DOI:
10.1039/B002905J
(Paper)
New J. Chem., 2001,
25, 40-54
Elucidation of the stereostructure of the annonaceous acetogenin (+)-montecristin
through total synthesis
Received 10th April 2000, Accepted 30th May 2000
First published on 7th December 2000
Abstract
Total syntheses of ent-5-epi-montecristin
(1a) and of (−)-montecristin (1b) were accomplished.
The stereocenters of compounds 1a and 1b were established
by asymmetric dihydroxylations of the trans-configurated β,γ-unsaturated
esters 6 ( →
4, up to 80% ee; Scheme 3; improved
procedure with up to 94% ee: Scheme 7) and 56 ( →
55, 97% ee: Scheme 9) while the stereogenic C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) C bonds
stem from the carbocuprations 48
→
49 and 50
→
51 (Scheme 9). Treating hydroxylactones 27 (Scheme 7),
3a (Scheme 12) and 3b (Scheme 13) with PPh3 and DEAD,
we found a racemization-free dehydration giving butenolide 26 and
epimerization-free dehydrations giving butenolides 2a and 2b
. Relating the [α]D values of synthetic
1a and 1b to the [α]D value
of natural (+)-montecristin, the absolute configuration of its side-chain
stereocenters was determined to be R.
C bonds
stem from the carbocuprations 48
→
49 and 50
→
51 (Scheme 9). Treating hydroxylactones 27 (Scheme 7),
3a (Scheme 12) and 3b (Scheme 13) with PPh3 and DEAD,
we found a racemization-free dehydration giving butenolide 26 and
epimerization-free dehydrations giving butenolides 2a and 2b
. Relating the [α]D values of synthetic
1a and 1b to the [α]D value
of natural (+)-montecristin, the absolute configuration of its side-chain
stereocenters was determined to be R.
Annonaceous acetogenins are an ever more numerous class of natural products
isolated from Annonaceae.1 They are γ-methylbutenolides
or γ-methylbutyrolactones with an unbranched C30 or C
32 side-chain at C-α. This side-chain is usually oxidized, exhibiting
one, two or three THF rings and/or hydroxy, acetoxy, epoxy or carbonyl
groups. The synthesis of such compounds has attracted much attention recently,2 in part because of their strong anti-tumor, anti-parasitic
and insecticide activities. A few annonaceous acetogenins contain cis
-configurated CC bonds in the place of of oxygenated side-chain
functions, such as muridienin3,4 or chatenaytrienin.4 These compounds are probably biogenetic precursors
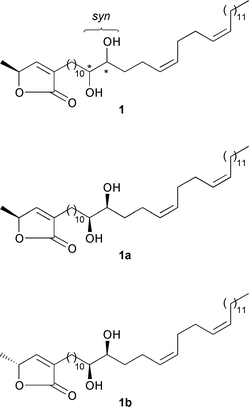
of the more heavily oxygenated annonaceous acetogenins.Given this background, the annonaceous acetogenin (+)-montecristin
(1; Scheme 1) isolated in 1997
from the roots of Annona muricata L.5
might be an intermediate en route between the less and the more oxygenated
acetogenins. Montecristin is an α,γ-disubstituted butenolide with
a C32 side-chain at C-α. The constitution of montecristin
was established by NMR spectroscopy, chemical derivatization and mass spectrometric
analysis.5 It has a side-chain which contains
a glycol moiety and two cis-configurated CC bonds. The relative
configuration of the glycol moiety was shown to be syn, while the
absolute configuration remained unknown. Montecristin also consists of a
butenolide moiety which possesses the S-configuration that is common
to all butenoide-containing annonaceous acetogenins.
 |
| | Scheme 1 | |
In recent years, we have prepared many kinds of γ-chiral butanolides
and butenolides by means of Sharpless' asymmetric dihydroxylation6 of trans-configurated β,γ-unsaturated
carboxylic esters.7 However, we had not yet
synthesized a γ-chiral butenolide having the substitution pattern displayed
by 1. Therefore, we chose this compound (or its enantiomer) as a
synthetic target. Since, however, the stereostructure of natural montecristin,
i.e. (+)-montecristin, was not known beyond formula
1 (Scheme 1, absolute configuration
shown), and since we also wished to determine the absolute configuration
of its side-chain, we had to synthesize two compounds rather than one; as
such, we chose structures 1a and 1b (
Scheme 1, absolute configurations shown), which are the
two possible diastereomers of structure 1 considering its relative
configuration. Considering absolute configurations, synthetic
1amight turn out to be identical with (+)-montecristin
because the former and the latter possess identically configurated
butenolide moieties. Conversely, synthetic 1bmight turn out
to be the enantiomer of (+)-montecristin (i.e. levorotatory
montecristin) because the former and the latter possess oppositely configurated
butenolide moieties. Accordingly, as soon as we would have synthesized both
1a and 1b we would know the complete stereostructure of (+)-montecristin.
However, we could not know beforehand whether at that point we would have
also synthesized (+)-montecristin or “only” (−)-montecristin.
It was evident that 1a and 1b would not be distinguishable
from one another spectroscopically since their stereocenters are 13 carbon
atoms apart. However, the specific rotations of 1a and 1b
would be distinct and therefore suitable for comparison with the specific
rotation reported for 1.
Results and discussion
Retrosynthesis
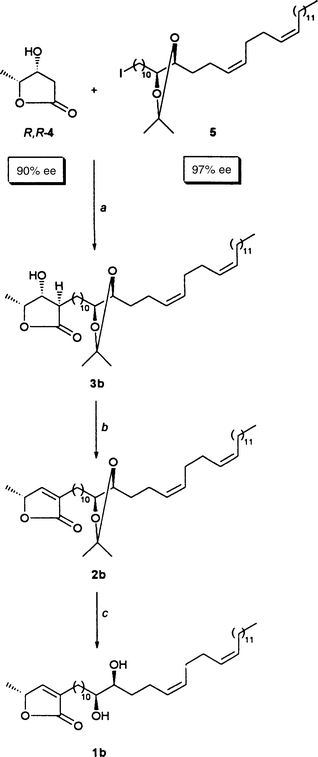
Our retrosynthetic analysis started by tracing back butenolides 1a
and 1b![[italic v]](https://www.rsc.org/images/entities/i_char_e0f5.gif) ia the acetonides 2a and
2b to the acetonide-containing hydroxylactones 3a and 3b
, respectively (Scheme 2). The latter
were thought to arise from the alkylation of the dilithio derivatives of hydroxylactones
S,S-4 and R,R-4, respectively,
with the acetonide-containing iodide 5. Hydroxylactones S,
S-4 and R,R-4 were alkylated in a
similar manner by simpler iodides than compound 5 in earlier work
of ours.7c–e,
g
ia the acetonides 2a and
2b to the acetonide-containing hydroxylactones 3a and 3b
, respectively (Scheme 2). The latter
were thought to arise from the alkylation of the dilithio derivatives of hydroxylactones
S,S-4 and R,R-4, respectively,
with the acetonide-containing iodide 5. Hydroxylactones S,
S-4 and R,R-4 were alkylated in a
similar manner by simpler iodides than compound 5 in earlier work
of ours.7c–e,
g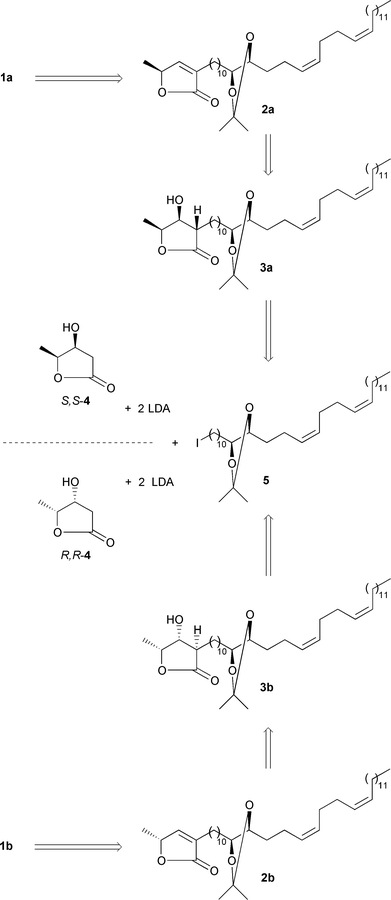 |
| | Scheme 2 | |
Our original preparation of hydroxylactone S,S-4
7c,8 was by the asymmetric
dihydroxylation of the commercially available pentenoic ester 6 (Scheme 3). The enantiomeric hydroxylactone R
,R-4 could be accessed analogously. Unfortunately,
these preparations suffered from low yields (⩽40%) and selectivities
(⩽80% ee). Since continuous extraction did not increase these yields,
we could exclude the possibility that some 4, because of its miscibility
with water, escaped our isolation procedure. The low ee values were probably
due to the smallness of the methyl substituent at the CC bond of our
dihydroxylation substrate 6. The adverse effect of too small substituents
was precedented.9
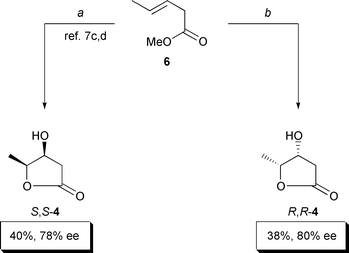 |
| | Scheme 3
a: AD-mix α (1.4 g mmol−1), ButOH–H
2O (1:1), 0°C, 4 days; 40%. b: AD-mix β (1.4
g mmol−1), ButOH–H2O (1:1), 0°C,
4 days; 38%. | |
The retrosynthetic simplification of iodide 5 (
Scheme 2) followed two strategies (Scheme
4). By “ strategy A”, we wanted to dihydroxylate enantioselectively
the CC bond of the enediynol precursor 9 and then hydrogenate
its CC bonds cis-selectively. Precursor 9 would
originate from an alkylation of a trans-configurated alkenyl metal
7 with the alkyl halide (or sulfonate) 8 or from a coupling
between a trans-configurated alkenyl iodide 7 and an organometallic
8. By “strategy B ”, iodide 5 would stem from a
saturated precursor 10 and from an unsaturated precursor 11
with two cis-CC bonds. We left open the question whether
10 should be the nucleophile and 11 the electrophile or
ice ersa. The glycol underlying compound 10
would be synthesized by the asymmetric dihydroxylation of an appropriate
trans-olefin.
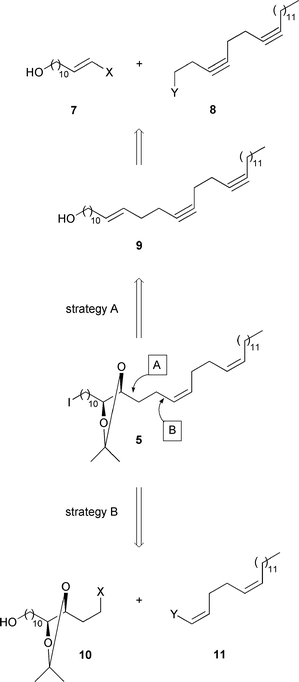 |
| | Scheme 4 | |
Synthesis of the lactone moiety
The discussion of Scheme 3 implied
that the desired hydroxylactones S,S-4 and
R,R-4 might be reached with >95% ee7 by modifying the β,γ-unsaturated ester
substrate of the dihydroxylation such that the small CH3 group
at C-γ would be made more voluminous by introducing one or several
bulky substituents. Scheme 5 shows our
vain efforts to prepare, in this sense, the tribromo analog 12 of
the previous dihydroxylation substrate 6. Tribromoacetaldehyde (
18) did not react with the ylide derived from zwitterion 17
by various deprotonating agents10 to give
the underlying tribromoacid 13; this was unexpected since tribromoacetaldehyde
forms an olefin with Ph3PCH–CHO.
11 Also, we could not add vinylmagnesium bromide to tribromoacetaldehyde
in order to attain alcohol 16. But 5% of this compound could
at least be obtained following a protocol for saturated aldehydes.12 This was insufficient for advancing to the next
step, which would have been the Buechi rearrangement
1316
→
14.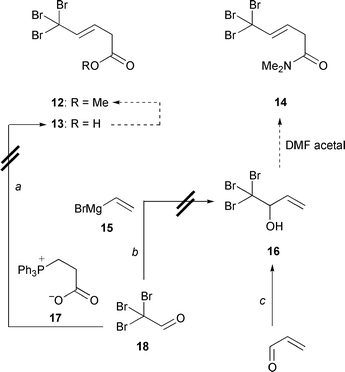 |
| | Scheme 5
a: (2-Carboxyethyl)triphenylphosphonium bromide (1.0 equiv.), NaH (2.0
equiv.) or KOBut (2.0 equiv.), THF–DMSO 1:1, room temperature,
20 h. b: CeCl3 (5 mol %), THF, room temperature,
1 h; 15 (1.1 equiv.), room temperature, 2 h; 0%. c:
Acrolein, CBr4 (3 equiv.), SnF2 (1 equiv.), DMSO, room
temperature, 5 min; SnF2 (1 equiv.), 5 min; 5%. | |
More modest size increases of the “too small methyl group”
of dihydroxylation substrate 6 were possible—now introducing
a single dummy substituent instead of three of them (
Scheme 6). We started from the known14
hydroxyester 20. It was converted into the phenylthio-containing
ester 23 by a Mukaiyama redox condensation.
15 Its asymmetric dihydroxylation gave the desired lactone 24
but the ee was 80% and thereby no better than the ee of the dihydroxylations
of Scheme 3. As an alternative, hydroxyester
20 was transformed into the chlorinated ester 22
16 under Corey’s conditions.17
The dihydroxylation of chloroester 22 gave only 19% of the
corresponding lactone 25, even when working in bicarbonate-buffered
solution as recommended for the asymmetric dihydroxylation of allyl halides.18 This 19% yield was too little to pursue
this approach. The only substituent tested that improved the yield of the
ester → lactone conversion and the enantioselectivity (92%
ee instead of 78% in the case of S,S-4)
was the ButMe2SiO group of ester 21.7c While 21 could be carried on
towards the silylated aglycone 19 of ranunculin as reported,7c there was no straightforward way for
converting it into the desired S,S-4.
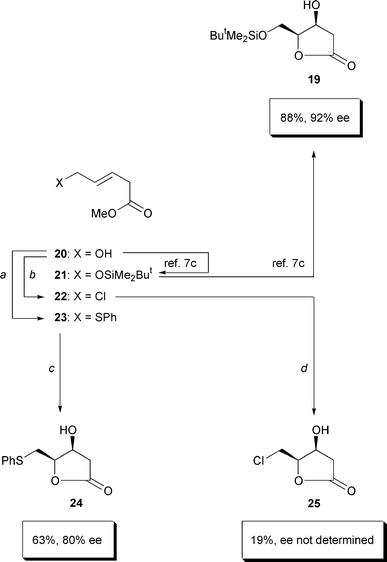 |
| | Scheme 6
a: Ph2S2 (3.0 equiv.), Bu3P (4.0 equiv.),
toluene, room temperature, 2 h; 81%. b: NCS (1.2 equiv.),
Me2S (1.2 equiv.), CH2Cl2, 0°C →
room temperature, 16 h; 83%. c: AD-mix α (1.4 g mmol
−1), MeSO2NH2 (1.0 equiv.), ButOH–H
2O (1:1), 0°C, 36 h; 63%. d: AD-mix α (1.4
g mmol−1), NaHCO3 (3.0 equiv.), MeSO2NH
2 (1.0 equiv.), ButOH–H2O (1:1), 0°C,
24 h; 19%. | |
Fortunately, we then found a chemically and stereochemically improved
synthesis of hydroxylactones S,S- and R,R-
4 (Scheme 7). An “ improved
asymmetric dihydroxylation” had been reported for a 1,1-disubstituted
olefin using 10 times more ligand and osmate;19
thereby, this olefin could be dihydroxylated with 97% ee rather than
with 85% ee under the standard conditions. Following the same procedure,
we dihydroxylated ester 6 with ees up to 86% (AD α, →
S,S-4) and 94% (AD β, →
R,
R-4). However, these values were not exactly reproducible. Rather,
they oscillated between 80 and 86% in the case of S,S
-4 and between 86 and 94% in the case of R,
R-4. (This random variation explains why starting material
4 of different ee was used in the reactions of
Schemes 7, 12 and 13). The improved dihydroxylation procedure
also conveniently reduced the reaction time to one-tenth (from 5 days to
16 h) and almost doubled the yields [from 40%
S,
S-4 (Scheme 3) to 73%
(Scheme 7) and from 38%
R,
R-4 (Scheme 3) to 69%
(Scheme 7)]. In addition, we recovered
the chiral ligand almost quantitatively (up to 94% yield) by extraction
into aqueous hydrochloric acid, addition of NaOH and back-extraction into
dichloromethane.
