Isomerization of allylic alcohols to carbonyl compounds by aqueous-biphase rhodium catalysis
Claudio Bianchini*, Andrea Meli and Werner Oberhauser
Istituto per lo Studio della Stereochimica ed Energetica dei Composti di Coordinazione, ISSECC-CNR, 50132, Firenze, Italy. E-mail: bianchin@fi.cnr.it
First published on UnassignedUnassigned14th June 2000
Abstract
Isomerization of allylic and homoallylic alcohols is catalyzed by the zwitterionic Rh(I) complex (sulphos)Rh(cod) in water–n-octane to give the corresponding aldehyde or ketone in high yields and chemoselectivity. A π-allyl metal hydride mechanism is proposed on the basis of various independent experiments in both homogeneous and biphasic systems [sulphos=−O3S(C6H4)CH2C(CH2PPh2)3].
The isomerization of allylic alcohols to carbonyl compounds is an important reaction that can be accomplished with homogeneous,1 heterogeneous2 and phase-transfer catalysts.3 Serious drawbacks have often been encountered in the application of this reaction to obtain synthetically useful quantities of either aldehydes or ketones, however. Most common drawbacks are: (i) difficult separation of the catalyst from product(s)/reagent(s);1,3 (ii) low chemoselectivity, generally due to competitive acetalization and dehydration reactions;1,2 (iii) sensitivity to the degree of substitution on the double bond;1 (i
![[italic v]](https://www.rsc.org/images/entities/i_char_e0f5.gif) ) possible catalyst deactivation ia decarbonylation of aldehydes to give stable carbonyl metal compounds.1,4
) possible catalyst deactivation ia decarbonylation of aldehydes to give stable carbonyl metal compounds.1,4The present paper constitutes a preliminary account of the first example of effective aqueous-biphase isomerization of allylic alcohols catalyzed by a water-soluble transition metal complex in a chemoselective manner. A comparative, in situ and reactor, study in the homogeneous phase has provided valuable mechanistic information, particularly on the pathways to catalyst deactivation. Prior to our work, the isomerization of geraniol to citronellal was attempted in toluene–water using Ni(cod)2–dppbts [tetrasulfonated 1,4-bis(diphenylphosphino)butane]. Very low conversion (1%) and selectivity (3%) were obtained, however.5
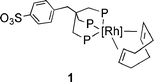
The zwitterionic rhodium complex (sulphos)Rh(cod) (1) is an efficient catalyst for the hydrogenation and hydroformylation of olefins in water–n-octane [sulphos=−O3S(C6H4)CH2C(CH2PPh2)3].6 We show here that the same catalytic system can successfully be employed for the chemoselective transformation of both allylic and homoallylic alcohols into the corresponding aldehyde or ketone. Selected results of aqueous-biphase and homogeneous reactions (in 1,2-dichloroethane) are reported in Table 1.
| Entry | Substrate | Products | Homogeneous yield(%) | Liquid-biphase yield(%) |
|---|---|---|---|---|
| a Reaction conditions: autoclave, 100 mL; catalyst, 0.021 mmol; substrate, 2.1 mmol; 1,2-dichloroethane (homogeneous) or 1:1 water–n-octane (liquid-biphase), 30 mL; 100°C; 1 h. | ||||
| 1 | 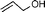 | 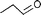 | 95 | 100 |
| 2 | 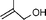 |  | 38 | 73 |
| 3 | 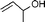 |  | 98 | 100 |
| 4 | 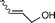 | 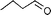 | 62 | 30 |
| 5 |  |  | 4 | <1 |
 | 4 | <1 | ||
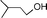 | 1 | <1 | ||
 | 4 | <1 | ||
 | 9 | <1 | ||
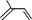
| 6 | 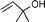 | none | ||
| 7 |  | 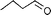 | 89 | 33 |
| 8 | 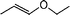 | 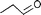 | 14 | |
 | <1 | |||
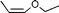 | 22 | |||
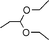 | 31 | |||
In biphasic conditions, 100 equiv. of allylic alcohol was converted to propanal within 1 h at 100°C (entry 1). By simple phase-separation, the aldehyde product (>99% in n-octane) was separated from the catalyst, which was recycled three times to give the following turn-over-frequencies [mol product (mol catalyst)−1 h−1]: 490, 430 and 390. Appreciable deactivation (ca. 10%) occurred after each run due to partial incorporation of active rhodium into the dicarbonyl complex (sulphos)Rh(CO)26 (2), which was independently shown to form upon reaction of 1 with propanal (and aldehydes in general) and to be inactive for allylic alcohol isomerization. In homogeneous phase, where the catalyst and the aldehyde product are in the same fluid system (entry 1), the formation of 2 was apparently much faster (NMR evidence) and the catalyst could not be re-used for a second run (90% formation of 2).
