. Naud and A. Tiripicchio, Chem. Commun., 2000, 897 RSC.
- T. G. Appleton, H. C. Clark and L. E. Manzer, Coord. Chem.
Re., 1973, 10, 335 Search PubMed.
- F. R. Hartley, Chem. Soc. Re., 1973, 2, 163 Search PubMed.
- K. L. Bray, C. P. Butts, G. C. Lloyd–Jones and M. Murray, J. Chem. Soc., Dalton Trans., 1998, 1421 RSC.
- P. Braunstein, L. Douce, F. Balegroune, D. Grandjean, D. Bayeul, Y. Dusausoy and P. Zanello, New J. Chem., 1992, 16, 925 Search PubMed.
- P. Braunstein, S. Coco Cea, A. DeCian and J. Fischer, Inorg. Chem., 1992, 31, 4203 CrossRef CAS.
- J. Bank, P. Steinert, B. Windmüller, W. Wolfsberger and H. Werner, J. Chem. Soc., Dalton Trans., 1996, 1153 RSC.
- G. Henig, M. Schulz and H. Werner, Chem. Commun., 1997, 2349 RSC.
- P. Braunstein, Y. Chauvin, J. Nähring, Y. Dusausoy, D. Bayeul, A. Tiripicchio and F. Ugozzoli, J. Chem. Soc., Dalton Trans., 1995, 851 RSC.
- P. Braunstein, Y. Chauvin, J. Nähring, A. DeCian and J. Fischer, J. Chem. Soc., Dalton Trans., 1995, 863 RSC.
- J. Andrieu, P. Braunstein and F. Naud, J. Chem. Soc., Dalton Trans., 1996, 2903 RSC.
- P. Braunstein, T. Stährfeldt and J. Fischer, C. R. Séances Acad. Sci., t. 2, Ser. IIc, 1999, 273 Search PubMed.
- P. Braunstein, M. D. Fryzuk, F. Naud and S. J. Rettig, J. Chem. Soc., Dalton Trans., 1999, 589 RSC.
- P. Braunstein, F. Naud, A. Pfaltz and S. J. Rettig, Organometallics, 2000, 19, 2676 CrossRef CAS.
- G. Zassinovich, G. Mestroni and S. Gladiali, Chem. Re., 1992, 92, 1051 Search PubMed.
- R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97 CrossRef CAS.
- M. J. Palmer and M. Willis, Tetrahedron: Asymmetry, 1999, 10, 2045 CrossRef CAS.
- A. Aranyos, G. Csjernyik, K. J. Szabó and J.-E. Bäckvall, Chem. Commun., 1999, 351 RSC.
- D. A. Alonso, P. Brandt, S. J. M. Nordin and P. G. Andersson, J. Am. Chem. Soc., 1999, 121, 9580 CrossRef CAS.
- (a) K.-J. Haack, S. Hashiguchi, A. Fujii, T. Ikariya and R. Noyori, Angew. Chem., Int. Ed. Engl., 1997, 36, 285 CrossRef CAS; (b) M. Yamakawa, H. Ito and R. Noyori, J. Am. Chem. Soc., 2000, 122, 1466 CrossRef CAS.
- Y. Jiang, Q. Jiang and X. Zhang, J. Am. Chem. Soc., 1998, 120, 3817 CrossRef CAS.
- Y. Nishibayashi, I. Takei, S. Uemura and M. Hidai, Organometallics, 1999, 18, 2291 CrossRef CAS.
- J. Takehara, S. Hashiguchi, A. Fujii, S.-I. Inoue, T. Ikariya and R. Noyori, Chem. Commun., 1996, 233 RSC.
- D. Carmona, C. Cativiela, S. Elipe, F. J. Lahoz, M.-P. Lamata, M.-P. López–Ram de Víu, L. A. Oro, C. Vega and F. Viguri, Chem. Commun., 1997, 2351 RSC.
- A. C. Cope and E. C. Friedrich, J. Am. Chem. Soc., 1968, 90, 909 CrossRef CAS.
- M. A. Bennett, T.-N. Huang, T. W. Matheson and A. K. Smith, Inorg. Synth., 1982, 21, 74 CAS.
- A. Sen and T.-W. Lai, Inorg. Chem., 1984, 23, 3257 CrossRef CAS.
- T. Y. Fu, Z. Liu, S. J. Rettig, J. R. Scheffer and J. Trotter, Acta Crystallogr., Sect. C, 1997, 53, 1577 CrossRef.
Footnotes |
| † Part of the Doctoral Thesis of F. N. |
| ‡ Deceased, October 27th 1998. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2001 |