
Self-aggregating cationic-chains enable alkaline stable ion-conducting channels for anion-exchange membrane fuel cells†
Jianjun
Zhang‡
,
Kaiyu
Zhang‡
,
Xian
Liang
,
Weisheng
Yu
,
Xiaolin
Ge
,
Muhammad A.
Shehzad
 ,
Zijuan
Ge
,
Zhengjin
Yang
,
Liang
Wu
* and
Tongwen
Xu
*
,
Zijuan
Ge
,
Zhengjin
Yang
,
Liang
Wu
* and
Tongwen
Xu
*
CAS Key Laboratory of Soft Matter Chemistry, Collaborative Innovation Centre of Chemistry for Energy Materials, School of Chemistry and Materials Science, University of Science and Technology of China, Hefei 230026, P. R. China. E-mail: liangwu8@ustc.edu.cn; twxu@ustc.edu.cn
First published on 26th November 2020
Abstract
Precise manipulation of the polyelectrolyte self-assembly process, to form the desired microstructure with ion-conducting channels, is of fundamental and technological importance to many fields, such as fuel cells, flow batteries and electrodialysis. To fabricate anion exchange membranes (AEMs) with highly conductive and alkaline stable ion-conducting channels, we hereby report a strategy for designing self-aggregating side chains with optimized alkaline stability, by inserting dipolar ethylene oxide (EO) spacers in the cationic side chain. Simulation and nano-scale microscopy analyses verify the self-assembly process of the flexible side chain with cation–dipole interaction to construct interconnected ionic highways for fast water and ion transportation. The resulting O-PDQA AEM exhibits higher hydroxide conductivity (106 mS cm−1 at 80 °C) and a competitive peak power density (1.18 W cm−2 at 70 °C) in alkaline H2/O2 single-cell fuel cells. Moreover, O-PDQA shows excellent alkaline stability with over 96% conductivity retention after storage in 2 M NaOH solution at 80 °C for 1080 h. This new concept of introducing dipolar moieties in the cationic side chain can accelerate the development of technologies that involve polyelectrolytes.
Introduction
Proton-exchange membrane fuel cells (PEMFCs) have aroused widespread interest after the launch of the first fuel cell vehicle.1 Currently, the commercial PEMFCs mainly rely on expensive platinum group metal (PGM) catalysts and perfluorinated proton exchange membranes (PEMs) due to the acidic working environment.2 In contrast, anion-exchange membrane fuel cells (AEMFCs) have enhanced oxygen reduction reaction kinetics in an alkaline environment; thus non-PGM catalysts (Ni-, Co-, Fe-, and Mn-based) and less expensive anion exchange membranes (AEMs) can be adopted.3,4 Currently, highly conductive and stable AEMs are urgently desired to make the AEMFC technology viable.5Generally, AEMs are less conductive than PEMs due to the lower diffusion coefficient of OH− than H+.6,7 Besides, the cationic groups (such as quaternary ammonium (QA), imidazolium, phosphonium, etc.) are usually randomly distributed along the hydrophobic polymer backbone in conventional AEMs. Therefore, OH− conduction will be inevitably blocked by the inert polymer backbone. Decreasing the inert backbone fraction through increasing the ion exchange capacity (IEC) can promote conductivity to some extent, but it also leads to an undesired high swelling ratio.8 An alternative solution to improve the conductivity is constructing unblocked OH− conducting paths, with clustered OH− conducting sites percolating the inert polymer matrix.9 The more ordered morphology can enable faster OH− conduction due to less energy dissipation.
The aforementioned OH− conducting path can be achieved by a well-designed polymer architecture of AEMs. For example, in blocked10–12 or densely functionalized AEMs,13–15 the cationic groups are selectively allocated to one particular segment of the polymer. Thus, the polarity discrimination between hydrophilic/hydrophobic segments can drive the self-aggregation of the hydrophilic components. This typical thermodynamic equilibration process will result in the formation of ion-conducting channels. Other strategies for improving self-aggregation include incorporating additional hydrophobic moieties (hydrophobic F-containing groups and alkyl side chains, etc.)16,17 to increase polarity discrimination, or allocating the cationic group to the AEM side chain18–22 to provide higher mobility for ion aggregation. It could be concluded that the general driving force for self-aggregation relies on the thermodynamical incompatibility between different polymer segments. It is speculated that the introduction of spontaneous intermolecular interactions like ion–dipole interaction, hydrogen bonding, and π–π stacking is expected to provide additional driving forces for self-assembly of the cationic groups in AEMs.23–25 Considering the abundant cationic sites within AEMs, cation–dipole interaction, i.e., the electrostatic attraction between a cation and a neutral dipolar molecule, is an ideal choice to regulate the self-assembled morphology of AEMs.
Ethylene oxide (EO) moiety is a typical dipolar molecule in which the positive and negative charges are non-uniformly distributed on the carbon and oxygen atoms, respectively. The lone pair of electrons of oxygen can interact with the cationic groups in AEMs. And, the created cation–dipole interaction can act as an additional driving force to manipulate the directional aggregation of the cationic groups.26,27 Furthermore, the highly rotatable C–O–C bond in EO groups can improve the flexibility for further aggregation.28 Therefore, we postulate that incorporating the EO group in the cationic side-chain will direct the self-assembly of cationic groups to form continuous ionic highways.29 Apart from the benefit of promoting ion aggregation, the hydrophilic EO groups can facilitate water or OH− transport via H-bonded networks.30 The electron-donating EO moieties are likely to weaken the electropositivity of the connected cationic groups, thus alleviating the OH− attack and improving the ion dissociation. EO containing cationic side chains have been previously reported for the fabrication of AEMs. Most studies aimed to increase the hydrophilicity of the ionic side chain by introducing a hydrophilic EO spacer.31–34 The enhanced hydrophilicity difference between the side chain and polymer backbone can act as a driving force to promote hydrophilic ionic group aggregation. The resulting AEMs show ordered hydrophilic ion channels and competitive ion conductivity. However, the roles of the potential cation–dipole interaction and the changed electronic environment of incorporating EO have been rarely investigated.
Alkaline stability is another primary concern of AEMs. This property is closely correlated with the chemical structure of polymer backbones and the anchored cationic groups.35 Aryl ether-based polyaromatics, including poly(phenylene oxide),36 poly(aryl ether sulfone),37,38 and poly(aryl ether ketone),39 are the most commonly used backbones due to their simple preparation procedure and balanced overall performance. However, Ramani and Kim et al. found that the cleavage of aryl-ether linkages in the polymer backbone occurred easily in a high pH environment.40,41 The recently reported aryl-ether free backbones, including poly(aryl piperidinium),42 poly(phenylene)43 and poly(fluorene-benzene)44 showed improved alkaline stability due to the absence of the aryl ether bond.45 As for the cationic groups, Marino and Kreuer46 reported that cyclic quaternary ammonium (QA) cations are exceptionally more stable than other cationic groups due to the inherent ring geometric constraints restraining the unfavorable Hoffman elimination and nucleophilic substitution degradation. For example, Jannasch and co-workers42 recently reported poly(aryl piperidinium) AEMs with durability for over 360 h in 2 M aq. NaOH at 60 °C. Yan et al.47 reported a partially fluorinated poly(aryl piperidinium) membrane, which can maintain the initial ionic conductivity after 2000 h in 1 M aq. KOH at 100 °C.48–50
Herein, we present an integrated approach to yield highly conductive and stable AEMs, as illustrated in Scheme 1. Self-aggregating side chains with alkaline stable piperidinium cations and ethylene oxide spacers were grafted onto an alkaline stable aryl-ether free poly(aryl piperidinium) (PBP). Additionally, a benchmark PDQA AEM without EO spacers was also prepared for comparison. Furthermore, density functional theory (DFT) and molecular dynamics (MD) simulations were conducted to reveal the mechanism of enhanced ion-aggregation and improved alkaline stability. The relationship between the structure and performance of these AEMs was experimentally investigated and discussed, in terms of nano-scale morphology, ion conductivity, alkaline stability, and H2/O2 single-cell fuel cell performance.