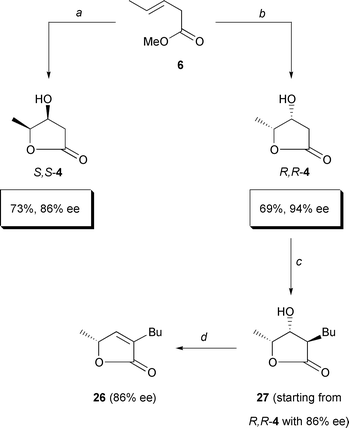 |
| | Scheme 7
a: K3Fe(CN)6 (3.0 equiv.), K2CO
3 (3.0 equiv.), (DHQ)2PHAL (10 mol %), K2OsO
4 (2.0 mol %), ButOH–H2O (1:1), 0°C,
16 h; 73%. b: K3Fe(CN)6 (3.0 equiv.),
K2CO3 (3.0 equiv.), (DHQD)2PHAL (10 mol %),
K2OsO4 (2.0 mol %), ButOH–H
2O (1:1), 0°C, 16 h; 69%. c: Pr2
iNH (2.5 equiv.), BuLi (2.5 equiv.), THF, − 78°C, 30 min;
addition of R,R-4, THF, − 78°C, 2 h;
BuI (1.2 equiv.), THF–DMPU (1:1), − 45°C, 20 h; 84%.
d: PPh3 (2.0 equiv.), DEAD (2.0 equiv.), THF, − 20°C →
room temperature, 3 h; 89%. | |
Repeating the completely trans-selective butylation previously
performed7c,d
with S,S-4 (78% ee) with the newly accessible
R,R-4 (here: of 86% ee) we obtained compound
2720 (ent-5-epi-blastmycinolactole;
86% ee). Compound 27 served to probe whether dehydration giving
butenolide 2621 would be possible
without partial racemization. This was of great interest since the penultimate
step of our synthesis of montecristin would be an exactly analogous dehydration
3
→
2 (cf. Scheme 2).
The problem is that butenolides such as compounds 26 or 2
can give up their stereochemical integrity even at room temperature when a
base as weak as diethylamine is present.22
Thus, we were afraid that the dehydration procedure appropriate for S
,S-4—treatment with MsCl and NEt3
in dichloromethane at 0°C, 30 min7d—would
put product stereochemistry at stake when applied to compound 27
(or later to 3) because starting from 27 it lasted several
hours at room temperature without even then having gone to completion. Therefore,
we were glad to find that the dehydration 27
→
26
could be brought about by treatment with 2 equivalents of both triphenylphosphine
and diethylazodicarboxylate.23 The reaction
proceeded in 89% yield and conserved the optical purity (86%
ee) completely. This was reassuring as concerned the envisaged approach to
the butenolide moiety of montecristin (Scheme 2).
Synthesis of the side-chain
Scheme 8 summarizes our efforts to
access iodide 5 by means of strategy A of
Scheme 4. The upper part of this scheme concerns the synthesis
of the diyne equivalent 35 of the diyne synthon 8 of Scheme 4. First, we protected
24 3-butynol (28) as the THP ether 29.25 This compound was less easy to alkylate than the
homologous THP ether 38 of propargyl alcohol. Deprotonating compound
29 with n-BuLi in a mixture of THF and DMSO
26 and adding dodecyl bromide thereafter led mainly to the formation
of 1-dodecene and only 7% of the alkylation product 30.27 Changing the solvent to DMPU increased the yield
of 30 to 17%. Replacing dodecyl bromide by dodecyl triflate
gave 48%
30. A 53% yield was finally obtained in THF–DMSO,
using the cheaper dodecyl bromide as the alkylating agent but deprotonating
the substrate with sodium amide. The resulting THP ether 30 was cleaved
in methanol through the action of TsOH (99% yield).
28 The alkynol 3129 thus
obtained was converted into a series of alkylating agents by treatment with
NBS–PPh330 ( → bromide
32, 88%) or PPh3–imidazole–I231 ( → iodide 33, 88%)
or Tf2O–NEt332
( → triflate 35, quantitative yield). However, none of them
could be introduced into THP ether 29 in satisfactory yield: the
sodium acetylide of compound 29 and bromide 32 gave the
desired diyne 35 in only 4% yield33
besides ca. 50% of the elimination product
36. Similarly, the lithium acetylide of compound 29 and iodide
33 furnished only trace amounts of diyne 35 besides 55%
of enyne 36. The same lithium acetylide and triflate 34
were a better combination (as expected34),
delivering diyne 35 as the major product and only traces of
36. However, the yield of 35 was 10–32% and never
became higher or reliable.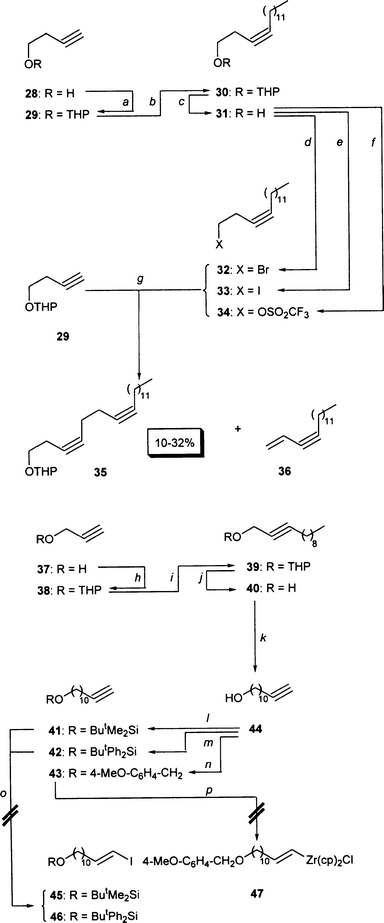 |
| | Scheme 8
a: Dihydro-2H-pyran (3.0 equiv.), camphor sulfonic acid (calatytic
amount), CH2Cl2, 0°C → room temperature, 3
h; 96%. b: NaNH2 (1.1 equiv.), THF, 0°C, 1
h; 1-bromododecane (1.1 equiv.), DMSO, room temperature, 2.5 h; 53%.
c: p-TsOH (0.4 equiv.), MeOH, room temperature, 30 min; 99%.
d: PPh3 (1.2 equiv.), NBS (1.1 equiv.), THF, − 20°C →
0°C, 4 h; 88%. e: PPh3 (1.1 equiv.), imidazole
(2.2 equiv.), I2 (1.1 equiv.), THF, 0°C, 1 h; 88%.
f: NEt3 (1.2 equiv.), Tf2O (1.2 equiv.), CH
2Cl2, 0°C, 2 h; quantitative. g: BuLi (1.1 equiv.),
THF, 0°C, 30 min; addition of alkylating agent (1.0 equiv.), THF, room
temperature, 16 h; 10–32%. Dihydro-2H-pyran (3.0 equiv.),
PPTS (cat.), CH2Cl2, 0°C → room temperature,
16 h; 84%. i: BunLi (1.2 equiv.), THF, 0°C,
30 min; Non-Br (1.1 equiv.), DMSO, room temperature, 24 h; 68%.
j: TsOH (cat.), MeOH, room temperature, 2 h; 84%. k:
Li (6.0 equiv.), 1,2-diaminopropane, reflux, 30 min; KOBut (4.0
equiv.), room temperature, 30 min; addition of 40, room temperature,
1 h; 74%. l: ButMe2SiCl (1.1 equiv.),
imidazole (2.1 equiv.), CH2Cl2, room temperature, 1
h; 97%. m: ButPh2SiCl (1.0 equiv.),
imidazole (2.1 equiv.), CH2Cl2, 0°C → room
temperature, 1 h; 99%. n: NaH (1.2 equiv.), para-methoxybenzyl
chloride (1.2 equiv.), DMF–THF 1:1, room temperature, 3 h; 96%.
o: DIBAL (1.05 equiv.), hexane, room temperature, 1 h; I2
(1.0 equiv.). p: Zr(cp)2ClH (0.95 equiv.), THF, room temperature,
1 h. | |
The lower part of Scheme 8 shows our
approach to the trans-olefin equivalents 45–
47 of synthon 7 of Scheme 4.
Protecting24 propargyl alcohol (37)
as the THP ether 38,35 alkylating
the latter ia its anion26
with nonyl bromide, deprotecting28 the resulting
chain-elongated THP ether 39 ( →
40), shifting its
C![[triple bond, length half m-dash]](https://www.rsc.org/images/entities/char_e007.gif) C bond to the end of the molecule36
( →
4437), and protecting the
OH group led to the terminal alkynes 41–43. DIBAL
reduction/iodination of compounds 41 and 42 seemed
to suffer from the sensitivity of the silyl ethers towards the reductant.
When the hydrozirconation of 41–43 started with unpromising
yields of the respective addition product 47, we stopped pursuing
strategy A of Scheme 4 towards iodide
5 and began to test strategy B. As shown in the upper part of Scheme 9, we synthesized the cis,cis
-dienyliodide 51 as a realization of synthon 11 of Scheme 4. To this end, dodecyl bromide (48)
was converted ia the corresponding Gilman cuprate into
a mixed cuprate (with a hexynyl group). It was added to acetylene whereupon
the resulting cis-vinylcuprate was transmetalated with hexynyllithium,
giving a mixed cuprate that was hydroxyalkylated with ethylene oxide, all
as described in the pionieering study of Alexakis et al.
38 Thus we advanced in a single step and 81% yield to homoallyl
alcohol 49.39 It was converted into
iodide 50 by treatment with PPh3–imidazole–I
2.31 Compound 50 was subjected
to a Li/I exchange reaction with ButLi.
40 Conversion into a Gilman cuprate followed by addition to acetylene
and iodiation of the resulting alkenylcopper intermediate
41 rendered the dienyliodide 51 with the desired cis
,cis-configuration in 66% yield. We found that the cuprate
precursor of compound 51 did not react well with iodide 5.
Quenching this cuprate as shown here and regenerating the organolithium derivative
later (first reaction of Scheme 10) worked
much better.
C bond to the end of the molecule36
( →
4437), and protecting the
OH group led to the terminal alkynes 41–43. DIBAL
reduction/iodination of compounds 41 and 42 seemed
to suffer from the sensitivity of the silyl ethers towards the reductant.
When the hydrozirconation of 41–43 started with unpromising
yields of the respective addition product 47, we stopped pursuing
strategy A of Scheme 4 towards iodide
5 and began to test strategy B. As shown in the upper part of Scheme 9, we synthesized the cis,cis
-dienyliodide 51 as a realization of synthon 11 of Scheme 4. To this end, dodecyl bromide (48)
was converted ia the corresponding Gilman cuprate into
a mixed cuprate (with a hexynyl group). It was added to acetylene whereupon
the resulting cis-vinylcuprate was transmetalated with hexynyllithium,
giving a mixed cuprate that was hydroxyalkylated with ethylene oxide, all
as described in the pionieering study of Alexakis et al.
38 Thus we advanced in a single step and 81% yield to homoallyl
alcohol 49.39 It was converted into
iodide 50 by treatment with PPh3–imidazole–I
2.31 Compound 50 was subjected
to a Li/I exchange reaction with ButLi.
40 Conversion into a Gilman cuprate followed by addition to acetylene
and iodiation of the resulting alkenylcopper intermediate
41 rendered the dienyliodide 51 with the desired cis
,cis-configuration in 66% yield. We found that the cuprate
precursor of compound 51 did not react well with iodide 5.
Quenching this cuprate as shown here and regenerating the organolithium derivative
later (first reaction of Scheme 10) worked
much better.
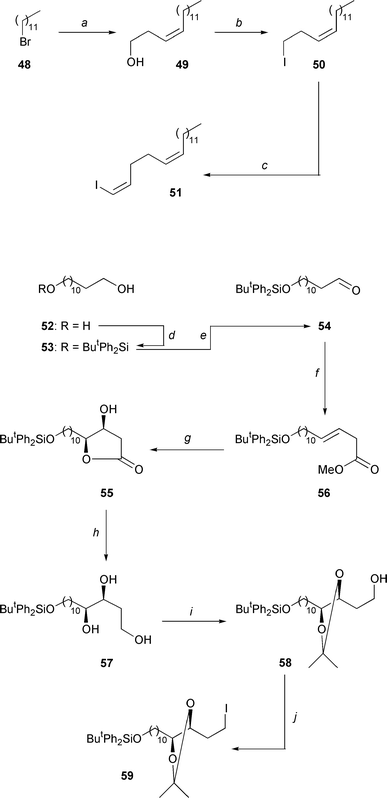 |
| | Scheme 9
a: Li (3.0 equiv.), Et2O, 0°C, 3 h; CuI (0.5 equiv.),
Et2O, − 35°C, 30 min; acetylene (1.0 equiv.), −
50°C →
− 25°C, 30 min; →
− 30°C; ethylene
oxide (1.0 equiv.); hexynyl lithium (0.5 equiv.), Et2O, −
15°C, 3 h; 81%. b: PPh3 (1.1 equiv.), imidazole
(2.2 equiv.), I2 (1.1 equiv.), THF, 0°C → room temperature,
30 min; 99%. c: ButLi (2.0 equiv.), Et2O–hexane
(1:1), − 20°C, 30 min; CuI (0.5 equiv.), Et2O, −
35°C, 30 min; acetylene (1.0 equiv.), − 50°C →
−
25°C, 1 h; I2 (1.0 equiv.), − 60°C →
−
10°C, 2 h; 66%. d: ButPh2SiCl (1.0
equiv.), imidazole (2.0 equiv.), DMF, room temperature, 15 h; 59%.
e: (ClCO)2 (1.1 equiv.), DMSO (2.2 equiv.), CH2Cl
2, − 78°C, 3 min; addition of 53, − 40°C,
1 h; NEt3 (5.0 equiv.), − 40°C → 0°C, 1 h; 83%.
f: HO2CCH2CO2Me (1.1 equiv.), NEt
3 (1.1 equiv.), 90°C, 12 h, 66%. g: K3Fe(CN)
6 (3.0 equiv.), K2CO3 (3.0 equiv.), (DHQ)
2PHAL (1.0 mol %), K2OsO4 (0.2 mol %),
MeSO2NH2 (1.0 equiv.), ButOH–H
2O (1:1), 0°C, 4 days; 68%. h: LiAlH4
(1.0 equiv.), THF, − 78°C → room temperature, 30 min; 98%.
i: 2,2-Dimethoxypropane (8.0 equiv.), Amberlyst 15, acetone, room temperature,
2 h. j: PPh3 (1.0 equiv.), imidazole (2.0 equiv.), I
2 (1.0 equiv.), THF, 0°C → room temperature, 15 min; 94%
for two steps. | |
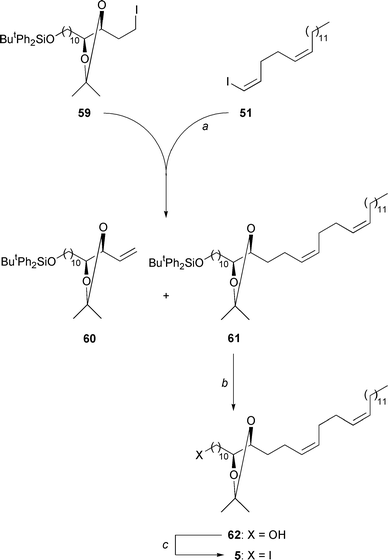 |
| | Scheme 10
a: 51, ButLi (2.2 equiv.), Et2O, −
50°C, 30 min; 59 (1.0 equiv.), THF, → room temperature,
4 h; 64%. b: TBAF (1.2 equiv.), THF, room temperature, 20
h; 99%. c: PPh3 (1.2 equiv.), imidazole (2.4 equiv.),
I2 (1.2 equiv.), THF, 0°C → room temperature, 30 min;
96%. | |
Next, we synthesized acetonide 59 (Scheme
9, bottom half) as an equivalent of synthon 10 of Scheme 4. We started by monosilylating 1,12-dodecanediol
(52) with ButPh2SiCl. Silylether 53
was formed in 59% yield. It was oxidized by the method of Omura and
Swern42 but at slightly higher temperature
(−40°C → 0°C) and longer than usual times in order to
make up for the poor solubility of the substrate in cold dichloromethane.
Aldehyde 54, obtained in 83% yield, was subjected to a deconjugating
decarboxylating Knoevenagel condensation with monomethyl malonate.43 It provided 66% of the trans-configurated
β,γ-unsaturated ester 56. The asymmetric dihydroxylation
of this compound7 using AD-mix α furnished
the hydroxylactone 55 in 68% yield. The enantiomeric purity
of this material was determined to be 97% ee by 1H-NMR
analysis of the methoxy singlets of its R-Mosher ester.
Hydroxylactone 55 was reduced with LiAlH4, giving
the 1,3,4-triol 57. After hydrolytic work-up, extraction by dichloromethane,
and evaporation of the solvent 98% of a white solid remained, which
was used without purification. Meyer's selective acetalizations of the butanetriol
obtained from optically active malic acid revealed that under thermodynamic
control acetone is preferentially taken up by the 1,2-diol moiety, giving
a dioxolane, rather than by the 1,3-diol moiety, which would give a 1,3-dioxane.44 Exploiting this effect, triol 57, excess
dimethoxypropane in acetone and Amberlyst as a catalyst provided, after 2
h at room temperature, essentially 1,3-dioxane 58. This compound
was so sensitive towards hydrolysis that we could not purify it even by flash
chromatography on deactivated (NEt3) silica gel. However, we could
use it crude and transform it by treatment with PPh3–imidazole–I
231 into iodide 59 (94%
yield from lactone 56). Thereby, the latter became accessible from
1,12-dodecanediol (52) in seven steps (20% overall yield).