In both homogeneous and biphase conditions, 2-methyl-2-propen-1-ol (entry 2) and 3-buten-2-ol (entry 3) were isomerized to 2-methylpropanal and methyl ethyl ketone, respectively. The biphasic reactions gave better results in general. Increasing the steric hindrance at the C-3 carbon atom of the allylic moiety as in 2-buten-1-ol (95:5 mixture of E and Z isomers) (entry 4) or 3-methyl-2-buten-1-ol (entry 5) resulted in a progressive decay of the catalytic performance.1 No catalytic activity was observed when two methyl groups were introduced in the C-1 position as in 1,1-dimethyl-2-propen-1-ol (entry 6). The homoallylic alcohol 3-buten-1-ol was isomerized by 1 to butanal in both phase systems (entry 7). Like the 2-buten-1-ol isomer, the conversion to aldehyde was higher in the homogeneous phase, however.
As a general feature, neither hydrates of aldehydes1 nor dehydration products1,2 were observed in the biphasic reactions, whereas some homogeneous reactions gave dehydration products in the form of free and coordinated butadiene or isoprene. Actually, the complexes (sulphos)Rh(η4-butadiene) (3) and (sulphos)Rh(η4-isoprene) (4) were either detected by in situ NMR experiments or isolated as termination products of the homogeneous reactions involving 2-buten-1-ol and 3-buten-1-ol or 3-methyl-2-buten-1-ol, respectively.† The transformation of homoallyl alcohol most likely involves its preliminary isomerization to the allylic isomer 2-buten-1-ol. Indeed, 1 is an efficient olefin isomerization catalyst as shown by the conversion of the cod ligand to free cycloocta-1,3-diene7 (1H NMR, GC-MS) as well as the isomerization of allyl ethyl ether to cis-propenyl ethyl ether (entry 8).
The two established pathways for transition metal-catalyzed isomerization of allylic alcohols are the p-allyl metal hydride1c,3 and the metal hydride addition-elimination mechanisms.1a,b A third variation is the “internal redox” mechanism suggested by Trost and Kulawiec that involves the coordination of the allylic alcohol as bidentate ligand.1b,f
The results from the catalytic reactions and from various
independent experiments are consistent wih the π-allyl metal hydride mechanism shown in Scheme 1, which is quite similar to that proposed by Bergens and Bosnich for [Rh(diphosphine)(solvent)2]+
catalyst precursors.1c Commonalities between the latter Rh systems and 1 are: (i) the interception of enol intermediates [Z- and E-CH(CH3)![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) CHOH]c along the isomerization of allylic alcohol to propanal; (ii) the selective incorporation of deuterium in the C-2 position of propanal when allylic alcohol is isomerized in D2O; (iii) the failure to isomerize
1,1-dimethyl-2-propen-1-ol. Unlike the cationic Rh-diphosphine systems, the zwitterionic Rh-sulphos fragment also converts homoallylic alcohol into butanal due to its peculiar ability to isomerize olefins.7
CHOH]c along the isomerization of allylic alcohol to propanal; (ii) the selective incorporation of deuterium in the C-2 position of propanal when allylic alcohol is isomerized in D2O; (iii) the failure to isomerize
1,1-dimethyl-2-propen-1-ol. Unlike the cationic Rh-diphosphine systems, the zwitterionic Rh-sulphos fragment also converts homoallylic alcohol into butanal due to its peculiar ability to isomerize olefins.7
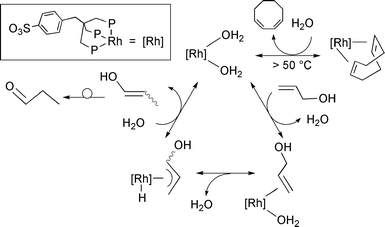 | ||
| Scheme 1 Proposed mechanism for the isomerization reaction of allyl alcohol to propanal catalyzed by 1. | ||
In conclusion, we have shown here that aqueous-biphase catalysis can be successfully employed for the isomerization of both allylic and homoallylic alcohols to carbonyl compounds. The main advantage of the present system over homogeneous systems is the facile catalyst recycling by simple phase separation with no loss of rhodium metal.