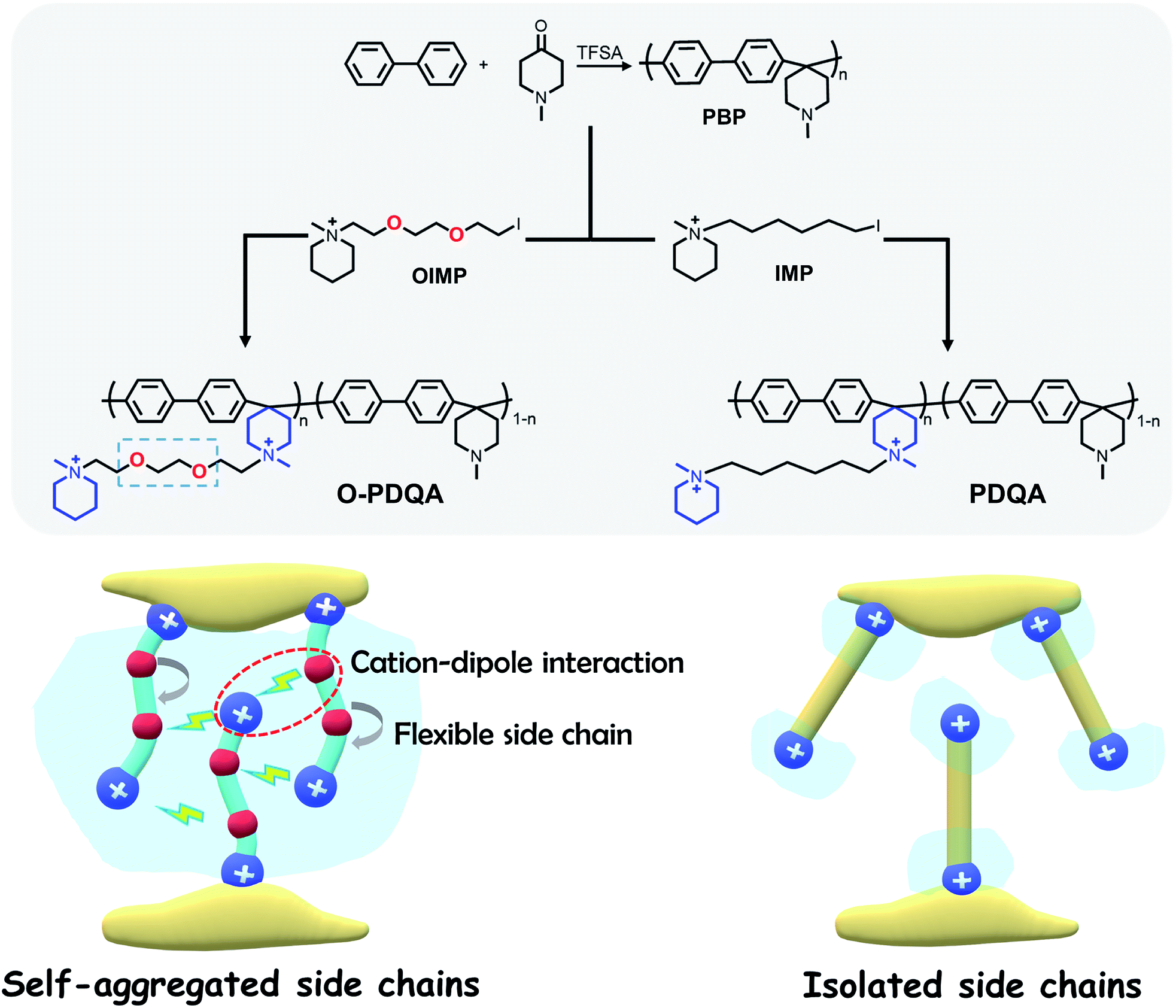 | ||
| Scheme 1 Synthetic routes of O-PDQA (with the self-aggregated side chain due to the cation–dipole interaction) and PDQA (isolated side chain). The schematic diagram shows the function of the self-aggregated side chain, which can promote the formation of the aggregated conducting region. | ||
Experimental
Materials
Biphenyl (99%), N-methyl-4-piperidone (98%), trifluoroacetic acid (TFA) (99%), trifluoromethanesulfonic acid (TFSA) (99%), 1,6-diiodohexane (97%), 1,2-bis(2-chloroethoxy)ethane (98%), methylene chloride, sodium iodide (99%), and N-methylpiperidine (98%) were purchased from Energy Chemical Co. Ltd (Shanghai, P. R. China). Sodium thiosulfate (98.5%), potassium carbonate (K2CO3) (99.0%), dimethyl sulfoxide (DMSO) (99%), tetrahydrofuran (THF) (99.0%), acetone (99.5%), N-methyl pyrrolidone (NMP) (99.5%), sodium hydroxide (NaOH) (96.0%) were all purchased from Sinopharm Chemical Co. Ltd (Shanghai, P. R. China).Synthesis of 1,2-bis(2-iodoethoxy)ethane
Sodium iodide (18.5 g, 124 mmol) and 1,2-bis(2-chloroethoxy)ethane (10.65 g, 57 mmol) were added to acetone (70 mL) and then stirred at reflux for 72 h. After filtering the mixture, the organic phase was concentrated under vacuum. The crude products were dissolved in methylene chloride followed by washing twice with 15% aq. sodium thiosulfate and drying over anhydrous MgSO4. Finally, the organic phase was concentrated under reduced pressure. The final product was obtained as a viscous pure liquid. The 1H NMR spectrum of 1,2-bis(2-iodoethoxy)ethane is given in Fig. S1 (ESI†).Synthesis of 1-(2-(2-(2-iodoethoxy)ethoxy)ethyl)-methylpiperidine (OIMP)
N-Methylpiperidine (1.04 mL, 10 mmol) was added dropwise to an excess amount of 1,2-bis(2-iodoethoxy)ethane (36.9 g, 100 mmol) in CH3CN (200 mL) at reflux for 24 h. After the removal of CH3CN under vacuum, the residue was added to ether (500 mL) to give a yellowish-brown precipitate. Then, the product was collected and washed with ether several times and dried. The final product OIMP was characterized by 1H-NMR, as shown in Fig. S2 (ESI†).Synthesis of 1-(6-iodohexyl)-1-methylpiperidine (IMP)
IMP was prepared using a similar procedure to that of OIMP. N-Methylpiperidine (1.04 mL, 10 mmol) was added dropwise to an excess amount of 1,6-diiodohexane (33.8 g, 100 mmol) in CH3CN (200 mL) at reflux for 24 h. After applying the same purification procedure as in the case of OIMP, the final product IMP was characterized by 1H-NMR, as shown in Fig. S3 (ESI†).Synthesis of poly(biphenyl N-methylpiperidine) (PBP)
The preparation of PBP was conducted according to the literature.42 PBP was synthesized by superacid-catalyzed step-growth polymerization. A typical synthesis procedure is as follows: biphenyl (2.5 g, 6.5 mmol) and N-methyl-4-piperidone (2.23 mL, 19.2 mmol) were added to methylene chloride (6 mL). And then, TFA (16.3 mmol, 1.21 mL) and TFSA (162.8 mmol, 14.4 mL) were added to the mixture dropwise at 0 °C. After reaction for 2 h, the extremely viscous mixture was poured slowly into 2 M NaOH and then washed with water several times. Finally, the yellow fibrous polymer was collected, filtered and dried. The 1H NMR spectrum of PBP is shown in Fig. S4 (ESI†).Synthesis of O-PDQA and PDQA
PBP (1.0 g, 4 mmol) was added to a mixture of DMSO (5 mL) and NMP (5 mL) at 80 °C, and a small amount of TFA was added to promote the dissolution of PBP. Then, OIMP (0.71 g, 1.52 mmol) and K2CO3 (0.42 g, 3.04 mmol) were added in batches. The mixture was stirred for 5 days and then poured into ethyl acetate (400 mL) to precipitate the product followed by washing with water and dried. The synthesis of PDQA is similar to that of O-PDQA. The 1H NMR spectra of O-PDQA and PDQA are shown in Fig. 2a and b, respectively.AEM preparation
O-PDQA and PDQA in the as-synthesized I− form were dissolved in NMP (10 wt%). The solution was filtered through a syringe filter (SCRC, φ = 13 mm × 0.45 μm) and then cast on a flat glass plate (10 cm × 10 cm) and heated at 40 °C for 72 h. The resulting AEMs were peeled from the glass by immersion in deionized water. AEMs in the OH− form (45 ± 4 μm) were obtained by immersing the AEMs in the I− form in NaOH aq. at room temperature for 1 day, and then thoroughly washed with deionized water.Simulation studies
We optimized the molecular structures of O-DQA and DQA using Gaussian 16 at the M06-2X/6-311g+(d, p) level with DFT-D3 dispersion correction.51 The restrained electrostatic potential (RESP) charge was performed at the same level. The electrostatic potential (ESP) and the molecular orbitals of cations were calculated using the Multiwfn program52 and visualized by visual molecular dynamics (VMD). We employed a polarizable continuum model (PCM) using water as the dielectric medium.The flexibility of the side chain was studied using GROMACS based on molecular dynamic (MD) simulation.53 The side chains were modeled using the OPLS-AA force field with RESP charges. We constructed the cubic simulation box, which included the corresponding cationic side chain and water molecules. The V-rescale method with a time constant of 0.1 ps and the Parrinello–Rahman method with a time constant of 2.0 ps were used to control the temperature and pressure. The above-mentioned cubic simulation boxes were obtained in the NPT ensemble (isothermal–isobaric ensemble), where the amount of particles (N), pressure (P) and temperature (T) are constant for 1 ns at 300 K to relax, followed by another 1 ns NPT dynamics for analysis. The coordinate in the trajectory was recorded and transformed into the probability density distribution map.