The ultimate steps to the key precursor 5 of montecristin are
shown in Scheme 10. To combine the alkyl
iodide 59 with the alkenyl iodide 51 we had the options
of performing a transition metal catalyzed coupling between an alkyl
metal and an alkenyl halide or of performing a nucleophilic
substitution of an alkenyl metal at an alkyl halide. Lithiation
of alkyl iodide 59 with tert-BuLi, followed by transmetalation
with MgBr2 and the addition of alkenyl iodide 51 and a
catalytic amount of NiCl2(PPh3)4
45 gave a ≈1:1 mixture of the desired coup ling product 61
and the β-hydride elimination product—which is a vinyl-substituted
dioxolane—of the alkyl nickel derivative of 59. Conversely,
clean C–C bond formation occurred when we switched polarities: lithiated
the alkenyl iodide 51, added the alkyl iodide 59, and isolated
the alkylation46 product 61 in 64%
yield. Desilylation liberated the underlying alcohol 62. It reacted
with PPh3–imidazole–I2
31 to give the key iodide 5.
One issue needed to be clarified before iodide 5 was fully qualified
for introducing the side-chain of montecristin (1) because the
ic-glycol moiety of 1 was protected as an acetonide
in 5. We wondered whether we would be able to release this acetonide
at the butenolide stage 2a/2b (cf. Scheme 2) without destroying the stereocenter C-γ.
We modelled the answer to this question by effecting the related acetonide
cleavage 5
→
63 with concentrated HCl–MeOH–CH
2Cl247 (
Scheme 11). What mattered in this context was hardly the 50%
yield of diol 63 but rather that the model butenolide 26,
which was present while the acetonide was cleaed, could
be re-isolated with undiminished enantiomeric purity (86%
ee; 77% yield). This meant that these conditions were suitable for
deprotecting butenolides 2a (Scheme 12)
and 2b (Scheme 13) without affecting
their stereostructures.
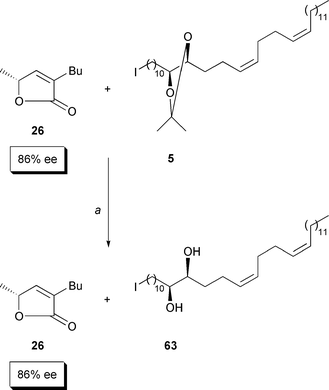 |
| | Scheme 11
a: HCl (4.0 equiv.), MeOH–CH2Cl2 (5:1), room
temperature, 24 h; 50%; 76% of reisolated 26. | |
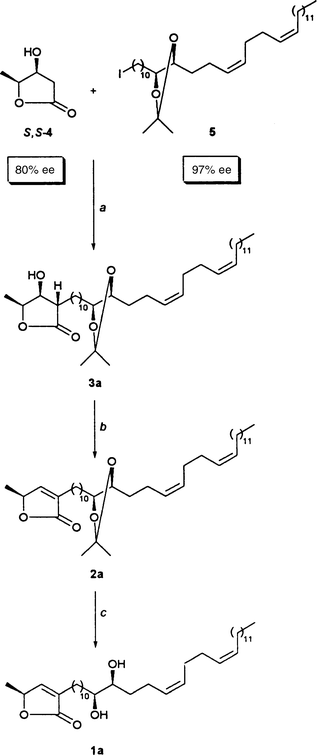 |
| | Scheme 12
a: Pr2iNH (2.5 equiv.), BuLi (2.5 equiv.), THF, −
78°C, 30 min; addition of S,S-4, THF, −
78°C, 2 h; 5 (1.0 equiv.), THF–DMPU (1:1), − 45°C,
20 h; 56%; 38% of reisolated 5. b: PPh
3 (2.0 equiv.), DEAD (2.0 equiv.), THF, − 20°C → room
temperature, 4 h; 94%. c: HCl (4.0 equiv.), MeOH–CH
2Cl2 (5:1), room temperature, 24 h; 88%. | |
 |
| | Scheme 13
a: Pr2iNH (2.5 equiv.), BuLi (2.5 equiv.), THF, −
78°C, 30 min; addition of R,R-4, THF, −
78°C, 2 h; 5 (1.0 equiv.), THF–DMPU (1:1), − 45°C,
20 h; 62%; 34% reisolated 5. b: PPh3
(2.0 equiv.), DEAD (2.0 equiv.), THF, − 20°C → room
temperature, 4 h; 97%. c: HCl (4.0 equiv.), MeOH–CH
2Cl2 (5:1), room temperature, 24 h; 83%. | |
Combining the building blocks
Reassured by the result from Scheme 11,
we performed the final steps of our syntheses in parallel experiments. We
headed for the stereoisomer 1a of montecristin as shown Scheme 12 and for its diastereomer 1b
as shown in Scheme 13.Each sequence lasted three steps. The start was deprotonating the enantiomeric β-hydroxylactones
S,S-4 (here 80% ee;
Scheme 12) and R,R-4 (here 90%
ee; Scheme 13) twice using 2.5 equiv.
of LDA. In HMPA-containing THF the α-alkylation of dilithiated β-hydroxy-γ-lactones
occurs such that the α-substituent is oriented exclusively trans
with respect to the β-OH group.48
Conveniently, alkylating the dilithiated hydroxylactones S,S
- and R,R-4 with iodide 5 (97%
ee) in 6:1 THF–DMPU49 delivered also
nothing but trans-alkylated hydroxylactones, namely compounds
3a (56% yield; 90% considering that 38%
5
was recovered) and 3b (62% yield; 94% considering that
34%
5 was recovered), respectively.
The ensuing β-eliminations followed the protocol developed for the
dehydration 27
→
26 of Scheme
7, that is under Mitsunobu conditions: 2 equiv. each of PPh
3 and DEAD were added to THF solutions of hydroxylactones 3a
(Scheme 12) and 3b (Scheme 13). The resulting mixtures were gradually
warmed from − 20°C to room temperature. Thereupon we isolated
94% of butenolide 2a and 97% of butenolide 2b,
respectively. The terminating steps were the acetonide cleavages. They were
effected under the “stereochemically benign ” conditions of Scheme 11. This led to diastereomers 1a
and 1b in yields of 88 and 83%, respectively.
As expected, the 1H-NMR, 13C-NMR and IR data of compounds
1a and 1b were identical with one another and identical to those
of montecristin. However, their specific rotations were distinct. They demonstrated
unequivocally50 that 1a is ent
-5-epi-montecristin while 1b is the enantiomer of
(+)-montecristin. This statement is justified even if our specimen of
1a must have been a ca. 90:10 mixture with 1b and our
specimen of 1b a ca. 95:5 mixture with 1a; the
occurrence of these mixtures is an inevitable consequence of the incomplete
optical purity of the building blocks S,S-4 (80%
ee), R,R-4 (90% ee) and 5: (97%
ee) incorporated into 1a and 1b.
In summary, we have accomplished the first total syntheses of ent
-5-epi-montecristin (1a) and (−)-montecristin
(1b). In the longest linear sequence 13 steps were needed, which
gave an overall yield of 6% ( = 81% per step). The side-chain
configuration of (+)-montecristin was established to be 11′
R,12′R.
Experimental
General
All reactions were performed in oven-dried (80°C) glassware under N
2. THF was freshly distilled from K, Et2O from Na, CH
2Cl2, DMSO, DMPU and 1,2-diaminopropane from CaH2.
Products were purified by flash chromatography51
on Macherey–Nagel silica gel 40–63 μm (eluents given in brackets).
Yields refer to analytically pure samples. Isomer ratios were derived from
suitable 1H NMR integrals. 1H NMR [CHCl3
(7.26 ppm) as internal standard in CDCl3 or C6HD
5 (7.16 ppm) as internal standard in C6D6]
and 13C NMR [CDCl3 (77.00 ppm) as internal standard
in CDCl3] were recorded on Varian VXR 200, Bruker AMX 300,
and Varian VXR 500S spectrometers. For 1H NMR spectra the integrals
were in accord with assignments; coupling constants are in Hz. In APT
13C NMR spectra the peak orientations were in accord with asssignments.
The assignments of 1H and 13C NMR resonances refer
to the IUPAC nomenclature with primed numbers belonging to the side-chains
in the order of their appearance in the IUPAC name. Combustion analyses were
obtained by F. Hambloch, (Institute of Organic Chemistry, University of Göttingen)
and MS by Dr. G. Remberg (Institute of Organic Chemistry, University of Göttingen).
IR spectra were recorded on a Perkin–Elmer 1600 Series FTIR spectrometer
neat or in KBr. Specific rotations were measured on a Perkin–Elmer
polarimeter 241 at 589 nm; the underlying rotational values were averaged
over 5 measurements (undertaken with a given solution of the respective sample).
Melting points were measured on a Dr. Tottoli apparatus (Büchi) and are
uncorrected.Syntheses
(5S,11′S,12′S)-
Z,Z-3-(10,11-Dihydroxy-15,19-dotriacontadienyl)-5-methyl-2(5
H)-furanone (ent-5-epi-montecristin) (1a).
HCl (12 M, 15 μl, 0.18 mmol, 4.0 equiv.) was added to a solution of the
acetonide 2a (27.0 mg, 0.0441 mmol) in MeOH–CH2Cl
2 (5:1, 0.6 ml) and the mixture was stirred for 24 h at room temperature.
Water (2 ml) was added and the organic phase extracted with ButOMe
(3 × 10 ml). The combined organic phases were dried over Na2SO
4 and the solvent was removed. The residue was purified by flash chromatography
(2.5 cm, petroleum ether–ButOMe 2:1 → fraction 13, 1:1 →
fraction 20, fractions 8–18) to give the title compound (22.3 mg, 88%)
as a white solid (mp 52°C). [α]D
25
=
+ 1.9 (c
= 0.4). Calculating the specific
rotation for the 100% enantiopure product, taking into account the
ees of the different building blocks (lactone S,S-4
80% ee; iodide 5 97% ee) as well as the specific
rotation measured for compound 1b (ide infra) leads
to [α]D25
=
+ 4.7
(c
= 0.4)52
{lit.5: [
α]D25
=
+ 25 for montecristin
(c
= 0.1, MeOH)}. 1H NMR (300 MHz): δ
= 0.88 (t, J32′,31′
=
6.8, 32′-H3), 1.24–1.38 (m, 2′-H2
to 9′-H2, 22′-H2 to 31′-H2),
1.41 (d, J5,5-Me
= 6.8, 5-Me), 1.44–1.60
(m, 10′-H2, 13′-H2), ca. 1.94 (very
br s, 2 OH), in part superimposed by 2.02 (td, J21′,22′
≈J21′,20′≈6.5, 21′-H
2), 2.11 (mc , 17′-H2, 18′-H
2), in part superimposed by 2.13–ca. 2.23 (m, 14′-H
2), in part superimposed by 2.26 (tdd, J1′,2′
= 7.8, 4J1′,4
=
5J1′,5
= 1.7, 1′-H2),
3.38–3.48 (m, 11′-H, 12′-H), 5.00 (qdt, J
5,5-Me
= 6.6, J5,4
=
5
J5,1′
= 1.8, 5-H), 5.30–5.47 (m, 15′-H,
16′-H, 19′-H, 20′-H), 6.99 (td, 4J
4,1′
=
J4,5
= 1.5, 4-H).
13C NMR (125.7 MHz, APT; slightly contaminated at δ
=
29.1): δ
= 14.05 (C-32′), 19.10 (5-Me), 22.61,
23.45, 25.06, 25.58, 27.19 (2-fold intensity), 27.29 (2-fold intensity), 29.07,
29.18, 29.25, 29.28, 29.37, 29.43, 29.46, 29.49, 29.58 (2-fold intensity),
29.60, 29.61, 29.65, 31.84, 33.39 and 33.51 (C-1′ to C-10′, C-13′,
C-14′, C-17′, C-18′, C-21′ to C-31′; 21 resonances
of 24-fold total intensity for 25 C atoms), 73.94 and 74.39 (C-11′,
C-12′), 77.42 (C-5), 128.88, 129.35, 129.99 and 130.45 (C-15′,
C-16′, C-19′, C-20′), 134.16 (C-3), 148.93 (C-4), 173.93
(C-2). IR (KBr): ν
= 3415, 3355, 3000, 2920, 2850, 1740,
1655, 1465, 1365, 1320, 1205, 1085, 1065, 1025, 930, 895, 720 cm−1
. C37H66O4 (574.9) calcd. C 77.30,
H 11.57; found C 77.27, H 11.33. (5R,11′S,12′S)-
Z,Z-3-(11,12-Dihydroxy-15,19-dotriacontadienyl)-5-methyl-2(5
H)-furanone (ent-montecristin) (1b). 1b was
prepared as for 1a using HCl (12 M, 20 μl, 0.22 mmol, 4.0 equiv.)
and acetonide 2b (33.0 mg, 0.0544 mmol). The residue obtained after
work-up was purified by flash chromatography (2.5 cm, petroleum ether–Bu
tOMe 2:1 → fraction 12, 1:1 → fraction 20, fractions 8–19)
to give the title compound (25.8 mg, 83%) as a white solid (mp 58°C,
lit.5: for the enantiomer 62°C). [
α]D25
=
− 24.2 (c
=
2.48). Calculating the specific rotation for the 100% enantiopure product,
taking into account the ees of the different building blocks (lactone
R,R-4 90% ee; iodide 5 97%
ee) as well as the specific rotation measured for compound 1a (
ide supra) leads to [α]D
25
=
− 25.9 (c
= 0.4)
52
{lit. 5: [α
]D25
=
+ 25 for montecristin (
c
= 0.1, MeOH)}. 1H NMR (300 MHz): δ
= 0.88 (t, J32′,31′
=
6.8, 32′-H3), 1.25–1.38 (m, 2′-H2
to 9′-H2, 22′-H2 to 31′-H2
), 1.41 (d, J5,5-Me
= 6.7, 5-Me), 1.43–1.60
(m, 10′-H2, 13′-H2), ca. 1.82 (very
br s, 2 OH), 2.02 (td, J21′,22′≈J
21′,20′≈6.5, 21′-H2), in part superimposed
by 2.11 (mc, 17′-H2, 18′-H2),
in part superimposed by 2.13–ca. 2.23 (m, 14′-H
2), in part superimposed by 2.26 (tdd, J1′,2′
= 7.8, 4J1′,4
=
5J1′,5
= 1.7, 1′-H2),
3.38–3.47 (m, 11′-H, 12′-H), 5.00 (qdt, J
5,5-Me
= 6.8, J5,4
=
5
J5,1′
= 1.8, 5-H), 5.30–5.47 (m, 15′-H,
16′-H, 19′-H, 20′-H), 6.98 (td, 4J
4,1′
=
J4,5
= 1.5, 4-H).
13C NMR (125.7 MHz, APT): δ
= 14.00 (C-32′),
19.05 (5-Me), 22.56, 23.40, 25.02, 25.56, 27.15 (2-fold intensity), 27.24
(2-fold intensity), 29.03, 29.15, 29.20, 29.23, 29.34, 29.40, 29.45, 29.53
(2-fold intensity), 29.56 (2-fold intensity), 29.61, 31.79, 33.36 and 33.47
(C-1′ to C-10′, C-13′, C-14′, C-17′, C-18′,
C-21′ to C-31′, 19 resonances of 23-fold total intensity for 25
C atoms), 73.86 and 74.31 (C-11′, C-12′), 77.40 (C-5), 128.85,
129.34, 129.86 and 130.36 (C-15′, C-16′, C-19′, C-20′),
134.08 (C-3), 148.93 (C-4), 173.90 (C-2). C37H66O
4 (574.9) calcd. C 77.30, H 11.57; found C 77.41, H 11.50. (5S,4″S,5″S)-
Z,Z-3-{10-[5-(3,7-Eicosadienyl)-2,2-dimethyl-1,3-dioxolan-4-yl]decyl}-5-methyl-2(5
H)-furanone (2a). A solution of the β-hydroxylactone
3a (42 mg, 0.066 mmol) in THF (2 ml) was treated with PPh3
(40.8 mg, 0.132 mmol, 2.0 equiv.) and DEAD (40% in toluene, 68 μl,
26 mg, 0.13 mmol, 2.0 equiv.) at − 20°C. The reaction mixture was
warmed to room temperature within 4 h. Water (2 ml) was added and the resulting
mixture was extracted with ButOMe (3 × 10 ml). The organic
phase was dried over MgSO4 and the solvent was removed. From the
residue the title compound (38.2 mg, 94%) was isolated as a colorless
oil by flash chromatography (2 cm, petroleum ether–ButOMe
10:1, fractions 4–9). [α]D
25
=
− 1.5 (c
= 1.4). 1H NMR
(300 MHz): δ
= 0.88 (t, J20‴,19‴
= 6.8, 20‴-H3), 1.24–ca. 1.38
(m, 2′-H2 to 9′-H2, 10‴-H2
to 19‴-H2), superimposed by 1.379 and 1.382 [2 s,
2″-(CH3)2], 1.41 (d, J5,5-Me
= 6.7, 5-Me), 1.47–1.60 (m, 10′-H2 ,
1‴-H2), 2.02 (td, J9‴,10‴≈
J9‴,8‴≈6.6, 9‴-H2), in
part superimposed by 2.10 (mc, 5‴-H2, 6‴-H
2), in part superimposed by 2.13–ca. 2.23 (m, 2‴-H
2), in part superimposed by 2.26 (tdd, J1′,2′
= 7.1, 4J1′,4
=
5J1′,5
= 1.7, 1′-H2),
3.60 (mc , 4″-H, 5″-H), 4.99 (qdt, J
5,5-Me
= 6.8, J5,4
=
5
J5,1′
= 1.7, 5-H), 5.30–5.46 (m, 3‴-H,
4‴-H, 7‴-H, 8‴-H), 6.98 (td, 4J
4,1′
=
J4,5
= 1.5, 4-H). IR
(neat): ν
= 2985, 2925, 2855, 1760, 1460, 1370, 1320,
1240, 1080, 1025, 950, 870, 725 cm−1. C40H
70O4 (615.0) calcd. C 78.12, H 11.47; found C 78.01, H 11.39.