Further modification of sulphos-based catalysts (different metals, introduction of stereocenters), as well as extension of the range of substrates and prospects for asymmetric induction8 remain exciting avenues to explore for the clean and selective transformation of allylic residues into aldehyde or ketone functional groups by aqueous-biphase catalysis.
Acknowledgements
MURST and CNR (legge 95/95) are gratefully acknowledged for financial support.Notes and references
- D. V. McGrath and R. H. Grubbs, Organometallics, 1994, 13, 224 CrossRef CAS; B. M. Trost and R. J. Kulawiec, J. Am. Chem. Soc., 1993, 115, 2027 CrossRef CAS; S. H. Bergens and B. Bosnich, J. Am. Chem. Soc., 1991, 113, 958 CrossRef CAS; C. Bianchini, E. Farnetti, M. Graziani, M. Peruzzini and A. Polo, Organometallics, 1993, 12, 3753 CrossRef CAS; F. P. Pruchnik, P. Smolenski and K. Wajda–Hermanowicz, J. Organomet. Chem., 1998, 570, 63 CrossRef CAS; C. Slugovc, E. Rüba, R. Schmid and K. Kirchner, Organometallics, 1999, 18, 4230 CrossRef CAS.
- C. Hoang–Van and O. Zegaoui, Appl. Catal. A, 1997, 164, 91 CrossRef CAS.
- H. Alper and K. Hachem, J. Org. Chem., 1980, 45, 2269 CrossRef CAS.
- (a) J. P. Collman and L. S. Hegedus, Principles and Applications of Organotransition Metal Chemistry, University Science Books, Mill Valley, CA, 1980, ch. 9, p. 496; Search PubMed; (b) C. M. Beck, S. E. Rathmill, Y. J. Park, J. Chen, R. H. Crabtree, L. M. Liable–Sands and A. L. Rheingold, Organometallics, 1999, 18, 5311; Search PubMed; (c) This work..
- H. Bricout, E. Monflier, J.-F. Carpentier and A. Mortreux, Eur. J. Inorg. Chem., 1998, 1739 CrossRef CAS.
- C. Bianchini, P. Frediani and V. Sernau, Organometallics, 1995, 14, 5458 CrossRef CAS; C. Bianchini, A. Meli, V. Patinec, V. Sernau and F. Vizza, J. Am. Chem. Soc., 1997, 119, 4945 CrossRef CAS.
- Complex 1 catalyzes the isomerization of ca. 90 equiv. of cycloocta-1,5-diene to cycloocta-1,3-diene in water–MeOH–n-octane (1:1:2; v:v:v) at 100°C in 1 h..
- C. Botteghi and G. Giacomelli, Gazz. Chim. Ital., 1976, 106, 1131 Search PubMed.
- Compounds 3 and 4 were isolated by column chromatography as yellow–orange solids from the catalytic solutions..
Footnote |
| † Satisfactory elemental analyses were obtained for compounds 3 and 4.9 Selected spectroscopic data for 3: 31P{1H} NMR (CD2Cl2, 81.01 MHz): 25°C, δ 12 (br, PA); −30°C, δ 14.4 [br d, J(PMRh)=100 Hz, PM], 4.5 [dt, J(PAP)=22.1 Hz, J(PARh)=153.2 Hz, PA]; −70°C, δ 22 (br, PQ), 8 (br, PM), 3.8 [br d, J(PARh)=150 Hz, PA]. 1H NMR (CD2Cl2, 200.13 MHz, 25°C): η4-butadiene hydrogens δ 5.61 (m, 2H, CH Hs), 2.0–1.8 (m, 4H, CH2). For 4: 31P{1H} NMR (CD2Cl2, 81.01 MHz): 25°C, δ 11 (br); −30°C, δ 13.4 (2nd order m, PA-PB), δ 1.2 [ddd, J(PMP)=23.8, 15.8 Hz, J(PMRh)=151.7 Hz, PM]. 1H NMR (CD2Cl2, 200.13 MHz, 25°C): η4-isoprene hydrogens δ 5.58 [t, 1H, J(HH)=6.9 Hz, H3 Hs], 2.15 (m, 2H, H1s+H4s), 1.81 (s, 3H, Me), 1.52 (m, 2H, H1a+H4a). |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2001 |