Results and discussion
Synthesis of O-PDQA and PDQA
As shown in Scheme 1, two AEMs abbreviated as O-PDQA and PDQA containing different cationic side chains were synthesized. O-PDQA has ethylene oxide spacers between the backbone and cationic groups, while PDQA has an aliphatic chain as the spacer. The same aryl-ether free poly(aryl piperidinium) backbone was employed as the main chain for O-PDQA and PDQA. Afterward, the EO containing OIMP precursor or EO-free IMP precursors were grafted onto the backbone. The OIMP and IMP precursors were synthesized by the Menshutkin reaction between N-methylpiperidine and an excess dihalogenated monomer. Their chemical structures were well confirmed by the 1H NMR spectra (Fig. S2 and S3 in the ESI†). The 1H NMR spectrum of the PBP backbone is shown in Fig. S4.† The arylene proton signals 1 and 2, piperidinium ring proton signals 3 and 4, and the methyl proton signal 5 are in good agreement with the previous literature.54PBP was further reacted with controlled amounts of cationic precursors (OIMP or IMP) to generate AEMs with various IECs. The PBP was partially quaternized by adding OIMP (or IMP) to produce O-PDQA (or PDQA) via the Menshutkin reaction. During the reaction process, a small amount of polymer precipitated in the solution when K2CO3 was added. As the reaction progressed, the polymer gradually redissolved, which indicates the success of the Menshutkin reaction. The reaction time was increased to 5 days to achieve a controlled quaternization degree. As verified by the 1H NMR spectra (Fig. 1a and b), the methyl proton signal at 2.7 ppm decreased, and the side chain signals were observed between 1.2 and 4.0 ppm, indicating the successful quaternization reaction. The IECs of the prepared AEMs were then determined by the Mohr titration method. The IECs were 1.33, 1.62, 1.93 mmol g−1 for O-PDQA, and 1.30, 1.59, 1.98 mmol g−1 for PDQA.
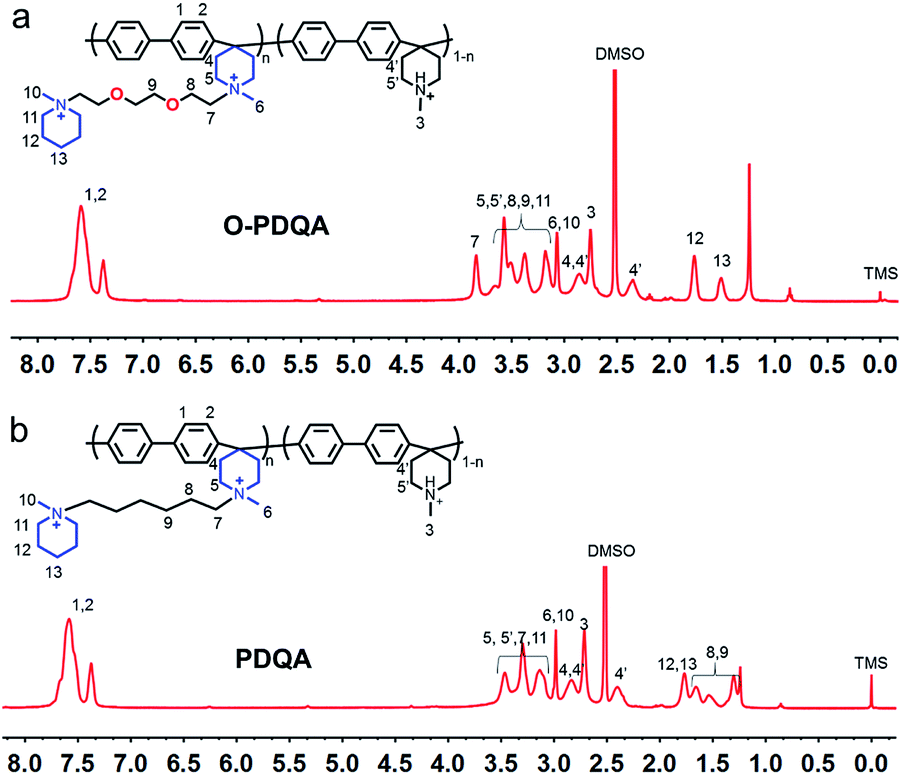 | ||
| Fig. 1 The representative 1H NMR spectra of (a) O-PDQA and (b) PDQA (DMSO-d6 containing 5 vol% of TFA as the solvent, TFA was applied to shift the water signal to above 8 ppm). | ||
Computational studies of the cationic side-chain with different spacers
Before evaluating the performance of the as-prepared AEMs, the computational studies of the dual-cationic side-chain O-DQA (connected with an EO spacer) and DQA (connected with a pure alkyl spacer) were initially performed to investigate the design feasibility.The AEM containing O-DQA is expected to form self-aggregated morphology through the intermolecular cation–dipole interaction between the cationic groups and EO moieties. As verification, the atomic charge distributions of O-DQA and DQA model compounds (Fig. 2a and b, respectively) were firstly calculated using DFT. In Fig. 2c, heterogeneous charge distribution can be observed along with the EO spacer of O-DQA. The oxygen atoms are electronegative (with a charge of −0.52), and other atoms are electropositive. In contrast, DQA shows negligible heterogeneity in charge distribution (Fig. 2d). As a result, the negatively charged O atom in O-DQA would lead to unique cation–dipole interaction, facilitating the self-aggregation of cationic side chains.