(5R,4″S,5″S)-
Z,Z-3-{10-[5-(3,7-Eicosadienyl)-2,2-dimethyl-1,3-dioxolan-4-yl]decyl}-5-methyl-2(5
H)-furanone (2b). 2b was prepared as for 2a
using β-hydroxylactone 3b (64 mg, 0.101 mmol) in THF (2 ml),
PPh3 (52.9 mg, 0.202 mmol, 2.0 equiv.) and DEAD (40% in
toluene, 102 μl, 38.6 mg, 0.202 mmol, 2.0 equiv.). From the residue of
the work-up the title compound (60.1 mg, 97%) was isolated as a colorless
oil by flash chromatography (2.5 cm, petroleum ether–ButOMe
10:1, fractions 4–10). [α]D
25
=
− 20.0 (c
= 2.25). 1H NMR
(300 MHz; contains 1.2 wt.% dihydro-DEAD; quartet at δ
=
4.99): δ
= 0.88 (t, J20‴,19‴
= 6.8, 20‴-H3), 1.24–ca. 1.38
(m, 2′-H2 to 9′-H2, 10″-H2
to 19‴-H2), superimposed by 1.38 [br s, (CH3
)2], 1.41 (d, J5,5-Me
= 6.8,
5-Me), 1.45–1.60 (m, 10′-H2, 1‴-H2),
2.02 (td, J9‴,10‴≈J9‴,8‴
≈6.4, 9‴-H2), in part superimposed by 2.10 (m
c, 5‴-H2, 6‴-H2), in part superimposed
by 2.13–ca. 2.23 (m, 2‴-H2), 2.26 (incompl.
res. tdd, J1′,2′
= 7.5, 4
J1′,4
=
5J1′,5
= 1.7, 1′-H2), 3.60 (mc , 4″-H,
5″-H), 4.99 (qdt, J5,5-Me
= 6.7, J
5,4
=
5J5,1′
= 1.7,
5-H), 5.30–5.45 (m, 3‴-H, 4‴-H, 7‴-H, 8‴-H),
6.98 (td, 4J4,1′
=
J
4,5
= 1.5, 4-H). IR (neat): ν
= 2980, 2925,
2855, 1760, 1460, 1370, 1320, 1240, 1080, 1025, 870, 725 cm−1.
C40H70O4 (615.0) calcd. C 78.12, H 11.47;
found C 78.00, H 11.29.
(3S,4S,5S,4″S,5″
S)-Z,Z-3-{10-[5-(3,7-Eicosadienyl)-2,2-dimethyl-1,3-dioxolan-4-yl]-decyl}-4,5-dihydro-4-hydroxy-5-methyl
-2(3H)-furanone (3a). n-BuLi (1.90 M in hexane, 0.66 ml,
1.3 mmol, 2.5 equiv.) was added to a solution of i-Pr2NH (0.16
ml, 0.13 g, 1.3 mmol, 2.5 equiv.) in THF (2.5 ml) at − 78°C. After
30 min a solution of the β-hydroxylactone S,S-
4 (58 mg, 0.50 mmol) in THF (2.5 ml) was added. After 2 h at −
78°C a solution of iodide 5 (322 mg, 0.500 mmol, 1.0 equiv.)
in THF–DMPU (1:1, 2 ml) was added dropwise. The mixture was stirred
for 20 h at − 45°C and worked up by the addition of HCl (2 M, 2.5
ml). After extraction with ButOMe (3 × 20 ml), drying over
MgSO4 and removal of the solvents the residue was separated by
flash chromatography (3 cm, petroleum ether–ButOMe 1:1) to
give unreacted 5 (fractions 2–3, 121 mg, 38%) and the
title compound (fractions 5–11, 175 mg, 56%; 90% based
on recovered starting material) as colorless oils. [α]
D25
=
− 21.8 (c
= 0.78).
1H NMR (300 MHz): δ
= 0.88 (t, J
20‴,19‴
= 6.8, 20‴-H3),
ca. 1.24–ca. 1.40 (m, 2′-H2 to 9′-H
2, 10‴-H2 to 19‴-H2), superimposed
by 1.379 and 1.383 [2 s, 2″-(CH3)2],
1.41 (d, J5,5-Me
= 6.7, 5-Me), 1.43–1.63
(m, 1′-H1, 10′-H2, 1‴-H2),
1.69–1.79 (m, 1′-H2), 2.02 (td, J9‴,10‴
≈J9‴,8‴≈6.5, 9‴-H
2), in part superimposed by 2.10 (mc, 5‴-H2,
6‴-H2), in part superimposed by ca. 2.13–2.26
(m, 2‴-H2), 2.54 (ddd, J3,1′-H(1)≈
J3,1′-H(2)≈7.1, J3,4
=
3.6, 3-H), 3.60 (mc, 4″-H, 5″-H), 4.20 (dd, J
4,5
= 4.7, J4,3
= 3.6, 4-H), 4.63
(qd, J5,5-Me
= 6.4, J5,4
=
4.9, 5-H), 5.30–5.46 (m, 3‴-H, 4‴-H, 7‴-H, 8‴-H).
IR (neat): ν
= 3445, 2985, 2925, 2855, 1760, 1460, 1375,
1240, 1185, 1100, 1055, 995, 725 cm−1. C40H
72O5 (633.0) calcd. C 75.90, H 11.47; found C 75.69, H 11.15.
(3R,4R,5R,4″S,5″
S)-Z,Z-3-{10-[5-(3,7-Eicosadienyl)-2,2-dimethyl-1,3-dioxolan-4-yl]-decyl}-4,5-dihydro-4-hydroxy-5-methyl-2(3
H)-furanone (3b). 3b was prepared as for 3a
using β-hydroxylactone R,R-4 (58 mg, 0.50
mmol). The residue after extraction and solvent removal was separated by flash
chromatography (3 cm, petroleum ether–ButOMe 1:1) to give
unreacted 5 (fractions 2–3, 111 mg, 34%) and the title
compound (fractions 6–12, 195 mg, 62%; 94% based on recovered
starting material) as colorless oils. [α]D
25
=
+ 6.02 (c
= 0.93). 1H NMR
(300 MHz, slightly contaminated around δ
= 0.9):
δ
= 0.88 (t, J20‴,19‴
=
7.2, 20‴-H3), 1.24–ca. 1.40 (m, 2′-H
2 to 9′-H2, 10‴-H2 to 19‴-H
2), superimposed by 1.38 [br s, 2″-(CH3)
2], 1.41 (d, J5,5-Me
= 6.4, 5-Me), 1.43–1.63
(m, 1′-H1, 10′-H2, 1‴-H2),
1.69–1.79 (m, 1′-H2), 2.02 (td, J9‴,10‴
≈J9‴,8‴≈6.5, 9‴-H
2), in part superimposed by 2.10 (mc, 5‴-H2,
6‴-H2), in part superimposed by 2.13–2.26 (m, 2‴-H
2), 2.54 (ddd, J3,1′-H(1)≈J
3,1′-H(2)≈7.0, J3,4
= 3.7, 3-H),
3.60 (mc, 4″-H, 5″-H), 4.20 (poorly res. dd, J
4,5
= 4.6, J4,3
= 3.6, 4-H),
4.63 (qd, J5,5-Me
= 6.4, J5,4
=
4.9, 5-H), 5.30-5.45 (m, 3‴-H, 4‴-H, 7‴-H, 8‴-H).
IR (neat): ν
= 3445, 2985, 2925, 2855, 1755, 1460, 1375,
1240, 1185, 1100, 1055, 995, 725 cm−1. C40H
72O5 (633.0) calcd. C 75.90, H 11.47; found C 75.80, H 11.31.
(4S,5S)-4,5-Dihydro-4-hydroxy-5-methyl-2(3
H)-furanone (S,S-4). Method A: Ref.
7d. Method B: At 0°C K3Fe(CN)6 (987 mg,
3.00 mmol, 3.0 equiv.), K2CO3 (414 mg, 3.00 mmol, 3.0
equiv.), (DHQ)2PHAL [
= 1,4-bis(dihydroquininyl)phthalazine;
78.0 mg, 0.100 mmol, 10.0 mol.%], K2OsO4
(6.6 mg, 0.020 mmol, 2.0 mol.%) and methyl trans-3-pentenoate
(123 μl, 114 mg, 1.00 mmol) were added to a 1:1 mixture of ButOH
and H2O (5 ml each). After this mixture had been stirred for 16
h the reaction was terminated by the addition of aqueous Na2SO
3 solution (10 ml). After extraction with CH2Cl2
(10 × 50 ml) the organic extracts were washed with diluted HCl. (DHQ)
2PHAL (74 mg, 95%) could be reisolated from this extract after
neutralization and extraction. The organic phase was dried over Na2SO
4. After removal of the solvent S,S-4 (85
mg, 73%) was obtained from the residue by flash chromatography (2.5
cm, ButOMe, fractions 6–12). Chiral capillary gas chromatography
revealed ee = 86%
[20% heptakis-(2,6-di-O-methyl-3-
O-pentyl-β-cyclodextrin) in 80% OV1701 (25 m), 70 kPa H
2, 110°C isothermal; RT 45.4 min, R
T of R,R enantiomer 44.3 min]. [α
]D25
=
− 62.2 (c
=
0.73) {lit.: [α]D25
=
−
73.7 (c
= 0.93, EtOH)}. 1H NMR (300 MHz):
δ
= 1.45 (d, J1′,5
= 6.8,
1′-H3), 2.49 (br s, OH), AB signal (δ
A
= 2.58, δB
= 2.82, J
AB
= 17.8, in addition split by JA,4
= 1.0, JB,4
= 5.7, 3-H2),
4.45 (br dd, J4,3-H(B)≈J4,5≈4,
4-H), 4.58 (qd, J5,1′
= 6.5, J
5,4
= 3.8, 5-H).
(4R,5R)-4,5-Dihydro-4-hydroxy-5-methyl-2(3
H)-furanone (R,R-4). Method A: AD-mix β
[14.0
g; containing 1,4-bis(dihydroquinidinyl)phthalazine (1 mol.%), K
3Fe(CN)6 (3 equiv.), K2CO3 (3 equiv.),
K2OsO4 (0.2 mol.%)] and methyl trans
-3-pentenoate (1.23 ml, 1.14 g, 10.0 mmol) were added to a 1:1 mixture
of ButOH and H2O (50 ml each) at 0°C. After stirring
for 4 days the reaction was terminated by the addition of aqueous Na
2SO3 solution (30 ml). After extraction with CH2Cl
2 (10 × 50 ml) the organic extracts were dried over Na2
SO4. Removal of the solvent yielded a residue that was purified
by flash chromatography (3 cm, ButOMe, fractions 12–24) to
give R,R-4 (441 mg, 38%). Chiral capillary
gas chromatography revealed ee = 80%
[20% heptakis-(2,6-di-
O-methyl-3-O-pentyl-β-cyclodextrin) in 80% OV1701
(25 m), 70 kPa H2, 110°C isothermal; RT
44.3 min, RT of S,S enantiomer 45.4
min].Method B: Same as for S,S-4 using (DHQD)
2 PHAL [
= 1,4- bis(dihydroquinidinyl)phthalazine; 78.0
mg, 0.100 mmol, 10.0 mol.%]. After extraction with CH2Cl
2 (10 × 50 ml) the organic phase was dried over Na2SO
4. After removal of the solvent R,R-4 (80
mg, 69%) was obtained from the residue by flash chromatography (2.5
cm, ButOMe, fractions 6–12). Chiral capillary gas chromatography
revealed ee = 94%
[20% heptakis-(2,6-di-O-methyl-3-
O-pentyl-β-cyclodextrin) in 80% OV1701 (25 m), 70 kPa H
2, 110°C isothermal; RT 44.3 min, R
T of S,S enantiomer 45.4 min].
(4S,5S)-Z,Z-4-(3,7-Eicosadienyl)-5-(10-iododecyl)-2,2-dimethyl-1,3-dioxolane
(5). At 0°C, PPh3 (605 mg, 2.31 mmol, 1.2 equiv.),
imidazole (315 mg, 4.63 mmol, 2.4 equiv.) and I2 (587 mg, 2.31
mmol, 1.2 equiv.) were added to a solution of alcohol 62 (1.03 g,
1.93 mmol) in THF (20 ml). The reaction mixture was warmed to room temperature
within 30 min, followed by the addition of water (20 ml). The organic phase
was separated and the aqueous phase extracted with ButOMe (50 ml).
The combined organic phases were dried over MgSO4 and the solvent
was removed. The residue was purified by flash chromatography (3 cm, deactivated
silica, petroleum ether–ButOMe 100:1, fractions 3–9).
The title compound (1.19 g, 96%) was obtained as a colorless liquid. [
α]D25
=
− 8.29 (c
=
0.76). 1H NMR (300 MHz): δ
= 0.88 (t,
J20′,19′
= 7.2, 20′-H3),
1.24–ca. 1.43 (m, 10′-H2 to 19′-H
2, 2″-H2 to 8″-H2), superimposed
by 1.38 [s, 2-(CH3)2], 1.45–1.60 (m,
1′-H2, 1″-H2), 1.82 (tt, J
9″,10″
=
J9″,8″
=
7.2, 9″-H2), 2.02 (br td, J9′,10′
≈J9′,8′≈6.4, 9′-H2
), in part superimposed by 2.10 (mc, 5′-H2,
6′-H2), in part superimposed by ca. 2.14–2.26
(m, 2′-H2), 3.19 (t, J10″,9″
=
6.9, 10″-H2), 3.60 (mc, 4-H, 5-H), 5.30–5.46
(m, 3′-H, 4′-H, 7′-H, 8′-H). 13C NMR (50.3
MHz, APT): δ
= 7.05 (C-10″), 14.04 (C-20′),
22.61, 23.83, 26.06, 27.19, 27.27, 28.44, 29.25, 29.29,* 29.30,* 29.38,
# 29.49, 29.58,# 29.62,# 29.65,*
30.41, 31.85, 32.89, 32.92 and 33.47 (19 resonances for 24 C atoms: C-1′,
C-2′, C-5′, C-6′, C-9′ to C-19′, C-1″
to C-9″), 27.24 [C(CH3)3],
80.30 and 80.86 (C-4, C-5), 107.75 [C(CH3)
3], 129.00, 129.19, 130.06 and 130.45 (C-3′, C-4′,
C-7′, C-8′); * increased but not 2-fold intensity;
#2-fold intensity. IR (neat): ν
= 2985, 2925,
2855, 1460, 1370, 1240, 1175, 1100, 1000, 875, 720 cm−1.
C35H65O2 (644.8) calcd. C 65.20, H 10.16,
found C 64.94, H 10.13.
1,1,1-Tribromo-2-hydroxy-3-butene (16). SnF
2 (0.237 g, 3 mmol, 1 equiv.) was added to a mixture of CBr4
(2.98 g, 9 mmol, 3 equiv.) and acrolein (0.2 ml, 3 mmol) in DMSO
(12 ml) and the solution was stirred for 5 min. Another portion of SnF
2 (0.237 g, 3 mmol, 1 equiv.) was added and stirring was continued
for a further 5 min. The mixture was diluted with CH2Cl2
(5 ml) and HCl (2 M, 5 ml). After 30 min the organic materials were
extracted with CH2Cl2. The combined organic layers
were dried over MgSO4, filtered and concentrated with a rotary
evaporator. The resultant crude product was purified by flash chromatography
(pentane–ether 8:2) to give the desired product (40 mg, 5%) as
a pale yellow oil. 1H NMR (300 MHz): δ
=
2.98 (br s, OH), 4.54 (d, J2,3
= 3.8, 2-H), 5.58
(dt, Jcis
= 10.5, 4J
Z-4,2
= 1.3, 4-HZ), 5.58 (dt, Jtrans
= 17.3, 4JE-4,2
= 1.3,
4-HE), 6.14 (ddd, Jtrans
= 17.0,
Jcis
= 10.5, J3,2
= 5.2,
3-H). 13C (50.3 MHz, APT, CDCl3 as internal standard):
δ
=
″
−
″ 52.96 (C-1), ″
+
″
84.50 (C-2), ″
−
″ 121.84 (C-4), ″
+
″
132.80 (C-3).
Methyl E-5-chloro-3-pentenoate (22
16). Hydroxyester 2014
(290 mg, 2.23 mmol) was added to a solution of NCS (356 mg, 2.68 mmol, 1.2
equiv.) and Me2S (0.20 ml, 0.17 g, 2.7 mmol, 1.2 equiv.) in CH
2Cl2 (10 ml) at 0°C. The reaction mixture was stirred
for 16 h at room temperature. After addition of water (10 ml) and extraction
with CH2Cl2 (3 × 20 ml) the organic phases were
dried over MgSO4. After removal of the solvent the title compound
(273 mg, 83%) was obtained by flash chromatography (3 cm, petroleum
ether–ButOMe 50:1 → fraction 12, 20:1 → fraction
22, fractions 13–22) as a colorless liquid. 1H NMR (300
MHz): δ
= 3.13 (br d, J2,3
=
6.7, 2-H2), 3.70 (s, OCH3), 4.06 (poorly res. dd,
J5,4
= 6.6, 4J5,3
=
1.0, 5-H2), 5.75 (dtt, Jtrans
= 15.0,
J3,2
= 6.8, 4J3,5
=
1.4, 3-H*), 5.90 (dt with shoulders, Jtrans
=
15.4, J4,5
= 6.8, 4-H*); * assignments
interchangeable. Methyl E-5-(phenylthio)-3-pentenoate (23). Ph2S2 (4.53 g, 20.8 mmol, 3.0 equiv.) and Bu
3P (6.89 ml, 5.60 g, 27.7 mmol, 4.0 equiv.) were dissolved in toluene
(40 ml). Hydroxyester 2014 (900
mg, 6.92 mmol) in THF (5 ml) was added after 2 h at room temperature. After
stirring overnight the solvent was removed. The residue was purified by flash
chromatography (6 cm, petroleum ether → fraction 10, petroleum ether–Bu
tOMe 10:1 → fraction 35, fractions 23–35). The title compound
(1.24 g, 81%) was isolated as a colorless liquid. 1H NMR
(300 MHz; with MeO-containing impurity at δ
= 3.63):
δ
= 3.03 (d, J2,3
= 3.8, 2-H
2), 3.54 (dmc , J5,4≈4, 5-H2
), 3.65 (s, OMe), 5.57–5.71 (m, 3-H, 4-H), 7.15–7.22 (m,
1 Ar-H), 7.24–7.36 (m, 4 Ar-H). IR (neat): ν
= 3055,
3000, 2950, 1735, 1585, 1480, 1435, 1355, 1290, 1255, 1195, 1170, 1025, 970,
740, 690 cm−1. No combustion analysis was performed. (4S,5R)-4,5-Dihydro-4-hydroxy-5-[(phenylthio)methyl]-2(3
H)-furanone (24). At 0°C AD-mix α
[2.80 g containing
1,4-bis(dihydroquininyl)phthalazine (1 mol%), K3Fe(CN)
6 (3 equiv.), K2OsO4 (0.2 mol%)],
methanesulfoneamide (190 mg, 2.00 mmol, 1.0 equiv.) and the β,γ-unsaturated
ester 23 (444 mg, 2.00 mmol) in ButOH (1 ml) were added
to a 1:1 mixture of ButOH and H2O (6 ml each). After
stirring for 36 h, the mixture was hydrolyzed by the addition of aqueous
Na2SO3 solution (2 ml) and water (10 ml). After extraction
with ButOMe (3 × 50 ml) the organic extracts were dried over
MgSO4 . After removal of the solvent the residue was purified
by flash chromatography (3 cm, petroleum ether–ButOMe 2:1 →
fraction 12, 1:1 → fraction 20, ButOMe → fraction 32,
fractions 14–30). 24 (283 mg, 63%) was isolated as a
colorless oil. Chiral capillary gas chromatography of traces of the subsequently
obtained (Raney Ni treatment in acetone–EtOH, 5 bar H2, room
temperature, 5 days), desulfurized material revealed ee = 80%
[20%
heptakis-(2,6-di-O-methyl-3-O-pentyl-β-cyclodextrin)
in 80% OV1701 (25 m), 70 kPa H2, 110°C isothermal;
RT 44.3 min, RT of R,R
enantiomer 43.2 min]. 1H NMR (300 MHz; contains 1.4 wt%
ButOMe): δ
= 2.42 (br s, OH), AB signal (
δA
= 2.58, δB
=
2.76, JAB
= 17.9, sp lit by JB,4
= 5.6, 3-H2), AB signal (δA
=
3.29, δB
= 3.44, JAB
=
13.6, split by JA,5
= 9.5, JB,5
=
5.3, 1′-H2), 4.45 (ddd, J5,1′-H(A)
=
9.8, J5,1′-H(B)
= 4.9, J5,4
= 3.4, 5-H), 4.64 (br dd, J4,5≈J
4,3-H(B)≈4.5, 4-H), 7.23–7.36 (m, 3 Ar-H), 7.41–7.46
(m, 2 Ar-H). No combustion analysis was performed.
(4S,5R)-5-Chloro-4,5-dihydro-4-hydroxy-2(3
H)-furanone (25). AD-mix α (1.72 g, 3.69 mmol, 3.0, equiv.),
NaHCO3 (309 mg, 3.69 mmol, 3.0 equiv.), methanesulfoneamide (117
mg, 1.23 mmol, 1.0 equiv.) and the β,γ-unsaturated ester 22
(182 mg, 1.23 mmol) in ButOH (1 ml) were added to a 1:1 mixture
of ButOH and H2O (6 ml each) at 0°C. After this
mixture had been stirred for 24 h the reaction was terminated by the addition
of aqueous Na2SO3 solution (2 ml) and water (10 ml).
After extraction with ButOMe (3 × 50 ml) the organic extracts
were dried over Na2SO4. Removal of the solvent yielded
a residue that was purified by flash chromatography (3 cm, petroleum ether–Bu
tOMe 2:1 → fraction 12, 1:1 → fraction 26, fractions 13–24)
to give 25 (53 mg, 19%). 1H NMR (300 MHz):
δ
= AB signal (δA
= 2.64,
δB
= 2.85, JAB
=
17.8, split by JA,4
= 1.2, JB,4
=
5.8, 3-H2), superimposed by 2.64 (br d, JOH,4
=
4.1, OH), 3.86 (d, J1′,5
= 7.2, 1′-H
2), 4.59 (td, J5,1′
= 7.2, J
5,4
= 3.7, 5-H), 4.72 (br ddd, J4,5≈
J4,3-H(B)≈J4,OH≈4.5, 4-H). No combustion
analysis was performed.
(5R)-3-Butyl-5-methyl-2(5H)-furanone (26)
. PPh3 (334 mg, 1.28 mmol, 2.0 equiv.) and DEAD (40%
in toluene, 0.58 ml, 0.22 g, 1.3 mmol, 2.0 equiv.) were added to a solution
of the β-hydroxylactone 27 (110 mg, 0.640 mmol; 86% ee)
in THF (10 ml) at − 20°C. The reaction mixture was allowed to warm
to room temperature within 3 h. Water (10 ml) was added, followed by extraction
with ButOMe (3 × 20 ml). The organic phase was dried over
MgSO4 and the solvent was removed. The residue yielded the title
compound (88.0 mg, 89%) as a colorless liquid after flash chromatography
(2.5 cm, petroleum ether–ButOMe 4:1, fractions 3–6). [
α]D25
=
− 48.3 (c
=
1.09); since the starting material 27 had 86% ee, this measured
specific rotation corresponds to − 48.3/0.86 =
− 56.1
for enantiopure 26
{lit.: [α]
D25
=
− 53.7 (c not given, CHCl
3)}. 1H NMR (300 MHz): δ
= 0.93
(t, J4′,3′
= 7.4, 4′-H3
), 1.29–1.44 (m, 3′-H2), superimposed by 1.41
(d, J5,5-Me
= 6.8, 5-Me), 1.49–1.60 (m,
2′-H2), 2.28 (tdd, J1′,2′
=
7.5, 4J1′,4
=
5
J1′,5
= 1.6, 1′-H2), 4.99 (qdt,
J5,5-Me
= 6.8, J5,4
=
5J5,1′
= 1.8, 5-H), 6.99 (td,
4J4,1′
=
J4,5
=
1.6, 4-H).
(3R,4R,5R)-3-Butyl-4,5-dihydro-4-hydroxy-5-methyl-2(3
H)-furanone (27). BunLi (1.90 M in hexane, 2.63 ml,
5.00 mmol, 2.5 equiv.) was added to a solution of Pr2iNH
(0.66 ml, 0.51 g, 5.0 mmol, 2.5 equiv.) in THF (20 ml) at − 78°C.
After 30 min a solution of the β-hydroxylactone R,R-
4 (232 mg, 2.00 mmol, 86% ee) in THF (5 ml) was added. After
stirring the mixture for 2 h at this temperature 1-iodobutane (0.27 ml, 0.44
g, 2.4 mmol, 1.2 equiv.) in THF–DMPU (1:1, 8 ml) was added. After stirring
at − 45°C for 20 h the mixture was hydrolyzed with HCl (1 M, 10
ml). After extracting the aqueous phase with ButOMe (3 ×
30 ml) the combined organic phases were dried over MgSO4 and the
solvent was removed. Flash chromatography (3 cm, petroleum ether–Bu
tOMe 4:1 → fraction 10, 2.5:1 → fraction 26, fractions 15–23)
of the residue yielded the title compound (298 mg, 84%) as a colorless
oil. [α]D20
=
+
58.4 (c
= 1.18) {lit.: [α]
D20
=
+ 71 (c
= 0.5, MeOH)};
since the starting material R,R-4 had 86%
ee this measured specific rotation corresponds to [α]
D20
=
+ 58.4/0.86 =
+ 67.9 for
enantiopure 27. 1H NMR (300 MHz): δ
=
0.92 (t, J4′,3′
= 7.2, 4′-H
3), 1.30–1.80 (m, 1′-H2, 2′-H2,
3′-H2), in part superimposed by 1.41 (d, J
5-Me,5
= 6.8, 5-Me), 2.21 (mc, OH), 2.55 (poorly res.
ddd, J3,1′-H(1)
= 7.8, J3,1′-H(2)
= 6.4, J3,4
= 3.4, 3-H), 4.21 (br
ddd, J4,5≈
J4,3≈J
4,OH≈4.5, 4-H), 4.64 (qd, J5,5-Me
= 6.4,
J5,4
= 4.9, 5-H).
1-[(2-Tetrahydro pyranyl)oxy]-3-butyne (29). A mixture of 3,4-dihydro-(2H)-pyran (27.4 ml, 25.2 g, 300 mmol,
3.0 equiv.) and 3-butyn-1-ol (9.30 ml, 8.41 g, 100 mmol) in CH2Cl
2 (100 ml) and a catalytic amount of camphor sulfonic acid was stirred
for 3 h at room temperature. Water (50 ml) was added and the phases were separated.
The aqueous phase was extracted with ButOMe (100 ml) and the combined
organic phases were dried over MgSO4. After removal of the solvent
the title compound (14.81 g, 96%) was isolated from the residue by
distillation under reduced pressure (bp 74°C at 20 mbar). 1H
NMR (300 MHz): δ
= 1.46–1.90 (m, 3′-H
2 , 4′-H2, 5′-H2), 1.98 (t,
4J4,2
= 2.7, 4-H), 2.50 (td, J
2,1
= 7.2, 4J2,4
= 2.7,
2-H2), ca. 3.52 (mc, 6′-H1),
heavily superimposed by A branch of AB signal (δA
=
3.57, δB
= 3.83, JAB
=
9.8, split by JA,2
= 7.0, JB,2
=
7.2, 1-H2), B branch severely superimposed by ca. 3.88
(mc , 6′-H2), 4.65 (dd, J2′,3′-H(1)
≈J2′,3′-H(2)
= 3.4, 2′-H).
1-[(2-Tetrahydropyranyl)oxy]-3-hexadecyne (30)
. Alkyne 29 (15.7 ml, 15.4 g, 100 mmol) was added dropwise
to a suspension of NaNH2 (4.29 g, 111 mmol, 1.1 equiv.) in THF
(50 ml) at 0°C. 1-Bromododecane (26.4 ml, 27.4 g, 110 mmol, 1.1 equiv.)
and DMSO (50 ml) were added to the yellowish solution after 1 h. The reaction
mixture was stirred for 2.5 h at room temperature and hydrolyzed with water
(50 ml). After extraction of the aqueous phase with ButOMe (3 ×
200 ml) the combined organic phases were dried over MgSO4. After
removal of the solvent flash chromatography (8 cm, petroleum ether →
fraction 8, petroleum ether–ButOMe 50:1 → fraction 12,
20:1 → fraction 20, fractions 8–18) yielded the title compound
(16.73 g, 53%) as a colorless liquid. 1H NMR (300 MHz):
δ
= 0.88 (t, J16,15
= 6.8, 16-H
3), 1.22–1.90 (m, 6-H2 to 15-H2 , 3′-H
2, 4′-H2, 5′-H2), 2.13 (tt, J
5,6
= 7.0, 5J5,2
=
2.3, 5-H2), 2.46 (tt, J2,1
= 7.4,
5J2,5
= 2.5, 2-H2), AB signal
(δA
= 3.52, δB
=
3.79, JAB
= 9.6, split by JA,2
=
JB,2
= 7.2, 1-H2), A branch superimposed
by ca. 3.50 (mc, 6′-HA), B branch of
AB signal (broadened, δB
= 3.89, J
AB
= 11.3, split by J6′-H(B),5′-H(1)
= 7.9, J6′-H(B),5′-H(2)
=
3.4, 6′-HB), 4.65 (dd, J2′,3′-H(1)
≈J2′,3′-H(2)≈3.4, 2′-H).
3-Hexadecyn-1-ol (31). While stirring, p-TsOH
(1.56 g, 8.22 mmol, 0.4 equiv.) was added to a solution of the THP ether
30 (6.62 g, 20.6 mmol) in MeOH (60 ml) at room temperature. After 30
min water (50 ml) and ButOMe (50 ml) were added and the phases
separated. After extraction of the aqueous phase with ButOMe (2 ×
50 ml) the combined organic phases were dried over MgSO4. After
removal of the solvent the alkynol 31 (4.90 g, 99%) was isolated
as a white solid (mp 41°C). 1H NMR (300 MHz): δ
=
0.88 (t, J16,15
= 6.8, 16-H3), 1.23–1.54
(m, 6-H2 to 15-H2), 1.81 (br s, OH), 2.16 (tt,
J5,6
= 7.0, 5J5,2
=
2.3, 5-H2), 2.43 (tt, J2,1
= 6.1,
5J2,5
= 2.3, 2-H2), 3.68 (br t,
J1,2
= 6.2, 1-H2). IR (neat): ν
=
3320, 2925, 2850, 1465, 1435, 1380, 1335, 1185, 1045, 720 cm−1
. C16H30O (238.4) calcd. C 80.61, H 12.68; found
C 80.88, H 12.45.
1-Bromo-3-hexadecyne (32). A solution of alkynol
31 (715 mg, 3.00 mmol) in THF (6 ml) at − 20°C was successively
treated with PPh3 (943 mg, 3.60 mmol, 1.2 equiv.) and NBS (587
mg, 3.30 mmol, 1.1 equiv.). The reaction mixture was warmed to 0°C. After
4 h NH4Cl solution (10 ml) was added and the organic phase was
separated. After extraction with petroleum ether (2 × 50 ml) the combined
organic phases were dried and the solvent removed. The residue was purified
ia flash chromatography (3 cm, petroleum ether, fractions 4–6)
to yield the title compound (794 mg, 88%) as a colorless liquid.
1H NMR (300 MHz, contains traces of Ph3P): δ
=
0.88 (t, J16,15
= 6.6, 16-H3), 1.22–1.54
(m, 6-H2 to 15-H2), 2.14 (tt, J5,6
=
7.0, 5J5,2
= 2.3, 5-H2),
2.71 (tt, J2,1
= 7.3, 5J
2,5
= 2.3, 2-H2), 3.41 (t, J1,2
=
7.4, 1-H2). No combustion analysis was performed. 1-Iodo-3-hexadecyne (33). A solution of alkynol
31 (1.90 g, 7.89 mmol) in THF (30 ml) at 0°C was successively treated
with PPh3 (2.30 g, 8.78 mmol, 1.1 equiv.), imidazole (1.19 g,
17.6 mmol, 2.2 equiv.) and I2 (2.23 g, 8.78 mmol, 1.1 equiv.).
After 1 h NH4Cl solution (10 ml) was added and the organic phase
was separated. After extraction with petroleum ether (2 × 100 ml) the
combined organic phases were dried and the solvent removed. The residue was
purified ia flash chromatography (4 cm, petroleum ether,
fractions 3–9) to provide the title compound (2.433 g, 88%)
as a colorless liquid. 1H NMR (300 MHz): δ
=
0.88 (t, J16,15
= 6.8, 16-H3), 1.24–1.54
(m, 6-H2 to 15-H2), 2.13 (tt, J5,6
=
6.8, 5J5,2
= 2.3, 5-H2),
2.73 (tt, J2,1
= 7.4, 5J
2,5
= 2.3, 2-H2), 3.21 (t, J1,2
=
7.4, 1-H2). IR (neat): ν
= 2925, 2850, 1465,
1435, 1250, 1170, 720 cm1. No combustion analysis was performed.
3-Hexynyl trifluoromethanesulfonate (34) and 1-[(2-tetrahydropyranyl)oxy]-3,7-eicosadiyne
(35). The alkynol 31 (770 mg, 3.24 mmol) was dissolved
in CH2Cl2 (10 ml); NEt3 (0.54 ml, 0.39 g,
3.9 mmol, 1.2 equiv.) and Tf2O (0.65 ml, 1.1 g, 3.9 mmol, 1.2
equiv.) were added at 0°C. After 2 h the solvent was removed and the residue
filtered over a short silica column (3 cm, petroleum ether–Bu
tOMe–CH2Cl2 5:1:1). The resulting crude
triflate 34 was dissolved in THF (2 ml). This solution was added
at 0°C to a solution prepared from alkyne 29 (505 mg, 3.24 mmol,
1.0 equiv.) and BunLi (1.50 M in hexane, 2.37 ml, 3.56 mmol, 1.1
equiv.) in THF (8 ml; deprotonation time 30 min). After stirring at room temperature
for 16 h an aqueous NH4Cl solution (10 ml) was added. After extracting
the aqueous phase with ButOMe (3 × 20 ml) the combined organic
phases were dried over MgSO4. After removal of the solvent flash
chromatography (3 cm, petroleum ether–ButOMe 50:1 →
fraction 10, 20:1 → fraction 20, fractions 9–18) provided
35 (383 mg, 32%) as a colorless liquid. 1H NMR (300
MHz): δ
= 0.88 (t, J20,19
=
6.8, 20-H3), 1.22–1.88 (m, 10-H2 to 19-H
2, 3′-H2 , 4′-H2, 5′-H
2), 2.13 (br t, J9,10
= 7.0, 9-H2),
2.33 (br s, 5-H2, 6-H2), 2.46 (t, J2,1
= 7.2, 2-H2), AB signal (δA
=
3.52, δB
= 3.79, JAB
=
9.8, split by JA,2
=
JB,2
=
7.2, 1-H2), A branch superimposed by ca. 3.50 (m
c, 6′-H1), B branch of AB signals (δ
B
= 3.88, JAB
= 11.2, split by
JB,5′-H(1)
= 7.5, JB,5′-H(2)
= 3.7, 6′-H2), 4.64 (dd, J2′,3′-H(1)
≈J2′,3′-H(2)≈3.4, 2′-H).
This compound was not characterized by combustion analysis.
1-Hexadecen-3-yne (36). The title compound was obtained
instead of the desired diyne 35 in the following experiment. At
0°C, BuLi (1.43 M in hexane, 5.61 ml, 8.03 mmol, 1.2 equiv.) was added
to the THP ether 29 (1.03 g, 6.69 mmol) in THF (20 ml). After 30
min iodide 33 (2.33 g, 6.69 mmol, 1.0 equiv.) in DMSO (50 ml) was
added and the mixture was allowed to warm to room temperature. After stirring
for 12 h water (50 ml) and ButOMe (50 ml) were added. The aqueous
phase was extracted with ButOMe (2 × 50 ml). The combined
organic phases were washed with brine and dried over MgSO4. After
removal of the solvent the residue was purified ia flash
chromatography (4 cm, petroleum ether, fractions 2–5) to yield enyne
36 (1.377 g, 93%). 1H NMR (300 MHz): δ
=
0.88 (t, J16,15
= 6.8, 16-H3), 1.24–1.57
(m, 6-H2 to 15-H2), 2.29 (td, J5,6
=
7.2, 5J5,2
= 1.9, 5-H2),
5.37 (dd, Jcis
= 11.0, Jgem
=
2.3, 1-HE), 5.54 (dd, Jtrans
= 17.3,
Jgem
= 2.3, 1-HZ), 5.78 (ddt,
Jtrans
= 17.3, Jcis
= 10.9,
5J2,5
= 2.1, 2-H). IR (neat): ν
=
2920, 2855, 1610, 1455, 1380, 1330, 970, 910, 720 cm−1. C
16H28 (220.4) calcd. C 87.19, H 12.81; found C 87.22, H 12.57.
1-(2-Tetrahydropyranyloxy)-2-dod ecyne (39). Bu
nLi (1.90 M, 11.7 ml, 22.2 mmol, 1.2 equiv.) was added to a solution
of the alkyne 3835 (2.60 g, 18.5
mmol) in THF at 0°C. After 30 min 1-bromononane (3.90 ml, 4.22 g, 20.4
mmol, 1.1. equiv.) and DMSO (50 ml) were added. After stirring at room temperature
for 24 h the reaction was terminated by the addition of water (50 ml). After
extraction with ButOMe (2 × 50 ml) the organic phase was
dried over MgSO4. After removal of the solvent flash chromatography
(5 cm, petroleum ether–ButOMe 50:1, fractions 12–17)
of the residue yielded the title compound (3.36 g, 68%) as a colorless
liquid. 1H NMR (300 MHz): δ
= 0.88 (t,
J12,11
= 6.8, 12-H3), 1.27 and 1.33–1.73
(mc and m, respectively, 5-H2–11-H2,
4′-H2, 5′-H2), 2.21 (tt, J
4,5
= 7.2, 5J4,1
= 2.2,
4-H2), 3.49–3.56 and 3.81–3.89 (2m, 6′-H
2), AB signal (δA
= 4.22, δ
B
= 4.27, JAB
= 15.5, split
by 5JA,4
= 2.2, 5J
B
= 2.3, 1-H2), 4.81 (t, J2′,3′
= 3.4, 2′-H). No IR spectrum was recorded. C17H
30O2 (266.4) calcd. C 76.64, H 11.35; found C 76.43, H 11.15.
2-Dodecyn-1-ol (40). A solution of THP ether
39 (3.18 g, 10.8 mmol) and TsOH monohydrate (0.830 g, 4.36 mmol, 0.4
equiv.) in methanol (80 ml) was stirred at room temperature for 2 h. Distribution
between water and ether, drying of the etheral phase over MgSO4,
concentration in acuo and flash chromatography (pentane–ether
8:1) yielded the title compound (1.92 g, 84%). 1H NMR (300
MHz): δ
= 0.88 (t, J12,11
=
6.7, 12-H3), 1.27–1.45 (m, 6-H2–11-H
2), 1.68 (br s, OH), 2.21 (tt, J4,5
= 7.2,
5J4,1
= 2.3, 4-H2), 4.25 (t,
5J1,4
= 2.3, 1-H2). 13C
NMR (50.3 MHz, APT): δ
= 14.09 (C-12), 18.72, 22.66,
28.59, 28.86, 29.13, 29.27, 29.46 and 31.85 (C-4–C-11), 51.38 (C-1),
78.23 and 86.61 (C-2, C-3). IR (KBr): ν
= 3175, 2955, 2945,
2850, 1470, 1400, 1135, 1025, 780, 715 cm−1. C12H
22O (182.3) calcd. C 79.06, H 12.16; found C 78.81, H 12.11. 12-(tert-Butyldiphenylsiloxy)-1-dodecyne (42). At 0°C ButPh2SiCl (3.47 ml, 3.67 g, 13.7 mmol,
1.0 equiv.) and imidazole (1.92 g, 28.1 mmol, 2.1 equiv.) were added to a
solution of alcohol 44 (2.43 g, 13.4 mmol) in CH2Cl
2 (50 ml). After stirring for 1 h at room temperature the mixture was
hydrolyzed with diluted HCl (1 M, 20 ml). The aqueous phase was extracted
with ButOMe (3 × 50 ml) and the combined organic phases were
dried. After removal of the solvent the title compound (5.58 g, 99%)
was obtained without the need for further purification. 1H NMR
(300 MHz): δ
= 1.05 (s, SiBut), 1.22–1.44
(m, 5-H2 to 10-H2), 1.46–1.60 (m, 4-H2,
11-H2), 1.94 (t, 4J1,3
=
2.6, 1-H), 2.18 (td, J3,4
= 7.0, 4
J3,1
= 2.7, 3-H2), 3.65 (t, J
12,11
= 6.4, 12-H2), 7.34–7.45 (m, 6 Ar-H),
7.64–7.70 (m, 4 Ar-H). C28H40SiO (420.7) calcd.
C 79.94, H 9.58; found C 79.85, H 9.45.
11-Dodecyn-1-ol (4437). Li (391 mg, 56.3 mmol, 6.0 equiv.) was added in small pieces to 1,2-diaminopropane
(40 ml). After 10 min the deep-blue solution was heated under reflux until
the blue color disappeared. After cooling the mixture to room temperature
KOBut (4.21 g, 37.6 mmol, 4.0 equiv.) was added. The olive-brown
suspension was stirred for 30 min and the alkyne 40 (1.71 g, 9.39
mmol) was added. After 1 h ice water was added and the mixture was extracted
with ButOMe (3 × 100 ml). The combined organic phases were
dried and the solvent was removed. The isolated residue was purified by flash
chromatography (4 cm, petroleum ether–ButOMe 3:1 →
fraction 8, 2:1 → fraction 12, fractions 5–11). Alkynol 44
(1.266 g, 74%) was isolated as a white solid (mp 25°C).
1H NMR (300 MHz): δ
= 1.24–1.45 (m, 3-H
2 to 8-H2), 1.46–1.62 (m, 2-H2, 9-H
2), 1.94 (t, 4J12,10
= 2.7, 12-H),
2.18 (td, J10,9
= 7.0, 4J
10,12
= 2.6, 10-H2), 3.64 (t, J1,2
= 6.8, 1-H2); the resonance of the OH was not detected.
13C NMR (50.3 MHz, APT): δ
= 18.36, 25.69, 28.44,
28.70, 29.05, 29.37, 29.49 and 32.74 (C-2-C-9), 62.97 (C-1), 68.02 (C-12),
84.73 (C-11). IR (KBr): ν
= 3285, 2920, 2850, 1470, 1060,
1030, 725 cm−1. Z-3-Hexadecen-1-ol (4938
). Li (2.07 g, 300 mmol, 3.0 equiv.) was cut into small
pieces and suspended in Et2O (50 ml). Within 1 h 1-bromododecane
(14.0 ml, 24.9 g, 100 mmol) in Et2O (50 ml) was added dropwise
at 0°C to this mixture. After stirring for an additional 2 h at 0°C
the concentration of the resulting 1-lithiododecane was determined by titration
of a hydrolyzed sample of known volume (1.0 ml) with HCl (0.1 M) using phenolphthalein
as an indicator. Subsequently, the solution of this organolithium compound
(103 ml, 0.95 M, 98.0 mmol) was transferred to a suspension of CuI (9.31 g,
49.0 mmol, 0.50 equiv.) in Et2O (100 ml) at − 35°C. After
30 min acetylene (2.4 l, 98 mmol, 1.0 equiv.) was introduced at − 50°C
into the dark-grey suspension. After another 30 min of stirring at −
25°C a green suspension was obtained. At − 30°C it was treated
with previously condensed ethylene oxide (4.9 ml, 4.3 g, 98 mmol, 1.0 equiv.)
and a previously prepared solution of hexynyllithium [from BuLi (2.05
M in hexane, 23.9 ml, 49.0 mmol, 0.50 equiv.) and 1-hexyne (5.62 ml, 4.02
g, 49.0 mmol, 0.50 equiv.)] in Et2O (100 ml). The black reaction
mixture was stirred for 3 h at − 15°C and then hydrolyzed with HCl
(6 M, 40 ml) and sat. NH4Cl solution (40 ml). After removing insoluble
material by filtration ButOMe (200 ml) was added, the organic phase
separated and the aqueous phase extracted with ButOMe (2 ×
100 ml). The organic phases were dried over MgSO4 and the solvent
was removed. Flash chromatography (8 cm, petroleum ether–ButOMe
10:1 → fraction 10, 5:1 → fraction 25, fractions 7–22) of
the residue provided the title compound (19.07 g, 81%) as a colorless
liquid. 1H NMR (300 MHz): δ
= 0.88 (t,
J16,15
= 6.6, 16-H3), 1.24–1.41 (m,
6-H2 to 15-H2), 1.49 (br s, OH), 2.06 (td, J
5,4
=
J5,6
= 6.7, 5-H2),
2.33 (td, J2,1≈J2,3≈6.6, 2-H
2), 3.65 (t, J1,2
= 6.6, 1-H2),
5.36 (br dtt,*
Jcis
= 10.8, J
3,2
= 7.4, 4J3,5
= 1.5,
4-H), 4.57 (br dtt,*
Jcis
= 10.8, J
4,5
= 7.5, 4J4,2
= 1.5,
3-H); *with additional peaks of the beginning of a transition to a higher
order signal. Z-1-Iodo-3-hexadecene (50). At 0°C
PPh3 (2.88 g, 11.0 mmol, 1.1 equiv.), imidazole (1.50 g, 22.0 mmol,
2.2 equiv.) and I2 (2.79 g, 11.0 mmol, 1.1 equiv.) were added
to a solution of the alcohol 49 (2.40 g, 10.0 mmol) in THF (100 ml).
The reaction mixture was warmed to room temperature within 30 min. Subsequently
water (100 ml) was added. The organic phase was separated and the aqueous
phase extracted with ButOMe (100 ml). The combined organic phases
were dried over MgSO4 and the solvent was removed. The residue
was purified by flash chromatography (4 cm, petroleum ether, fractions 4–6).
The title compound (3.48 g, 99%) was obtained as a colorless liquid.
1H NMR (300 MHz): δ
= 0.88 (t, J
16,15
= 6.8, 16-H3), 1.24–1.38 (m, 6-H
2 to 15-H2), 2.02 (br td, J5,6
=
J5,4
= 6.9, 5-H2), 2.63 (br td, J
2,1
=
J2,3
= 7.0, 2-H2),
3.13 (t, J1,2
= 7.2, 1-H2), 5.31 (dtt,
Jcis
= 10.9, Jvic
= 7.2,
Jallyl
= 1.5, 4-H*), 5.53 (dtt, J
cis
= 10.6, Jvic
= 7.5, J
allyl
= 1.5, 3-H*); *assignments interchangeable. IR
(neat): ν
= 3010, 2925, 2850, 1695, 1460, 1375, 1300, 1240,
1170, 970, 720 cm−1. C16H31I (350.3)
calcd. C 54.86, H 8.92; found C 54.97, H 8.99.
Z,Z-1-Iodo-1,5-octadecadiene (51). Iodide 50 (6.20 g, 17.7 mmol) was dissolved in Et2O–hexane
(1:1, 40 ml), and at − 20°C ButLi (1.52 M in Et
2O, 23.3 ml, 35.4 mmol, 2.0 equiv.) was added. After 30 min the resulting
solution was transferred to a − 35°C suspension of CuI (1.68 g,
8.85 mmol, 0.5 equiv.) in Et2O (20 ml). The dark-grey suspension
was stirred for another 30 min at − 35°C. At − 50°C acetylene
(425 ml, 17.7 mmol, 1.0 equiv.) was introduced. The mixture was allowed to
warm to − 25°C and stirred again for 1 h. At − 60°C powdered
I2 (4.49 g, 17.7 mmol, 1.0 equiv.) was added to the greenish-black
suspension. The reaction mixture was warmed to − 10°C within 2
h and hydrolyzed thereafter at the same temperature with water (10 ml) and
sat. NH4Cl solution (10 ml). After separation from insoluble material
by filtration the filtrate was extracted with petroleum ether (100 ml). The
organic phase was washed with diluted NH3 solution (10 ml) and
Na2S2O3 solution (0.5 M, 10 ml). The now
colorless solution was dried and the solvent removed. Flash chromatography
(6 cm, petroleum ether, fractions 4–6) yielded the title iodide (4.41
g, 66%) as a colorless liquid. 1H NMR (300 MHz, C6
D6; in CDCl3 the olefinic signals superimpose
each other): δ
= 0.92 (t, J18,17
=
6.4, 18-H3), 1.29–1.38 (m, 8-H2 to 17-H2
), 1.96–2.16 (m, 3-H2, 4-H2, 7-H2),
AB signal (δA
= 5.35, δ
B
= 5.45, JAB
= 10.9, A branch split
by Jvic
= 7.0, B branch split by J
vic
= 7.2, 5-H, 6-H), 5.80 (td, J2,3≈
Jcis≈6.8, 2-H), 5.92 (br d, Jcis
=
7.5, 1-H). 13C NMR (50.3 MHz, APT, CDCl3): δ
= 14.15 (C-18), 22.72, 25.65, 27.29, 29.34, 29.39, 29.60, 29.70
(3-fold intensity), 31.96 and 34.83 (C-3, C-4, C-7 to C-17), 82.56 (C-1),
127.99, 131.20 and 140.70 (C-2, C-5, C-6). IR (neat): ν
=
3005, 2925, 2850, 1610, 1460, 1285, 1240, 720 cm−1. C
18H33I (376.4) calcd. C 57.44, H 8.84; found C 57.27, H 8.69.
12-(tert-Butyldiphenylsiloxy)-1-dodecanol (53). At room temperature imidazole (10.2 g, 150 mmol, 2.0 equiv.) and Bu
tPh2SiCl (19.3 ml, 20.6 g, 75.0 mmol, 1.0 equiv.) were added
to a solution of 1,12-dodecanediol (15.2 g, 75.0 mmol) in DMF (150 ml). After
15 h the reaction was terminated by the addition of water (100 ml) and EtOAc
(100 ml). After extraction of the aqueous phase with EtOAc (3 × 200
ml) the combined organic phases were washed with water (50 ml) and dried over
MgSO4. After removal of the solvent the residue was purified by
flash chromatography (8 cm, petroleum ether–ButOMe 20:1 →
fraction 15, 10:1 → fraction 25, 5:1 → fraction 40, 2:1 →
fraction 52, fractions 22–46) to yield the title compound (19.60 g,
59%). 1H NMR (300 MHz): δ
= 1.05 (s,
ButSi), 1.22–1.40 (m, 3-H2 to 10-H2),
1.50–1.61 (m, 2-H2, 11-H2), 3.63 (t, J
1,2
= 6.6, 1-H2),* in part superimposed by 3.65
(t, J12,11
= 6.4, 12-H2),* 7.34–7.44
(m, 6 Ar-H), 7.64–7.70 (m, 4 Ar-H); *assignments interchangeable.
IR (neat): ν
= 3340, 3070, 2930, 2855, 1465, 1430, 1390,
1360, 1190, 1110, 825, 740, 705 cm−1. C28H
40O2Si (440.7) calcd. C 76.30, H 10.06; found C 76.39, H
9.91.
12-(tert-Butyldiphenylsiloxy)dodecanal (54). At − 78°C DMSO (6.72 ml, 7.39 g, 94.8 mmol, 2.2 equiv.) was added
dropwise to a solution of oxalylic chloride (4.15 ml, 6.02 g, 47.4 mmol, 1.1
equiv.) in CH2Cl2 (120 ml). After 3 min alcohol
53 (19.0 g, 43.1 mmol) in CH2Cl2 (10 ml) was added.
After stirring for 1 h at − 40°C NEt3 (29.9 ml, 21.8
g, 216 mmol, 5.0 equiv.) was added. Within 1 h the mixture was allowed to
warm to 0°C and water (150 ml) was added. The organic phase was separated,
washed with HCl (2 M, 100 ml) and NaHCO3 solution (10 ml) and
dried over MgSO4. After removal of the solvent the residue was
purified by flash chromatography (8 cm, petroleum ether–ButOMe
15:1, fractions 7–12) to yield the title compound (15.43 g, 83%).
1H NMR (300 MHz, contains traces of petroleum ether): δ
=
1.05 (s, ButSi), 1.23–1.38 (m, 4-H2 to 10-H
2), 1.55 (mc, 3-H2),* in part superimposed
by 1.63 (mc, 11-H2),* 2.41 (td, J
2,3
= 7.4, J2,1
= 1.9, 2-H2),
3.65 (t, J12,11
= 6.6, 12-H2), 7.34–7.44
(m, 6 Ar-H), 7.64–7.70 (m, 4 Ar-H), 9.76 (t, J1,2
=
1.9, 1-H); *assignments interchangeable. IR (neat): ν
=
3070, 2930, 2855, 1710, 1465, 1430, 1390, 1360, 1185, 1110, 825, 740, 705,
615 cm−1. C28H42O2Si (438.7)
calcd. C 76.65, H 9.65; found C 76.51, H 9.69.
(4S,5S)-5-[10-(tert-Butyldiphenylsiloxy)decyl]-4,5-dihydro-4-hydroxy-2(3
H)-furanone (55). At 0°C K3Fe(CN)6
(22.1 g, 67.3 mmol, 3.0 equiv.), K2CO3 (9.29 g, 67.3
mmol, 3.0 equiv.), (DHQ)2PHAL (174 mg, 0.224 mmol, 1.0 mol.%),
K2OsO4 (17 mg, 0.045 mmol, 0.2 mol.%), methanesulfoneamide
(2.13 g, 22.4 mmol, 1.0 equiv.) and the unsaturated ester 56 (11.1
g, 22.4 mmol) were added to a 1:1 mixture of ButOH and H2
O (110 ml each). This mixture was stirred for 4 days. The reaction was
worked up by the addition of aqueous Na2SO3 solution
(100 ml). After extraction with EtOAc (3 × 200 ml) the organic extracts
were dried over Na2SO4. Removal of the solvent yielded
a residue that was purified by flash chromatography (6 cm, petroleum ether–Bu
tOMe 3:1 → fraction 8, 1:1 → fraction 18, 1:2 → fraction
50, fractions 5–24) to yield 55 (7.56 g, 68%) as a colorless
liquid. [α]D25
=
−
17.9 (c
= 0.84). The R-Mosher ester of 55
revealed ee = 97% by δMeO
=
3.48 s. δMeO in the diastereomer =
3.54. 1H NMR (300 MHz, contains 11 wt.% ButOMe):
δ
= 1.05 (s, ButSi), 1.22–1.60 (m, 2′-H
2 to 9′-H2), 1.65–1.92 (m, 1′-H2),
2.11 (d, JOH,4
= 4.5, OH), AB signal (δ
A
= 2.54, δB
= 2.78,
JAB
= 17.7, split by JB,4
=
5.3, 3-H2), 3.65 (t, J10′,9′
=
6.6, 10′-H2), 4.35 (ddd, J5,1′-H(1)
= 8.8, J5,1′-H(2)
= 5.7,
J5,4
= 3.6, 5-H), 4.46 (ddd, J4,3-H(B)
≈J4,5≈J4,OH≈4.5, 4-H),
7.34–7.45 (m, 6 Ar-H), 7.64–7.70 (m, 4 Ar-H). IR (neat):
ν
= 3435, 3070, 2930, 2855, 1765, 1465, 1430, 1390, 1360, 1200,
1170, 1110, 1015, 825, 740, 705, 610 cm−1. C30H
44SiO4 (496.8) calcd. C 72.53, H 8.96; found C 72.30, H
9.33. Methyl E-14-(tert-butyldiphenylsiloxyl)-3-tetradecenoate
(56). A mixture of aldehyde 54 (15.0 g, 34.2 mmol), monomethylmalonate
(4.44 g, 37.6 mmol, 1.1 equiv.) and NEt3 (5.20 ml, 3.80 g, 37.6
mmol, 1.1 equiv.) was heated at 90°C overnight. After the addition of
ice (100 ml), diluted HCl (2 M, 40 ml) and ButOMe (100 ml) the
organic phase was separated and dried over MgSO4. After removal
of the solvent the residue was purified by flash chromatography (6 cm, petroleum
ether–ButOMe 50:1 → fraction 7, 20:1 → fraction
15, fractions 7–13) to give the title compound (11.15 g, 66%).
1H NMR [300 MHz; contains 6 mol.% methyl trans-14-(
tert-butyldiphenylsiloxy)-2-dodecenoate as determined from the integral
intensity of its CO2Me group at δ
= 3.72]:
δ
= 1.05 (s, ButSi), 1.20–1.40 (m, 6-H
2 to 12-H2), 1.55 (tt, J13,14≈
J13,12≈6.9, 13-H2), 2.02 (dt, J
5,4≈J5,6≈6.4, 5-H2), 3.03 (d,
J2,3
= 5.3, 2-H2), 3.65 (t, J
14,13
= 6.4, 14-H2), in part superimposed by 3.68
(s, OMe), extreme AB signal with additional peaks indicating transition to
higher order signal (δA
= 5.50, δ
B
= 5.57, JAB
= 15.5, split
by JA,2
= 5.6,*
JB,5
=
5.6,* 3-H, 4-H; *assignments of coupling partners interchangeable),
7.34–7.44 (m, 6 Ar-H), 7.64–7.70 (m, 4 Ar-H). IR (neat):
ν
= 3050, 2930, 2855, 1740, 1465, 1430, 1390, 1360, 1255,
1165, 1110, 970, 825, 740, 705, 610 cm−1.( C31H
46SiO3 (494.8) calcd. C 75.25, H 9.37; found C 75.41, H 9.51.
(3S,4S)-14-(tert-Butyldiphenylsiloxy)-1,3,4-tetradecanetriol
(57). At − 78°C a solution of hydroxylactone 55
(5.67 g, 11.4 mmol) in THF (25 ml) was slowly added to a suspension of LiAlH
4 (433 mg, 11.4 mmol, 1.0 equiv.) in THF (25 ml). The reaction mixture
was warmed to room temperature and after 30 min hydrolyzed with diluted H
2SO4 (3 wt.%, 40 ml). The mixture was extracted with
EtOAc (4 × 100 ml). The combined organic extracts were thoroughly dried
over Na2SO4. After removal of the solvent the title
compound (5.59 g, 98%) was obtained as a colorless liquid. [
α]D25
=
− 4.4 (c
=
0.45). 1H NMR (300 MHz): δ
= 1.05 (s, Bu
t), 1.20–1.38 (m, 6-H2 to 12-H2), 1.39–1.58
(m, 5-H2, 13-H2), 1.68–1.83 (m, 2-H2),
ca. 2.80 (very br s, 2 × OH), ca. 3.30 (very br s, 1 ×
OH), 3.45 (mc, 4-H), 3.65 (t, J14,13
=
6.6, 14-H2), superimposed by ca. 3.68 (mc,
3-H), 3.86 (mc, 1-H2), 7.33–7.44 (m, 6 Ar-H),
7.64–7.70 (m, 4 Ar-H). The H–C–O signals in the
1H NMR spectrum were assigned as in the related compound (3S,4
S)-14-(tert-butyldimethylsiloxy)-1,3,4-tetradecanetriol
in which the correponding signals are less superimposed. IR (neat):
ν
= 3380, 3070, 2930, 2855, 1465, 1430, 1390, 1110, 825, 740,
705 cm−1. C30H48SiO4 (500.8)
calcd. C 71.95, H 9.66; found C 71.72, H 9.86.
(4′S,5′S)-2-{5-[10-(
tert-Butyldiphenylsiloxy)decyl]- 2,2-dimethyl-1,3-dioxolan-4-yl}ethanol
(58). Triol 57 (3.10 g, 6.20 mmol) was dissolved in acetone
(25 ml); 2,2-dimethoxypropane (6.12 ml, 5.20 g, 50.0 mmol, 8.0 equiv.) and
Amberlyst 15 ion exchange resin (120 mg) were added. After stirring the mixture
for 2 h at room temperature the resin was removed by filtration and the solvent
evaporated. The residue obtained was used in the next reaction without further
purification. [α]D25
=
−
12 (c
= 0.81). 1H NMR (500 MHz): δ
=
1.05 (s, But), 1.24–1.38 (m, 2″-H2 to 8″-H
2), 1.40 [s, C(Me)2], 1.45–1.59 (m, 1′-H
2 , 9″-H2), AB signal (δA
=
1.74, δB
= 1.84, JAB
=
14.4, split by JA,4′
= 9.0, J
A,1-H(1)
= 6.6, JA,1-H(2)
= 5.2,
JB,1-H(1)
= 5.6,*
JB,1-H(2)
=
5.2,*
JB,4′
= 3.2, 2-H2),
2.41 (br t, JOH,1
= 4.7, OH), 3.65 (t, J
10″,9″
= 6.5, 10″-H2), superimposed
by 3.68 (probably interpretable as ddd, J5′,4′
=
8.3, J5′,1″-H(1)
= 7.1, J
5′,1″-H(2)
= 4.2, 5′-H**), 3.77 (ddd,
J4′,5′
=
J4′,2-H(A)
= 8.6, J4′,2-H(B)
= 3.0, 4′-H),
3.83 (br td, J1,2≈J1,OH≈5.2,
1-H2), 7.36–7.45 (m, 6 Ar-H), 7.66–7.70 (m, 4 Ar-H); *assignments
interchangeable; **analysis of the coupling constants performed as
in (4′S,5′S)-2-{5-[10-(tert-butyldi
methylsiloxy)decyl]- 2,2-dimethyl-1,3-dioxolan-4-yl}ethanol
where the 5-H signal and the 10″-H2 signal do not coincide.
IR (neat): ν
= 3425, 3070, 2930, 2855, 1465, 1430, 1375,
1240, 1110, 875, 825, 740, 705 cm−1. C33H
52SiO4 (540.9) calcd. C 73.28, H 9.69; found C 73.34, H
9.98.
(4S,5S)-4-[10-(tert-Butyldiphenylsiloxy)decyl]-2,2-dimethyl-5-(2-iodoethyl)-1,3-dioxolane
(59). At 0°C PPh3 (1.62 g, 6.20 mmol, 1.0 equiv.),
imidazole (843 mg, 12.4 mmol, 2.0 equiv.) and I2 (1.58 g, 6.20
mmol, 1.0 equiv.) were added to a solution of alcohol 58 (crude product
from 3.10 g of triol 57, 6.20 mmol) in THF (50 ml). The reaction
mixture was allowed to warm to room temperature within 15 min. Water (100
ml) was added, the organic phase separated and the aqueous phase extracted
with ButOMe (100 ml). The combined organic phases were dried over
MgSO4 and the solvent was removed. The residue was purified by
flash chromatography (4 cm, deactivated silica, petroleum ether–Bu
tOMe 50:1, fractions 3–10). The title compound (3.814 g, 94%
over the two steps from triol 57) was obtained as a colorless liquid. [
α]D25
=
− 20.0 (c
= 1.09). 1H NMR (500 MHz): δ
=
1.05 (s, But), 1.23–1.38 (m, 2′-H2 to 8′-H
2), 1.37 and 1.39 [2 s, C(Me)2], 1.42–1.58
(m, 1′-H2, 9′-H2), AB signal (δ
A
= 2.03, δB
= 2.08,
JAB
= 14.3, split by JA,4
=
JA,2″-H(A)
= 8.3, JA,2″-H(B)
= 5.3, JB,2″-H(B)
=
J
B,2″-H(A)
= 8.3, JB,4
= 3.0,
1″-H2), AB signal (δA
=
3.24, δB
= 3.34, JAB
=
9.6, split by JA,1″-H(A)
=
J
A,1″-H(B)
= 8.0, JB,1″-H(B)
=
7.3, JB,1″-H(A)
= 5.1, 2″-H2),
3.66 (t, J10′,9′
= 6.7, 10′-H
2), completely superimposed by (mc, 4-H, 5-H), 7.36–7.45
(m, 6 Ar-H), 7.66–7.70 (m, 4 Ar-H); *assignments and analysis of
coupling constants as in the 1H NMR spectrum of the alcohol precursor
58. 13C NMR (50.3 MHz, APT): 1.87 (C-2″), 19.22 [
C(CH3)3], 25.76, 26.03, 29.35, 29.50 (2-fold
intensity), 29.57, 29.73, 32.58, 32.78 and 37.45 (C1′ to C-9′
C-1″), 26.87 [C(CH3)3], 27.23
and 27.30 (2 × Me), 63.98 (C-10′), 80.32 and 80.57 (C-4, C-5),
108.31 (C-2), 127.52, 129.42 and 135.52 (4 ortho, 4 meta
and 2 para C), 134.12 (2 ipso C). IR (neat): ν
= 3070, 2930, 2855, 1465, 1430, 1375, 1235, 1175, 1110, 825,
740, 705 cm−1. C33H51SiO3I
(650.8) calcd. C 60.91, H 7.90; found C 61.09, H 7.79.
(4S,5S)-Z,Z-4-[10-(
tert-Butyldiphenylsiloxy)decyl]-5-(3,7-eicosadienyl)-2,2-dimethyl-1,3-dioxolane
(61). Vinyl iodide 51 (94.0 mg, 0.250 mmol) was dissolved
in Et2O (1 ml) and ButLi (1.52 M in Et2O,
0.36 ml, 0.55 mmol, 2.2 equiv.) was added at − 50°C. After 30 min
alkyl iodide 59 (163 mg, 0.250 mmol, 1.0 equiv.) in THF (1 ml) was
added. The mixture was warmed to room temperature and stirred for another
4 h. The reaction was terminated by the addition of HCl (1 M, 2 ml). After
extraction with ButOMe (2 × 20 ml) the combined organic phases
were dried over MgSO4. After removal of the solvent flash chromatography
(2.5 cm, petroleum ether–ButOMe 100:1, fractions 6–14)
yielded the title compound (123 mg, 64%). [α]
D25
=
− 7.4 (c
= 0.64).
1H NMR (300 MHz): δ
= 0.88 (t, J
20′,19′
= 6.6, 20′-H3), 1.05 (s,
But), 1.24–1.38 (m, 10′-H2 to 19′-H
2 , 2′-H2 to 8″-H2), 1.39 [br
s, 2-(CH3)2], 1.47–1.60 (m, 1′-H
2, 1″-H2, 9″-H2), 2.02 (td, J
9′,10′≈J9′,8′≈6.3,
9′-H2), in part superimposed by 2.10 (mc, 5′-H
2, 6′-H2), in part superimposed by 2.14–2.26
(m, 2′-H2), 3.61 (mc, 4-H, 5-H), in part superimposed
by 3.65 (t, J10″,9″
= 6.4, 10″-H
2), 5.30–5.46 (m, 3′-H, 4′-H, 7′-H, 8′-H),
7.34–7.46 (m, 6 Ar-H), 7.65–7.70 (m, 4 Ar-H). IR (neat):
ν
= 3050, 2925, 2855, 1465, 1430, 1370, 1240, 1110, 825, 740,
705 cm−1. C51H84SiO3 (773.3)
calcd. C 79.21, H 11.95; found C 79.73, H 11.94.
(4′S,5′S)-Z,Z
-10-[5-(3,7-Eicosadienyl)-2,2-dimethyl-1,3-dioxolan-4-yl]-1-decanol
(62). At room temperature silylether 61 (1.64 g, 2.12 mmol)
in THF (20 ml) was treated with Bu4NF (1.0 M in THF, 2.55 ml,
2.55 mmol, 1.2 equiv.). Stirring was continued for 20 h. After hydrolysis
with water (20 ml) the aqueous phase was extracted with ButOMe
(3 × 50 ml). The organic phases were dried over MgSO4 and
the solvent was evaporated. Flash chromatography (3 cm, petroleum ether–Bu
tOMe 10:1 → fraction 12, 2:1 → fraction 30, fractions 17–26)
yielded 62 (1.133 g, 99%). [α]
D25
=
− 9.33 (c
= 1.20).
1H NMR (300 MHz, slightly contaminated around δ
=
0.9): δ
= 0.88 (t, J20″,19″
= 6.5, 20″-H3), 1.24–ca. 1.38
(m, 10″-H2 to 19″-H2, 3-H2 to
9-H2), 1.38 [s, 2′-(CH3)2],
1.45–1.62 (m, 2-H2, 10-H2, 1″-H2),
2.02 (td, J9″,10″≈J9″,8″
≈6.4, 9″-H2), in part superimposed by 2.10 (m
c, 5″-H2, 6″-H2), in part superimposed
by 2.13–2.26 (m, 2″-H2), 3.60 (mc, 4′-H,
5′-H), in part superimposed by 3.64 (t, J1,2
=
6.6, 1-H2), 5.30–5.46 (m, 3″-H, 4″-H, 7″-H,
8″-H). IR (neat): ν
= 3360, 2925, 2855, 1460, 1375,
1240, 1170, 1090, 1060, 875, 725 cm−1. C35H
66O3 (534.9) calcd. C 78.59, H 12.44; found C 78.71, H 12.52.
(11S,12S)-Z,Z-1-Iodo-15,19-dotriacontadien-11,12-diol
(63). HCl (12 M, 10 μl, 0.10 mmol, 4.0 equiv.) was added to a
solution of the acetonide 5 (30.0 mg, 0.0466 mmol) and the butenolide
26 (40.0 mg, 0.260 mmol) in MeOH–CH2Cl2 (5:1,
0.6 ml) and the mixture was stirred for 24 h at room temperature. Water (2
ml) was added and the organic phase extracted with ButOMe (3 ×
10 ml). The combined organic phases were dried over Na2SO
4 and the solvent was removed. The residue was purified by flash chromatography
(2.5 cm, petroleum ether–ButOMe 10:1 → fraction 12,
5:1 → fraction 18, 2.5:1 → fraction 34). The unconsumed butenolide
26
{fractions 10–15, 30.6 mg, 76%; [α
]D25
=
− 48.4 (CHCl3,
c
= 0.8)} and the title compound (fractions 28–34,
14.1 mg, 50%) were obtained as colorless liquids. 1H NMR
(300 MHz): δ
= 0.88 (t, J32,31
=
6.8, 32-H3), 1.24–ca. 1.43 (m, 3-H2 to
9-H2, 22-H2 to 31-H2), ca. 1.45–1.60
(m, 10-H2, 13-H2), 1.82 (tt, J2,1
=
J2,3
= 7.1, 2-H2), 2.02 (br td, J
21,22≈J21,20≈6.5, 21-H2), in part
superimposed by 2.11 (mc, 17-H2, 18-H2),
in part superimposed by ca. 2.14–2.25 (m, 14-H2),
3.19 (t, J1,2
= 7.0, 1-H2), 3.36–3.47
(m, 11-H, 12-H), 5.30–5.47 (m, 15-H, 16-H, 19-H, 20-H). No combustion
analysis was performed.
Acknowledgements
We are grateful to Magali Slozsek–Pinaud [Laboratoire
de Pharmocognosie, (CNRS URA 1843 BIOCIS), Faculté de Pharmacie, Chatenay-Malabry,
France] for her participation in the exploratory phase of this work
and to the European Union for financing her stay through a stipend of the
ERASMUS Program. Moreover, we are indebted to the Metallgesellschaft GmbH
(Langelsheim) and BASF AG (Ludwigshafen) for donating chemicals.References and notes
- J. K. Rupprecht,
Y.-H. Hui and J. L. McLaughlin, J. Nat. Prod.,
1990, 53, 237 CrossRef CAS; X.-P. Fang, M. J. Rieser, Z. M. Gu, G. X. Zhao and J. L. McLaughlin
, Phytochem. Anal., 1993,
4, 27 and 49 CrossRef CAS; A. Cavé, B. Figadère,
A. Laurens and D.
Cortes, Prog. Chem. Nat. Prod., 1997
, 70, 81 Search PubMed; F. Q. Alali, X.-X. Liu and
J. L. McLaughlin, J. Nat. Prod., 1999, 62, 504 CrossRef.
-
B. Figadère, Acc. Chem. Res.
, 1995, 28, 359 CrossRef CAS; U. Koert
, Synthesis, 1995,
115 CrossRef CAS; R.
Hoppe and H.-D. Scharf, Synthesis, 1995, 1447 CrossRef CAS; G. Casiraghi
, F. Zanardi, L. Battistini, G. Rassu and G. Appendino, Chemtracts, 1998
, 11, 803 Search PubMed.
- C.
Gleye, A. Laurens, R. Hocquemiller, B. Figadère and A. Cavé, Tetrahedron
Lett., 1996, 37, 9301 CrossRef CAS.
- C. Gleye, S. Raynaud, R. Hocquemiller
, A. Laurens, C. Fourneau,
L. Sérani, O. Laprévote, F.
Roblot, M. Leboeuf, A. Fournet, A. R. De Arias,
B. Figadère and A. Cavé, Phytochemistry,
1998, 47, 749 CrossRef CAS.
- C.
Gleye, A. Laurens, R. Hocquemiller, A. Cavé, O. Laprévote and
L. Serani, J. Org. Chem., 1997, 62, 510 CrossRef CAS.
- Reviews:
(a) R. A.
Johnson and K. B. Sharpless
, Asymmetric Catalysis in Organic Synthesis,
ed. I. Ojima, VCH, New York
, 1993, pp. 227–272 Search PubMed;
(b) H. C. Kolb
, M. S. VanNieuwenhze and K. B. Sharpless, Chem. Re
![[italic v]](https://www.rsc.org/images/entities/char_e0f5.gif) ., 1994, 94, 2483 Search PubMed.
., 1994, 94, 2483 Search PubMed. -
(a) Z.-M. Wang
, X.-L. Zhang, K. B. Sharpless, S. C. Sinha,
A. Sinha–Bagchi and
E. Keinan, Tetrahedron Lett., 1992, 33, 6407 CrossRef CAS;
(a) Y. Miyazaki
, H. Hotta and F. Sato, Tetrahedron
Lett., 1994, 35, 4389 CrossRef CAS;
(b) C. Harcken
and R. Brückner, Angew. Chem., 1997, 109, 2866;
C. Harcken and R.
Brückner, Angew. Chem. Int., Ed. Engl., 1997, 36, 2750 CrossRef CAS;
(c) C. Harcken
, E. Rank and R. Brückner,
Chem. Eur. J., 1998, 4,
2342 (corrigendum: 2390) CrossRef CAS;
(d) T. Berkenbusch
and R. Brückner, Tetrahedron, 1998, 54, 11461 CrossRef CAS;
(e) T. Berkenbusch
and R. Brückner, Tetrahedron, 1998, 54, 11471 CrossRef CAS;
(f) C.
Harcken, T. Berkenbusch
, S. Braukmüller
, A. Umland, K. Siegel, F. Görth,
F. von der Ohe and R.
Brückner, in Current Trends
in Organic Synthesis, ed. C. Scolastico and
F. Nicotra, Plenum Press, New York,
1999, pp. 153–161. Search PubMed.
- Previous preparation of compound
4: S.-Y. Chen and M. M. Joullié, J.
Org. Chem., 1984, 49, 2168. Search PubMed; Specific rotation of 4: C. A. M. Afonso, M. T.
Barros, L. S. Godinho
and C. D. Maycock, Tetrahedron, 1993, 49,
4283. Search PubMed.
- Asymmetric dihydroxylations of disubstituted
olefins trans-Me–CHCH–R showed 72% ee
in the case of 2-butene (“unpublished results” in ref. 6b, 73% ee in the case
of 1-phenyl-3-penten-1-yne ( K.-S. Jeong, P. Sjö
and K. B. Sharpless, Tetrahedron Lett., 1992, 33, 3833 CrossRef CAS ) and 95% ee
in the cases of 1-chloro-2-butene ( K.
P. M. Vanhessche, Z.-M.
Wang and K. B.
Sharpless, Tetrahedron Lett., 1994, 35, 3469 CrossRef CAS
) or 4,4-dimethyl-2-pentene (“unpublished results” in ref. 6b)..
- Method using NaH or LiHMDS: B. E. Maryanoff,
A. B. Reitz and B. A. Duhl–Emswiler, J. Am. Chem. Soc., 1985, 107, 217 CrossRef CAS; , , ; using NaH: R.
Beugelmans, J. Chastanet
, H. Ginsburg, L. Quintero–Cortes and G. Roussi,
J. Org. Chem., 1985, 50,
4933 Search PubMed using dimsyl-Li: S. R. Baker,
D. W. Clissold and A. McKillop, Tetrahedron Lett.,
1988, 29, 991 CrossRef CAS using KOBut: F. Ozaki, M. Matsukura
, Y. Kabasawa, K. Ishibashi and M. Ikemori, Chem.
Pharm. Bull., 1992, 40,
2735 CrossRef CAS.
- S. Zahr and
I. Ugi, Synthesis, 1979
, 266 CrossRef CAS.
- Method: T. Mukaiyama, M.
Yamaguchi and J.-i. Kato, Chem. Lett., 1981, 1505 CAS.
- G. Buechi, M. Cushman and H. Wüest
, J. Am. Chem. Soc., 1974,
96, 5563 CrossRef CAS.
- C. S. Pak, E. Lee and G. H. Lee, J. Org. Chem., 1993, 58, 1523 CrossRef CAS.
- Procedure: F. Kido, K. Yamaji, S. C.
Sinha, T.
Abiko and M. Kato, Tetrahedron, 1995, 51, 7697 CrossRef CAS.
- E. J. Corey, C. U. Kim and
M. Takeda, Tetrahedron Lett., 1972, 4339 CrossRef.
- This compound was first prepared by: J. Tsuji,
J. Kiji and S.
Hosaka, Tetrahedron Lett., 1964, 605 CrossRef CAS.
- K. P. M. Vanhessche, Z.-M. Wang and K. B. Sharpless, Tetrahedron
Lett., 1994, 35, 3469 CrossRef CAS.
- P. Blundell, A. K.
Ganguly and V. M. Girijavallabhan, Synlett, 1994
, 263 CrossRef CAS.
- Earlier preparation:
J. Mulzer, T.
Schulze, A. Strecker and W. Denzer,
J. Org. Chem., 1988, 53,
4098 CrossRef CAS.
-
Earlier preparation: J.
Corbera, J. Font, M. Monsalvatje, R. M. Ortuñ and F. Sánchez–Ferrando, J. Org. Chem., 1988, 53, 4393 CrossRef CAS.
- P. Duret, B. Figadère,
R. Hocquemiller and A. Cavé, Tetrahedron Lett.,
1997, 38, 8849 CrossRef CAS.
- Procedure: R. H. Bradbury and
K. A. M. Walker, J. Org. Chem., 1983, 48, 1741 CrossRef CAS.
- Procedure: D. Savoia, E. Tagliavini, C. Trombini and A. Umani–Ronchi, J. Org. Chem., 1981, 46, 5340 CrossRef CAS.
- This compound was previously
described by: E. R. H.
Jones, T. Y. Shen and M. C. Whiting, J. Chem. Soc., 1950, 230 RSC.
- Procedure: L. Brandsma, Preparatie Acetylenic Chemistry,
Elsevier, Amsterdam, 1988,
pp. 42–43. Search PubMed.
- This compound was previously described
by: A. B ranner and H. Budzikiewicz, Org. Mass. Spectrom., 1983, 18, 324 Search PubMed.
- Procedure: E. J.
Corey, H. Niwa and J. Knolle,
J. Am. Chem. Soc., 1978, 100, 1942 CrossRef CAS.
- This compound was previously described by: R. E. Doolittle,
D. G. Patrick and R. H. Heath, J. Org. Chem., 1993
, 58, 5063 CrossRef CAS.
- Method: H. Hayashi, K. Nakanishi, C.
Brandon and J. Marmur, J. Am. Chem. Soc., 1973, 95, 8749 CrossRef CAS.
- Procedure: U.
Berlage, J. Schmidt, U. Petres and P. Welzel, Tetrahedron Lett.
, 1987, 27, 3091 CrossRef CAS.
- Procedure:
J. Bao, W. D. Wulff,
V. Dragisich, S. Wenglowsky and R. G.
Ball, J. Am. Chem. Soc., 1994, 116, 7616 CrossRef CAS.
- Alkylation of an acetylide with a homopropargyl bromide: ref. 26..
- Alkylation of an acetylide with a homopropargyl triflate: ref. 32..
- This compound was described by: H. B. Henbest, E.
R. H. Jones and I. M. S. Walls, J. Chem. Soc., 1950, 3646 RSC.
- Method: C. A.
Brown and A. Yamashita, J. Am. Chem. Soc., 1975, 97, 891 CrossRef CAS procedure: S. R. Abrams and A. C. Shaw, Org.
Synth., 1993, VIII, 146
.
- This
compound is known: R.
Rossi and A. Carpita, Tetrahedron, 1983, 39, 287 CrossRef CAS.
- A. Alexakis, G. Cahiez and
J. F. Normant, Tetrahedron, 1980, 36, 1961 CrossRef CAS.
- This compound was used
by: M. Horiike, G. Yuan and C.-S. Kim, Org. Mass. Spectrom.
, 1992, 27, 944 Search PubMed.
- W. F. Bailey, R.
P. Gagnier and J. J. Patricia, J. Org. Chem., 1984
, 49, 2098 CrossRef CAS.
- Method: A. Alexakis, G. Cahiez and J. F.
Normant, J. Organomet. Chem., 1979, 177, 293 CrossRef CAS.
- K. Omura and D. Swern,
Tetrahedron Lett., 1978, 1651 CAS.
- Method: H. Yamanaka,
M. Yokoyama, T. Sakamoto, T. Shiraishi
, M. Sagi and M. Mizugaki, Heterocycles
, 1983, 20, 1541 Search PubMed.
- A. I. Meyers and J.
P. Lawson, Tetrahedron Lett., 1982, 23, 4883 CrossRef CAS.
- Ni-catalyzed coupling
between alkyl Grignard reagents and alkenyl iodides: T. Hayashi,
M. Konishi, Y.
Kobori, M. Kumada, T. Higuchi and K. Hirotsu, J. Am. Chem. Soc.
, 1984, 106, 158 CrossRef CAS.
- Method: G. Linstrumelle, Tetrahedron Lett., 1974, 3809 CrossRef CAS.
- Procedure: U. Berlage, J. Schmidt,
U. Petres and P.
Welzel, Tetrahedron Lett., 1987, 27, 3091 CrossRef CAS.
- Method: H.-M. Shieh and G. D. Prestwich
, J. Org. Chem., 1981,
46, 4319 CrossRef CAS; H.-M. Shieh and G. D.
Prestwich, Tetrahedron Lett., 1982, 23, 4643 CrossRef CAS.
- Organic synthesis applications
of DMPU: D. Seebach, Chimia, 1985, 39, 147 Search PubMed;
D. Seebach, A. K. Beck and A.
Studer, Modern Synthetic Methods 1995,
ed. B. Ernst and C. Leumann, VCH
, Weinheim, 1995, pp.
1–178. Search PubMed.
- Cf. the detailed
calculations in Experimental..
- W. C. Still, M. Kahn
and A. Mitra, J. Org. Chem., 1978, 43, 2923 CrossRef CAS.
- This value is based on the following calculation, wherein
[α]D25 lactone is not the
specific rotation of compound S,S-4 but the “molar
contribution of a 100% enantiopure left-hand moiety of compound
1a to the [α]D25 value
of 1a”. Analogously, −
[α]
D25 lactone is not the specific rotation of compound
R,R-4 but the “molar contribution of a 100%
enantiopure left-hand moiety of compound 1b to the [α
]D25 value of 1b”. Similarly, [
α]D25 iodide is not the specific rotation
of compound 5 but the “molar contribution of a 100%
enantiopure right-hand moiety of compounds 1a or 1b to
the [α]D25 values of 1a
or 1b, respectively. One starts from the equation 0.80 ×
[
α]D25 lactone + 0.97 ×
[
α]D25 iodide =
+ 1.9 ( =
[
α]D25 obtained sample of 1a)
and the equation 0.90 × (−[α]D
25 lactone) + 0.97 ×
[α]D
25 iodide =
− 24.2 ( =
[α
]D25 obtained sample of 1b). Separation
of the unknowns delivers [α]D25
lactone = 15.3 and [α]D
25 iodide =
− 10.6. Inserting these values into the equations
1.00 ×
[α]D25 lactone +
1.00 ×
[α]D25 iodide =
[
α]D25 sterically pure sample of
1a and 1.00 × (−[α]D
25) lactone + 1.00 ×
[α]D
25 iodide =
[α]D25
sterically pure sample of 1b), respectively, leads to [
α]D25 sterically pure sample of
1a
= 1.00 × 15.3 + 1.00 × (−10.6) =
+
4.7 and to [α]D25 sterically
pure sample of 1b
= 1.00 × ( × 15.3) + 1.00 ×
(−10.6) =
− 25.9..
Footnotes |
| † Previous address: Institut für Organische Chemie der Georg-August-Universität,
Tammannstr. 2, D-37077, Göttingen, Germany. |
| ‡ Present address: Department of Chemistry and Biochemistry,
University of Texas at Austin, Texas 78712, USA. |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2001 |
Click here to see how this site uses Cookies. View our privacy policy here.