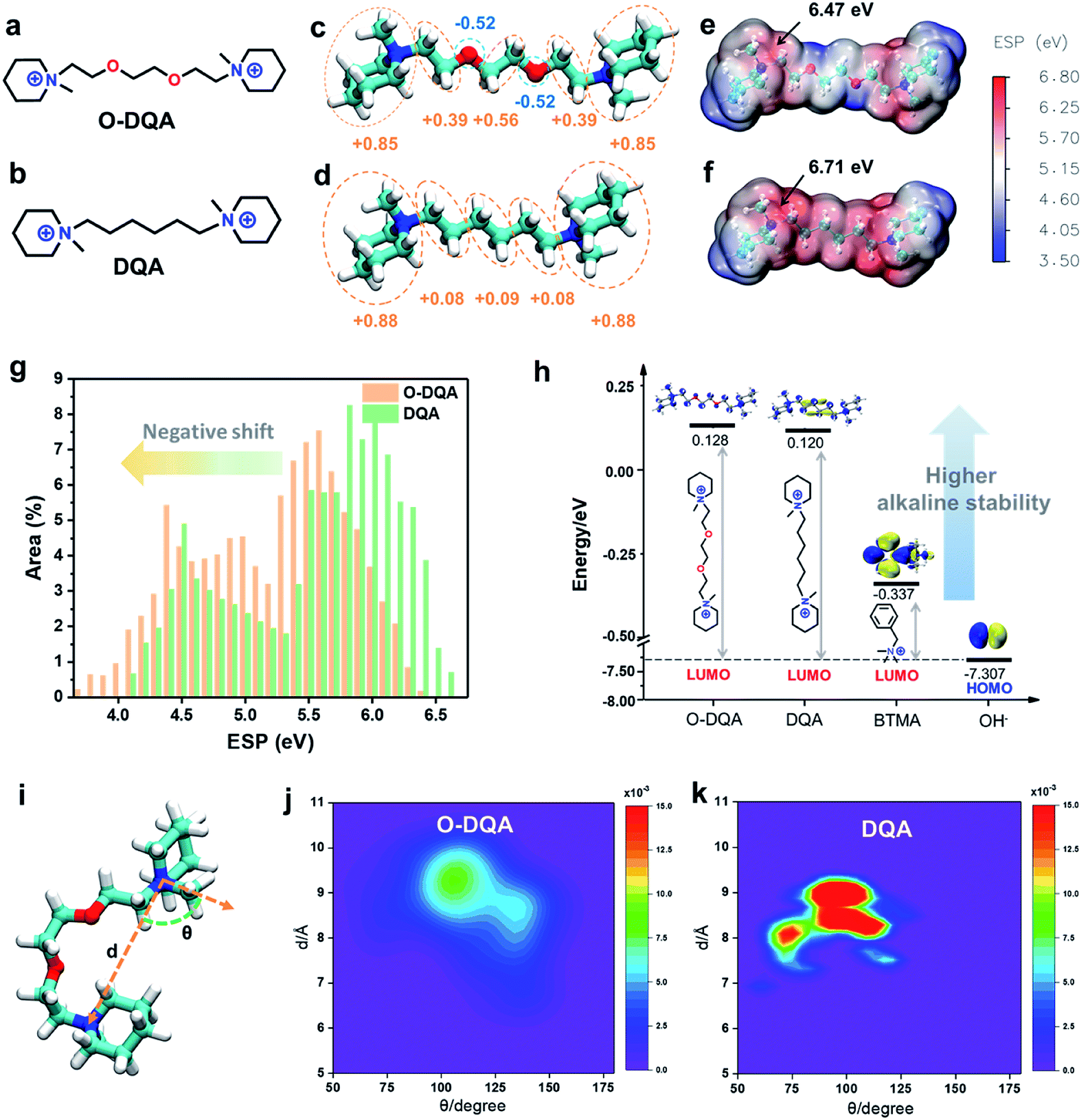 | ||
| Fig. 2 Computational studies. The chemical structure of (a) O-DQA (with EO spacer) and (b) DQA (without EO spacer). The charge distribution analysis of (c) O-DQA and (d) DQA, the atomic charge on O-DQA shows that oxygen atoms are electronegative, which are expected to interact with the cationic group by cation–dipole interaction, while the atomic charges on DQA are all electropositive. Electrostatic potential (ESP) mapping of ρ = 0.001 e bohr−3 isodensity surface of (e) O-DQA and (f) DQA. (g) The regional ESP distribution of O-DQA and DQA, the X-axis serves as the ESP value of the molecule (eV), the Y-axis serves as the surface area ratio (%) of the corresponding value in that molecule. (h) The calculated LUMO energies of O-DQA, DQA, and BTMA (benzyl trimethylammonium, the most commonly used cationic group in AEMs) and HOMO energy of OH−; higher the LUMO energy, higher the alkaline stability of the molecule. (i) The vectors d and θ of the model compound were applied to describe the molecule's conformation in the simulation box. d is defined as the length of the side-chain while θ describes the extent and bending of the side-chain. The joint probability density distribution of d and θ with their bars showing conformational distribution probability of (j) O-DQA side chain and (k) DQA side chain, respectively. | ||
Electrostatic potential (ESP) calculations were then performed to provide insight into the surface charge environment of O-DQA and DQA. In Fig. 2e and f, the maximum ESP value was observed at the nitrogen atom in the cationic groups for DQA and O-DQA. Besides, O-DQA shows a maximum ESP value of 6.47 eV, which is 0.24 eV lower than the control DQA sample. The decreased ESP value is beneficial for weakening the binding force between the cationic group and OH− and thus enabling better OH− dissociation ability. As for the regional ESP in Fig. 2g, visible negative shifts can be observed in O-DQA due to the introduction of the EO spacer. The resulting weaker interaction between OH− and cationic side chains would decrease the activation energy for OH− hopping and promote its transfer along the adjacent sites.7
DFT calculations further investigated the impact of the cationic side chain on alkaline stability according to the frontier molecular orbital theory. As we know, LUMO stands for the lowest unoccupied molecular orbital, which can easily accept electrons compared with other vacant orbitals. HOMO stands for the highest occupied molecular orbital, which can donate electrons to other types of molecular orbitals. When OH− attacks cationic molecules, the electron transfer from the HOMO of the OH− to the LUMO of the cationic molecules happens. Hence, higher the LUMO energy of the cationic molecule, harder it is for OH− to attack. Thus, in this work, the LUMO energy of the molecule can be regarded as the design guideline for alkaline stable cationic side chains.31,55 In Fig. 2h, the LUMO energies of the O-DQA, DQA and BTMA (benzyl trimethylammonium, the most commonly used cationic groups in AEMs) were compared. Both O-DQA and DQA show much higher LUMO energies than BTMA (−0.337 eV), suggesting the better alkaline stability of O-DQA and DQA. The piperidinium cation is responsible for the higher LUMO energy, since the piperidinium cation's geometric constraint avoids unfavorable conformations with distorted bond angles, increasing the transient energy for potential elimination and ring-opening substitution reactions.56 Also, the incorporation of the EO spacer imparts a slightly higher LUMO energy to O-DQA than DQA (0.128 eV vs. 0.120 eV). The EO spacer reduces the electropositivity of cations in O-DQA (Fig. 2e and f). Furthermore, the flexible EO spacer promotes the ring strain relaxation, thus mitigating ring distortion.42 Therefore, the combination of alkaline stable piperidinium cations and the synergistic advantages of the EO spacer lead to high alkaline stability of the O-DQA based AEM.
The flexibility and mobility of cationic chains affect the self-assembly process and play a vital role in the construction of the ion channels.29 Therefore, molecular dynamics (MD) simulations were conducted to investigate the flexibility of O-DQA and DQA. The spatial distributions of O-DQA and DQA were transferred to the joint probability density distribution contour maps. As shown in Fig. 2i, the vectors d and θ describe the extension and bending of the cationic side chain. DQA shows a deeper red zone than O-DQA, implying a more concentrated spatial probability density distribution. This demonstrates that the more rigid alkyl side chains in DQA lead to undesired constraints and inhibit the self-assembling process. Conversely, the EO spacer in O-DQA provides a less concentrated probability of chain distribution (Fig. 2j and k), which can trap the adjacent cationic side chains more efficiently, thus promoting the self-assembling process of the O-DQA containing membrane.
Benefits of self-aggregated side-chain for membrane morphology, ion transfer, and alkaline stability
The nano-scale morphologies of the prepared AEMs were characterized by small-angle X-ray scattering (SAXS) and microscopy analysis (TEM and AFM). The SAXS profile of O-PDQA in Fig. 3a shows a distinct scattering peak at q = 1.45 nm−1, which corresponds to a Bragg spacing of 4.33 nm (d = 2π/q). This quantitatively confirms the nano-scale self-aggregated morphology. In contrast, the SAXS profile of PDQA showed no obvious patterning structure (Fig. 3b). The inefficient ion aggregation in PDQA is probably due to the less flexible side chain and the rigid polymer backbone. This phenomenon is consistent with the previous report.48,50,57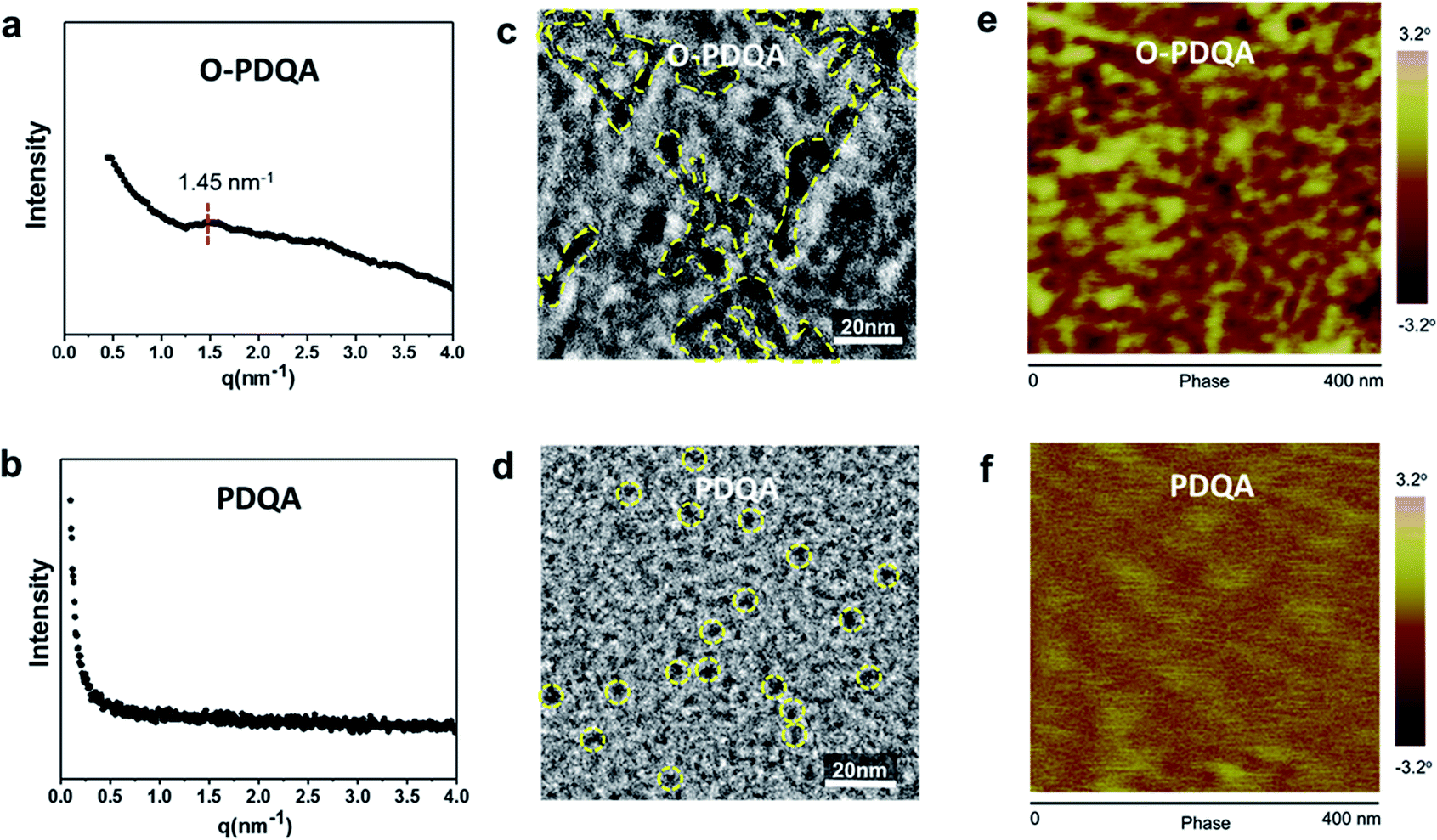 | ||
| Fig. 3 Microstructural characterization of exemplar AEMs. SAXS spectra of (a) O-PDQA (IEC = 1.93 mmol g−1) and (b) PDQA (IEC = 1.98 mmol g−1) AEMs. TEM images of (c) O-PDQA and (d) PDQA, where the bright and dark regions correspond to the hydrophobic backbone and hydrophilic QA ionic clusters, respectively. AFM phase images of (e) O-PDQA and (f) PDQA membrane. | ||
TEM micrograph and AFM phase image of PDQA provide visible evidence for the presence of ionic clusters. In the TEM image of O-PDQA (Fig. 3c), the aggregated ionic clusters (dark regions) penetrated the hydrophobic phase of the polymer backbone (light regions). The hydrophilic dyed micro-domains are around 7 nm in diameter and interconnected to each other. Such a microstructure is similar to the state of the art Nafion®.58 The tapping mode AFM phase image also shows a similar phase-separated morphology with interconnected ionic clusters (ca. d = 7 nm) (Fig. 3e).
In contrast, there is no apparent micro-phase separation for PDQA due to the poor self-assembling ability (Fig. 3d). The ionic clusters are dispersed in the polymer matrix and isolated from each other. Also, in the AFM image of PDQA (Fig. 3f), no distinct phase difference was found. These results highlight the role of EO segments in facilitating phase separation by providing cation–dipole interaction and improving side-chain mobility.
To investigate the benefits of the self-aggregated side-chain to ion transport, the hydroxide conductivity of O-PDQA was compared with that of PDQA, as shown in Fig. 4a and b. At similar IECs, O-PDQA exhibits higher ion conductivities than PDQA at the whole temperature range (30–80 °C). Notably, the preponderance is more evident at higher IEC values. For example, the conductivity of O-PDQA (IEC = 1.93 mmol g−1) is 106 mS cm−1 at 80 °C. In comparison, the conductivity of PDQA (IEC = 1.98 mmol g−1) is 90 mS cm−1 at 80 °C. The O-PDQA with EO spacers shows improved ion conductivity. The well-defined channels in O-PDQA undoubtedly promote facile hydroxide ion transport across the membrane. Besides, the introduction of EO spacers also optimizes the hydration environment for ion transport in the channel. EO spacers can serve as additional hydrated OH− hopping sites by forming a hydrogen-bonded network with water. And the increased WU (Fig. 5a) of the EO containing AEM would lead to a good hydration structure of the cationic groups and facilitates the dissociation of OH− from them. Overall, the well-defined ion pathway combined with the hydrophilic EO spacer-involved hydrogen-bonding network contributes to the fast ion and water transport.
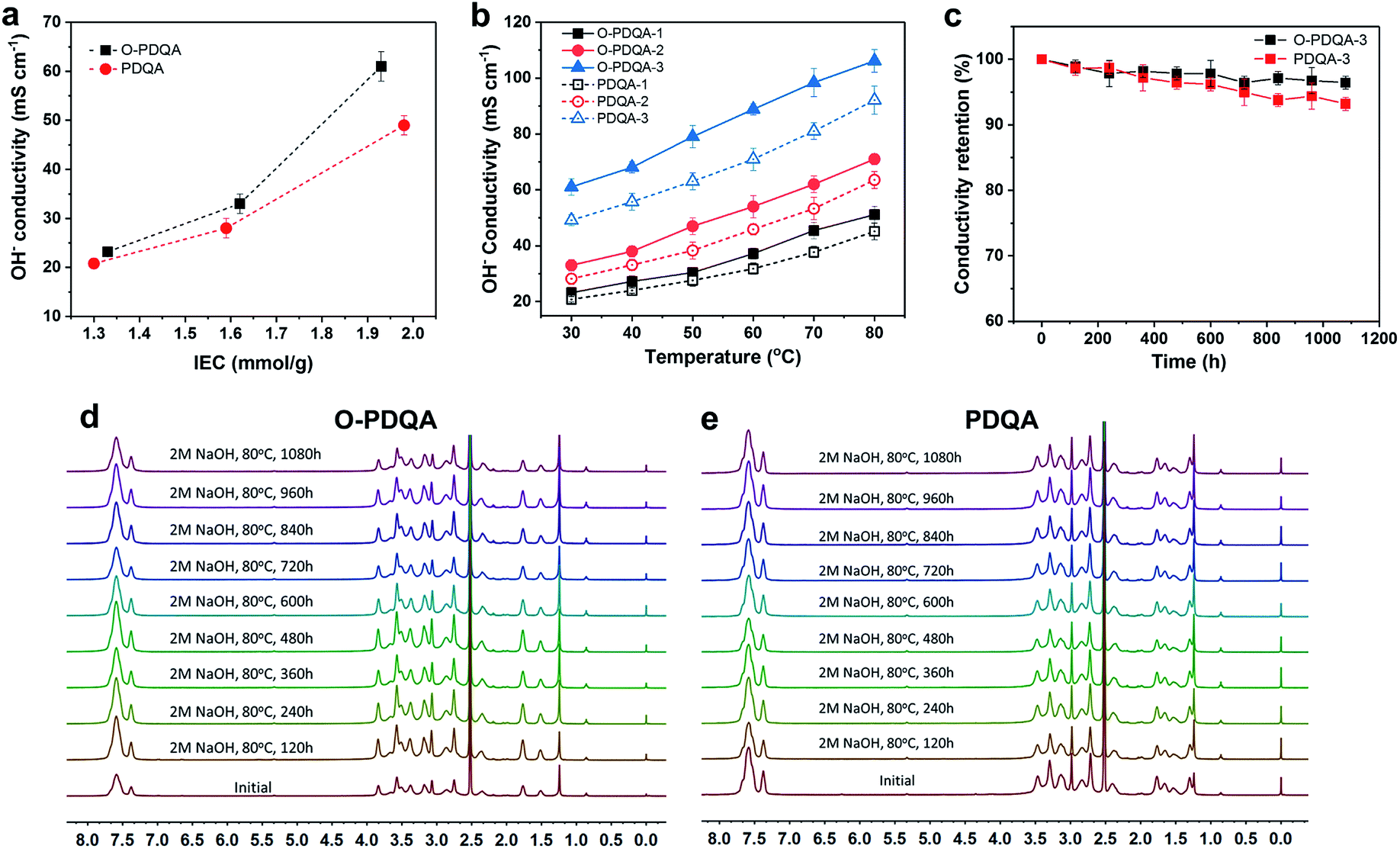 | ||
| Fig. 4 (a) The OH− conductivities as a function of IEC of the O-PDQA and PDQA membranes at 30 °C. (b) The OH− conductivities of O-PDQA and PDQA at increasing temperatures. (c) Conductivity retention of the O-PDQA and PDQA membranes when immersed at 80 °C in 2 M aq. NaOH for different time intervals. 1H NMR spectra of the (d) O-PDQA and (e) PDQA membranes for different alkali treatment times in 2 M aq. NaOH, 80 °C. | ||
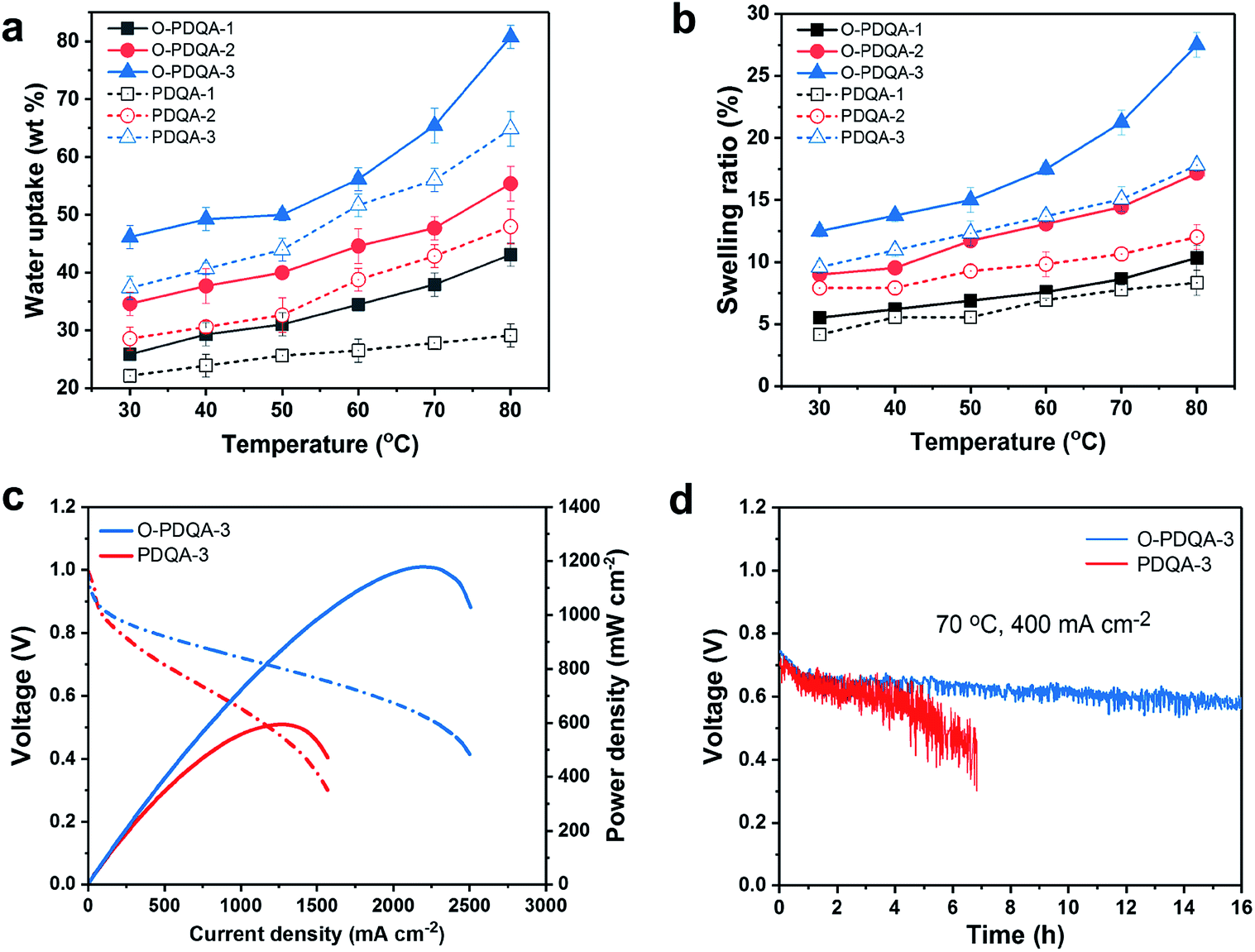 | ||
| Fig. 5 Temperature-dependent (a) water uptake and (b) swelling ratio (in plane) of O-PDQA and PDQA. (c) I–V polarization curves(dash) and power density curves (solid) of O-PDQA-3 and PDQA in H2/O2 AEMFC tests at 70 °C (anodes: PtRu/C, cathodes: Pt/C, both with 0.5 mg cm−2 of metal loading, 1 L min−1 of gas flow at 100% RH without back pressure). (d) AEMFC durability of O-PDQA and PDQA operating at 400 mA cm−2, 70 °C, 0.5 L min−1 of gas flow at 100% RH. | ||
Another problem of AEMs is the severe degradation under alkaline conditions, especially at elevated temperatures and high pH. The degradation mechanisms mainly include direct α-C nucleophilic substitution, β-hydrogen elimination (Hofmann degradation) of the cationic groups, and chain scission on the polymer backbone.45 To evaluate the long-term alkaline stability of the as-prepared AEMs, O-PDQA and PDQA membranes were treated in 2 M aq. NaOH at 80 °C for 1080 h. Both O-PDQA and PDQA maintain high hydroxide conductivity (Fig. 4c) after 1080 h, highlighting the excellent alkaline stability of piperidinium groups and aryl ether free backbone. Specifically, conductivity retention >96% was observed for the O-PDQA membrane, which is slightly higher than that of PDQA (93.2%). In order to further study the alkaline stability of the membranes, the IECs of the aging AEMs were also evaluated, the results are quite similar to the ion conductivity tests. The IEC of the O-PDQA-3 decreased to 97.2% after 1080 h, while 94.9% for PDQA-3 membranes (Fig. S6†). This result is consistent with the previous simulation prediction (ESP and LUMO energy in Fig. 2). The electron-donating and flexible EO spacer reduces the possibility of OH− attack at piperidinium groups. And the 1H NMR spectra of both alkaline treated O-PDQA and PDQA show negligible change in pre-existing signals, and no new signal could be observed.
Benefits of self-aggregated side-chain for fuel cell-related performance
Apart from the ionic conductivity and chemical stability, dimensional stability and mechanical strength are also important criteria for fuel cell application. Fig. 5a and b indicate that the water uptake and swelling ratio increase with the IECs and temperature. Also, the EO spacers in O-PDQA lead to slightly higher water uptake and swelling ratio than PDQA (O-PDQA-3, IEC = 1.93 mmol g−1, WU = 46.1 wt%, SR = 12.5%; PDQA-3, IEC = 1.98 mmol g−1, WU = 37.4 wt%, SR = 9.6%). The H-bonding between H2O and EO spacers is the reason behind it.30 But the dimensional stability of PDQA and O-PDQA is still superior to that of most of the aryl ether-based AEMs (e.g. QPE-X16Y11, IEC = 2.05 mmol g−1, WU = 112 wt%, SR = 32.2%; BTMA30, IEC = 2.10 mmol g−1, WU = 59 wt%, SR = 13%).7,17 This may benefit from the higher rigidity of the aryl ether free backbone. Fig. S7 (ESI†) shows the mechanical properties (tensile strength (Ts) and elongation at break (Eb)) of the AEMs in this study. The robust polymer backbone endows the as-prepared membranes with sufficient mechanical properties for practical application. Compared with PDQA, O-PDQA demonstrates higher flexibility and similar tensile strength, which is derived from the cation–dipole interactions between cationic chains. This is quite different from other high-IEC designs, where high ionic conductivity is achieved by sacrificing dimensional and mechanical stability. Moreover, thermogravimetric analysis demonstrates that our membranes have sufficient thermal stability for practical application (Fig. S8 and S9†). All the above properties fulfil the requirements of the AEMFC application.The benefits of the self-aggregating side chains were further investigated by evaluating the performance of H2/O2 single-cell AEMFCs (Fig. 5c). Both membrane electrode assemblies (MEAs) share the same electrocatalyst, ionomeric binder, and MEA fabrication process, but different AEMs (O-PDQA vs. PDQA). Open-circuit voltages for both MEAs are about 1 V, which reveals their excellent gas barrier properties. The AEMFC containing O-PDQA produces a peak power density of 1.18 W cm−2 at 70 °C without back pressure, which is competitive to other previously reported AEMFCs and about 2-fold higher than PDQA (0.60 W cm−2). To shed light on the reason for the increased output of the AEMFCs, in situ electrochemical impedance spectroscopy (EIS) analysis of the O-PDQA-3 and PDQA MEAs was also performed. In Fig. S10,† the intercept in the high-frequency region represents the ohmic resistance, mainly consisting of membrane resistances and contact resistance.59 O-PDQA-3 shows a lower ohmic resistance than PDQA-3, which is consistent with the ion conductivity data in Fig. 4a and b. Therefore, O-PDQA-3 with higher conductivity reduced the internal resistance of the cell. Furthermore, the arc in the high- and low-frequency regions (the semicircle diameter) indicates the charge-transfer resistance associated with the electrochemical reaction.60 O-PDQA-3 shows reduced charge-transfer resistance, thus accelerating the electrochemical reaction in the fuel cell, and giving a higher power density. This is due to the fast ion transport and water back-diffusion provided by the well-connected hydrophilic channel. The results further highlight the benefit of the self-aggregation of the EO containing side chain in O-PDQA AEMs.
To further evaluate the fuel cell device durability, the MEA containing O-PDQA was operated at a constant current density of 400 mA cm−2 (70 °C, 0.5 L min−1 of gas flow at 100% RH) for 16 h (Fig. 5d). In the first 1 h, the cell voltage declined rapidly from ≈0.75 to 0.65 V. In the following 15 h, the cell voltage is relatively stable at ≈0.60 V, while the PDQA membrane experiences a rapid decrease of cell voltage, from ≈0.70 to 0.42 V over 6.8 h. The obvious difference in device durability can be attributed to the different water balance and mass transport processes at the catalyst–AEM interface, since the highly hydrophilic ion-channel in O-PDQA allows for enhanced ion transport and water back-diffusion at the interface. As a result, anode flooding was alleviated and water supply to the cathode was improved. Thus, continuous electrochemical reactions on both anode and cathode could thus be achieved.61,62
The device durability in this study still falls behind the current advanced level. To the best of our knowledge, only a few previous studies have presented AEMFC durability,7,13,16,18,34,47,49,62–65 and device durability >100 h is rarely reported. As the most significant challenge in this field, AEMFC durability is codetermined by many factors, including the degradation of MEA components (AEMs, electrocatalyst, ionomeric binder), MEA fabrication process, water management, carbonation and so on.66 The ex situ alkaline stabilities of O-PDQA and PDQA membranes are unquestionably excellent, but the in situ device durability is far from satisfactory. This might be due to the MEA fabrication or the ionomer/binder degradation. In future studies, we will focus on optimizing the ionomer structure and MEA fabrication technology to improve the AEMFC durability.
Conclusions
In summary, we proposed a new concept of self-aggregating cationic chains with dipolar EO spacers and alkaline stable piperidinium cations. Both the computational and experimental studies demonstrated the role of cation–dipole interaction in facilitating the formation of ion-conducting channels. The resultant O-PDQA membranes delivered a higher ion conductivity of 106 mS cm−1 at 80 °C and a superior peak power density of 1.18 W cm−2 in AEMFCs. More importantly, the membranes presented excellent alkaline stability (over 96% of hydroxide conductivity remained in 2 M NaOH, 80 °C for 1080 h). This concept of constructing ion conduction highways using self-aggregating cationic chains will benefit many fields, which involve anion conductive electrolytes (fuel cells, redox flow batteries, electrolyzers, etc.).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Key R&D Program of China (No. 2020YFB1505601, 2018YFB1502301), the National Natural Science Foundation of China (No. 22038013, 21875233, 21720102003, 21706247), Key Technologies R&D Program of Anhui Province (No. 18030901079), and the USTC Super Computing Center.References
- Z. P. Cano, D. Banham, S. Ye, A. Hintennach, J. Lu, M. Fowler and Z. Chen, Nat. Energy, 2018, 3, 279–289 CrossRef.
- D. W. Shin, M. D. Guiver and Y. M. Lee, Chem. Rev., 2017, 117, 4759–4805 CrossRef CAS.
- D. R. Dekel, J. Power Sources, 2018, 375, 158–169 CrossRef CAS.
- J. R. Varcoe, P. Atanassov, D. R. Dekel, A. M. Herring, M. A. Hickner, P. A. Kohl, A. R. Kucernak, W. E. Mustain, K. Nijmeijer, K. Scott, T. Xu and L. Zhuang, Energy Environ. Sci., 2014, 7, 3135–3191 RSC.
- S. Noh, J. Y. Jeon, S. Adhikari, Y. S. Kim and C. Bae, Acc. Chem. Res., 2019, 52, 2745–2755 CrossRef CAS.
- J. Pan, C. Chen, Y. Li, L. Wang, L. Tan, G. Li, X. Tang, L. Xiao, J. Lu and L. Zhuang, Energy Environ. Sci., 2014, 7, 354–360 RSC.
- Y. Kim, Y. Wang, A. France-Lanord, Y. Wang, Y. M. Wu, S. Lin, Y. Li, J. C. Grossman and T. M. Swager, J. Am. Chem. Soc., 2019, 141, 18152–18159 CrossRef CAS.
- T. P. Pandey, H. N. Sarode, Y. Yang, Y. Yang, K. Vezzù, V. D. Noto, S. Seifert, D. M. Knauss, M. W. Liberatore and A. M. Herring, J. Electrochem. Soc., 2016, 163, H513–H520 CrossRef CAS.
- N. Li and M. D. Guiver, Macromolecules, 2014, 47, 2175–2198 CrossRef CAS.
- M. Tanaka, K. Fukasawa, E. Nishino, S. Yamaguchi, K. Yamada, H. Tanaka, B. Bae, K. Miyatake and M. Watanabe, J. Am. Chem. Soc., 2011, 133, 10646–10654 CrossRef CAS.
- Y. A. Elabd and M. A. Hickner, Macromolecules, 2011, 44, 1–11 CrossRef CAS.
- C. G. Arges, Y. Kambe, M. Dolejsi, G.-P. Wu, T. Segal-Pertz, J. Ren, C. Cao, G. S. W. Craig and P. F. Nealey, J. Mater. Chem. A, 2017, 5, 5619–5629 RSC.
- X. Q. Wang, C. X. Lin, F. H. Liu, L. Li, Q. Yang, Q. G. Zhang, A. M. Zhu and Q. L. Liu, J. Mater. Chem. A, 2018, 6, 12455–12465 RSC.
- Y. He, J. Zhang, X. Liang, M. A. Shehzad, X. Ge, Y. Zhu, M. Hu, Z. Yang, L. Wu and T. Xu, J. Membr. Sci., 2018, 559, 35–41 CrossRef CAS.
- E. A. Weiber and P. Jannasch, ChemSusChem, 2014, 7, 2621–2630 CrossRef CAS.
- L. Zhu, X. Peng, S. Shang, M. T. Kwasny, T. J. Zimudzi, X. Yu, N. Saikia, J. Pan, Z. Liu and G. N. Tew, Adv. Funct. Mater., 2019, 29, 1902059 CrossRef.
- N. Li, Y. Leng, M. A. Hickner and C. Y. Wang, J. Am. Chem. Soc., 2013, 135, 10124–10133 CrossRef CAS.
- L. Liu, X. Chu, J. Liao, Y. Huang, Y. Li, Z. Ge, M. A. Hickner and N. Li, Energy Environ. Sci., 2018, 11, 435–446 RSC.
- J. Pan, Y. Li, J. Han, G. Li, L. Tan, C. Chen, J. Lu and L. Zhuang, Energy Environ. Sci., 2013, 6, 2912–2915 RSC.
- L. Liu, G. Huang and P. A. Kohl, J. Mater. Chem. A, 2018, 6, 9000–9008 RSC.
- A. M. Ahmed Mahmoud and K. Miyatake, J. Mater. Chem. A, 2018, 6, 14400–14409 RSC.
- D. Liu, M. Xu, M. Fang, J. Chen and L. Wang, J. Mater. Chem. A, 2018, 6, 10879–10890 RSC.
- N. A. Wasio, R. C. Quardokus, R. P. Forrest, C. S. Lent, S. A. Corcelli, J. A. Christie, K. W. Henderson and S. A. Kandel, Nature, 2014, 507, 86–89 CrossRef CAS.
- E. B. Trigg, T. W. Gaines, M. Marechal, D. E. Moed, P. Rannou, K. B. Wagener, M. J. Stevens and K. I. Winey, Nat. Mater., 2018, 17, 725–731 CrossRef CAS.
- L. H. Wang, Z. D. Zhang, C. Y. Hong, X. H. He, W. You and Y. Z. You, Adv. Mater., 2015, 27, 3202–3207 CrossRef CAS.
- E. Bortel and A. Kochanowski, Makromol. Chem., 1984, 185, 1409–1417 CrossRef CAS.
- Y. V. Oza, D. R. MacFarlane, M. Forsyth and L. A. O'Dell, Electrochim. Acta, 2015, 175, 80–86 CrossRef CAS.
- H. Zhang, C. Li, M. Piszcz, E. Coya, T. Rojo, L. M. Rodriguez-Martinez, M. Armand and Z. Zhou, Chem. Soc. Rev., 2017, 46, 797–815 RSC.
- C. X. Lin, X. L. Huang, D. Guo, Q. G. Zhang, A. M. Zhu, M. L. Ye and Q. L. Liu, J. Mater. Chem. A, 2016, 4, 13938–13948 RSC.
- H. Lai and P. Wu, Polym. Chem., 2013, 4, 3323–3332 RSC.
- N. A. Qaisrani, L. Ma, J. Liu, M. Hussain, L. Li, P. Li, S. Gong, F. Zhang and G. He, J. Membr. Sci., 2019, 581, 293–302 CrossRef CAS.
- C. Cheng, X. He, S. Huang, F. Zhang, Y. Guo, Y. Wen, B. Wu and D. Chen, Int. J. Hydrogen Energy, 2020, 45, 19676–19690 CrossRef CAS.
- N. A. Qaisrani, L. Ma, M. Hussain, J. Liu, L. Li, R. Zhou, Y. Jia, F. Zhang and G. He, ACS Appl. Mater. Interfaces, 2020, 12, 3510–3521 CrossRef CAS.
- Y. Zhu, L. Ding, X. Liang, M. A. Shehzad, L. Wang, X. Ge, Y. He, L. Wu, J. R. Varcoe and T. Xu, Energy Environ. Sci., 2018, 11, 3472–3479 RSC.
- Y.-K. Choe, C. Fujimoto, K.-S. Lee, L. T. Dalton, K. Ayers, N. J. Henson and Y. S. Kim, Chem. Mater., 2014, 26, 5675–5682 CrossRef CAS.
- J. Wang, S. Gu, R. Xiong, B. Zhang, B. Xu and Y. Yan, ChemSusChem, 2015, 8, 4229–4234 CrossRef CAS.
- W. Chen, M. Hu, H. Wang, X. Wu, X. Gong, X. Yan, D. Zhen and G. He, J. Mater. Chem. A, 2017, 5, 15038–15047 RSC.
- X. Li, Y. Yu, Q. Liu and Y. Meng, ACS Appl. Mater. Interfaces, 2012, 4, 3627–3635 CrossRef CAS.
- A. Jasti and V. K. Shahi, J. Mater. Chem. A, 2013, 1, 6134–6137 RSC.
- C. Fujimoto, D.-S. Kim, M. Hibbs, D. Wrobleski and Y. S. Kim, J. Membr. Sci., 2012, 423–424, 438–449 CrossRef CAS.
- C. G. Arges and V. Ramani, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2490–2495 CrossRef CAS.
- J. S. Olsson, T. H. Pham and P. Jannasch, Adv. Funct. Mater., 2018, 28, 1702758 CrossRef.
- L. J. Small, H. D. Pratt, C. H. Fujimoto and T. M. Anderson, J. Electrochem. Soc., 2015, 163, A5106–A5111 CrossRef.
- W.-H. Lee, A. D. Mohanty and C. Bae, ACS Macro Lett., 2015, 4, 453–457 CrossRef CAS.
- E. J. Park and Y. S. Kim, J. Mater. Chem. A, 2018, 6, 15456–15477 RSC.
- M. G. Marino and K. D. Kreuer, ChemSusChem, 2015, 8, 513–523 CrossRef CAS.
- J. Wang, Y. Zhao, B. P. Setzler, S. Rojas-Carbonell, C. Ben Yehuda, A. Amel, M. Page, L. Wang, K. Hu, L. Shi, S. Gottesfeld, B. Xu and Y. Yan, Nat. Energy, 2019, 4, 392–398 CrossRef CAS.
- S. Zhang, X. Zhu and C. Jin, J. Mater. Chem. A, 2019, 7, 6883–6893 RSC.
- H. Peng, Q. Li, M. Hu, L. Xiao, J. Lu and L. Zhuang, J. Power Sources, 2018, 390, 165–167 CrossRef CAS.
- H. Zhu, Y. Li, N. Chen, C. Lu, C. Long, Z. Li and Q. Liu, J. Membr. Sci., 2019, 590, 117307 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS.
- J. S. Olsson, T. H. Pham and P. Jannasch, J. Membr. Sci., 2019, 578, 183–195 CrossRef CAS.
- Z. Sun, J. Pan, J. Guo and F. Yan, Adv. Sci., 2018, 5, 1800065 CrossRef.
- T. H. Pham, J. S. Olsson and P. Jannasch, J. Am. Chem. Soc., 2017, 139, 2888–2891 CrossRef CAS.
- T. H. Pham, J. S. Olsson and P. Jannasch, J. Mater. Chem. A, 2018, 6, 16537–16547 RSC.
- B. Bae, T. Yoda, K. Miyatake, H. Uchida and M. Watanabe, Angew. Chem., Int. Ed., 2010, 49, 317–320 CrossRef CAS.
- J. E. Park, S. Y. Kang, S.-H. Oh, J. K. Kim, M. S. Lim, C.-Y. Ahn, Y.-H. Cho and Y.-E. Sung, Electrochim. Acta, 2019, 295, 99–106 CrossRef CAS.
- S. Siracusano, V. Baglio, N. Van Dijk, L. Merlo and A. S. Aricò, Appl. Energy, 2017, 192, 477–489 CrossRef CAS.
- T. J. Omasta, A. M. Park, J. M. LaManna, Y. Zhang, X. Peng, L. Wang, D. L. Jacobson, J. R. Varcoe, D. S. Hussey, B. S. Pivovar and W. E. Mustain, Energy Environ. Sci., 2018, 11, 551–558 RSC.
- L. Wang, X. Peng, W. E. Mustain and J. R. Varcoe, Energy Environ. Sci., 2019, 12, 1575–1579 RSC.
- H. Ono, T. Kimura, A. Takano, K. Asazawa, J. Miyake, J. Inukai and K. Miyatake, J. Mater. Chem. A, 2017, 5, 24804–24812 RSC.
- D. P. Leonard, S. Maurya, E. J. Park, S. Noh, C. Bae, E. D. Baca, C. Fujimoto and Y. S. Kim, J. Mater. Chem. A, 2020, 8, 14135–14144 RSC.
- G. Huang, M. Mandal, X. Peng, A. C. Yang-Neyerlin, B. S. Pivovar, W. E. Mustain and P. A. Kohl, J. Electrochem. Soc., 2019, 166, F637–F644 CrossRef CAS.
- W. E. Mustain, M. Chatenet, M. Page and Y. S. Kim, Energy Environ. Sci., 2020, 13, 2805–2838 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H NMR spectra of the synthesized compounds, experimental details and a summary of the general properties of the prepared AEMs. See DOI: 10.1039/d0ta11011f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |