
Crystalline red phosphorus for selective photocatalytic reduction of CO2 into CO†
Zhuofeng
Hu
 *a,
Yinglong
Lu
a,
Minghao
Liu
a,
Xiaoyue
Zhang
a and
JunJie
Cai
*b
*a,
Yinglong
Lu
a,
Minghao
Liu
a,
Xiaoyue
Zhang
a and
JunJie
Cai
*b
aSchool of Environmental Science and Engineering, Guangdong Provincial Key Laboratory of Environmental Pollution Control and Remediation Technology, Sun Yat-sen University, Guangzhou 510275, China. E-mail: huzhf8@mail.sysu.edu.cn
bSchool of Materials and Energy, Guangdong University of Technology, Guangzhou 510006, China
First published on 13th November 2020
Abstract
Huge emissions of CO2 have caused serious environmental problems. Photocatalytic CO2 reduction is an important strategy to solve this problem by using sustainable solar energy. Elemental photocatalysts remain underexplored in this context relative to metals or metal oxides. Herein, we discover that amorphous red P is able to selectively reduce CO2 to CO. Furthermore, a crystalline fibrous phase of red P with two parallel polyphosphide chains (consisting of 21 P atoms) has been synthesized and was used for CO2 reduction for the first time. Compared with amorphous P, the fibrous P with a periodical P atom arrangement exhibits a 10.5 times enhancement in CO2 reduction and an apparent improvement in charge carrier dynamics, supported by femtosecond and nanosecond transient absorption spectra. The CO2 reduction process at the gas–solid interface on elemental P is investigated by density functional calculations and the nudged elastic band method for the first time. The results indicate that CO2 can be easily activated by the P atoms on the surface. The breaking of the C–O bond is the key step and a proton-involving process is preferred during CO2 reduction. This work innovatively investigates CO2 reduction on elemental photocatalysts and helps to broaden the applications of elemental red P.
1 Introduction
The widespread application of fossil fuels and the fast development of industry have caused huge emissions of CO2, a typical greenhouse gas.1,2 Based on research by the World Meteorological Organisation, the average content of CO2 in the atmosphere has increased by about 46% compared with the 20th Century. It reached a very high level of 415 ppm last year. This causes a serious environmental problem and global warming.3–7 The summer of 2019 is considered to be one of the hottest summers.8 Many cities in Europe and China suffered from extremely hot temperatures above 40 °C for a long period of time. Recently, it was reported that a record high temperature of >20 °C was recorded in the Antarctic, which leads to the irreversible melting of ice in the Antarctic. Therefore, the reduction of CO2 levels is of great significance to all of us.The reduction of CO2 into valuable chemical fuels such as CH4, CH3OH, and HCOOH, is a very hot topic.3–7,9–11 Traditionally, a high temperature or high voltage is needed to activate the stable chemical bonds of CO2 for its reduction. In the past decade, solar reduction of CO2 by using sustainable solar energy has attracted more and more attention because of its mild operating conditions, sustainability, and environmental-friendliness. To achieve high-efficiency photocatalytic CO2 reduction, an elaborately designed photocatalyst is crucial.12,13 Currently, metal oxides and sulfides are the most popular photocatalysts. For example, TiO2, a typical photocatalyst, has been successfully used for CO2 reduction. C3N4 is a well-known metal-free photocatalyst for CO2 reduction. However, their absorption range is often lower than 500 nm and they often suffer from serious charge recombination, which will lead to low activity.14 Besides, selectivity is another important factor to consider.14–16 The selectivity can be influenced by the adsorption of light, CB position, surface catalytic active sites and so on.3–7
Phosphorus is one of the most abundant elements in the Earth’s crust. There are about 100 billion tons of phosphorus on the Earth.17–20 Plenty of research has been carried out on it since its discovery in the 1660s,21–23 especially when new types of phosphorus were discovered. For example, type V violet Hittorf P (monoclinic) was discovered in 1969 by Thurn and Krebs.24 Pfitzner et al.25 and Ruck et al.23 have prepared further tubular P allotropes like fibrous P crystals (triclinic).23,26,27 Recently, it has received more and more attention after the discovery of its semiconductive properties.28,29 Under solar illumination, it produces photoexcited electron–hole pairs. The electrons can induce water reduction to produce hydrogen as a clean energy.30,31 The holes can transform water into OH radicals for environmental pollutant degradation.31,32 Elemental P contains several allotropes including white P, black P, and red P.23 In addition, many P allotropes have been theoretically predicted by Haeser et al.33 Among them, white P is often used as a raw material for organic chemistry. Red P is used for igniters, flame retardants, rechargeable batteries, and supercapacitors. Black P is one of the hottest materials for electronic devices, energy storage and catalysis.21–23 There are some reports about the application of black P for photocatalytic CO2 reduction.34–37 However, most of them need to couple black P with other photocatalysts to construct a heterojunction, and most of the black P needs to be at a small size.
It is well-known that the bandgap of black P depends on its size or the number of layers. Bulk black P has a very small bandgap of about 0.3 eV,38 which is not large enough to drive photocatalytic CO2 reduction. To achieve a bandgap larger than 1.5 eV, black P needs to have a thickness of one or two layers,39,40 which is very difficult to achieve. Compared with black P, red P has a bandgap of 1.6 eV and this is not affected by its size. It is very easy to prepare red P with a suitable bandgap for photocatalytic CO2 reduction. However, there are hardly any reports about the application of red P for photocatalytic CO2 reduction.
It is reported that the efficiency of solar-driven CO2 reduction is affected by the photoabsorption, CO2 activation on the surface and the transport of photoexcited electron–hole pairs inside the photocatalyst.10–13 A good photocatalyst should have a wide solar energy absorption range, photoexcited electrons with enough energy and fast charge transfer efficiency.15,16 Elemental red P has a bandgap of 1.6 eV, indicating that red P can theoretically absorb most visible light up to the near-infrared light region of 770 nm. This is superior to many metal oxide photocatalysts. Red P is expected to be a good candidate for CO2 reduction.
Herein, we discover that CO2 reduction can be achieved on commercial elemental phosphorus (Scheme 1). A CO generation rate of 2.1 μmol g−1 h−1 is obtained on amorphous red P (AP), which is 3.50 times higher than that obtained on a typical CdS photocatalyst (0.6 μmol g−1 h−1). Furthermore, although amorphous red P has a large solar absorption capacity and high CB, the internal charge transfer between the different grains in an individual particle greatly inhibits its wider application. As shown in Fig. S1,† there are plenty of interfaces between amorphous P grains. The presence of particle interfaces easily blocks the transfer of photoexcited carriers (Scheme S1†), and this means the potential of elemental phosphorus has not been fully realized. We believe this should be considered for the further application of elemental phosphorus. Therefore, in this study, we for the first time propose a strategy to achieve CO2 reduction on large-scale crystalline fibrous phase red P (FP). Compared with commercial amorphous P, the crystalline red P has a much better charge transfer efficiency, as confirmed by femtosecond and nanosecond transient absorption spectra. Besides, it is discovered that CO2 can be easily activated on crystalline red P according to density functional calculations. As a result, the CO2 reduction efficiency (22.0 μmol g−1 h−1) increases by about 10.5 times compared to amorphous P. Electrochemical measurements confirm the faster charge transfer in fibrous P. Theoretical calculations using the nudged elastic band method clearly demonstrate the CO2 reduction process of fibrous P, and indicate that the breaking of the C–O bond is the crucial step for CO2 reduction.
 | ||
| Scheme 1 Schematic diagram showing the concept of using elemental crystalline fibrous red P for photocatalytic CO2 reduction. | ||
2 Experimental section
2.1 Preparation of purified amorphous red P and fibrous phase red P
Amorphous red P was purified from commercial red phosphorus via a hydrothermal method. Commercial red P was dispersed in pure water, put into a Teflon-lined stainless steel autoclave, and heated at 180 °C for 6 h. After washing with D.I. water and ethanol, amorphous red P was obtained. The amorphous P was dried in a vacuum oven to remove the residual water and ethanol. To prepare fibrous phase red P, 300 mg of high-purity dried amorphous red phosphorus was transferred to a SiO2 capsule, evacuated to vacuum, and sealed afterward. The following temperature program was applied using a muffle furnace: the capsule was heated to 550 °C for 8 h, held at this temperature for 1 h, cooled to 275 °C for 75 h, held at this temperature for 1 h, and finally cooled to room temperature for 75 h. The product was collected by breaking the capsule, washed with water and ethanol several times, and dried in vacuum at 50 °C overnight. In addition, the typical photocatalyst CdS was purchased from Sigma-Aldrich (powder, 99.995%) as a reference.2.2 Characterization
SEM images and the corresponding EDX spectra were obtained using an FEI Quanta 400 microscope. The TEM and HRTEM images were recorded using a Philips Tecnai F20 instrument and a CM-120 microscope (Philips, 120 kV). TEM images were processed with Digital Micrograph (Gatan, USA). The powder XRD patterns were collected on a Rigaku SmartLab X-ray diffractometer using Cu Kα radiation (λ = 1.5406 Å). The accelerating voltage and applied current were 40 kV and 40 mA. UV-vis diffuse reflectance spectra were obtained using a UV-vis spectrophotometer (Cary 100 scan spectrophotometers, Varian). Raman spectra were recorded using a Renishaw RM3000 Micro-Raman system. FT-IR spectra were recorded on a Perkin Elmer Frontier spectrometer on samples embedded in KBr pellets. X-Ray photoelectron spectroscopy (XPS) was performed using a Thermo ESCALAB 250Xi spectrometer equipped with an anode emitting Al Kα radiation (1486.6 eV) as an X-ray source. The spectra were calibrated using C1s (284.8 eV). The Brunauer–Emmett–Teller (BET) surface area was measured by a Surface Area and Porosity Analyzer (ASAP 2460).2.3 Photocatalytic CO2 reduction
Photocatalytic CO2 reduction was carried out in a 100 mL homemade sealed glass reactor. Typically, 0.05 g of the photocatalysts were uniformly dispersed on a glass substrate and put into the reactor. Then 0.5 g of KHCO3 was also put into the bottom of the reactor. The air in the reactor was removed by evacuating the reactor. Carbon dioxide and water vapor were generated by injecting 1–2 mL of 6 M H2SO4 into the reactor to mix with the KHCO3 at the bottom by using a syringe injector. The photocatalytic CO2 reduction was carried out in a solid–gas system. The substrate loaded catalysts were not in contact with the KHCO3 and H2SO4. Herein, we used the reaction of KHCO3 and H2SO4 to produce high-purity CO2, because commercial CO2 gas in a steel cylinder often contains CO or CH4 impurities that can be detected by gas chromatography. This will affect the accuracy of the experiment. A 300 W xenon lamp was used as the radiation source. At intervals of 30 minutes, 1 mL of the gas was extracted and injected into a Shimadzu GC 2014C gas chromatograph equipped with a flame ionization detector (FID) and a thermal conductivity detector (TCD). High purity argon gas was used as a carrier gas. The GC curve showing the formation of CO and the standard curve are shown in Fig. S7.†2.4 Photoelectrochemical measurements
The powder sample was deposited onto a FTO conducting glass substrate by a drop-coating method. 2.5 mg of the photocatalysts were ultrasonically dispersed in 0.05 mL 0.05 wt% Nafion solution. Subsequently, the solution was dropped onto a FTO glass substrate as a working electrode. All the photoelectrochemical measurements were performed in a three-electrode cell with a saturated potassium chloride–silver chloride electrode (Ag/AgCl) as a reference electrode, a graphite rod (diameter = 0.6 cm) as a counter electrode, and the amorphous P or fibrous P electrodes as working electrodes. The electrolyte was 1 M KHCO3 saturated with Ar or CO2. A 300 W xenon arc lamp coupled with an AM 1.5G global filter (100 mW cm−2) and a UV cut-off filter (λ > 420 nm) was used as a radiation source. Linear sweep voltammetry and transient photocurrent were performed using a CHI 760E electrochemical workstation. The measured potential versus the Ag/AgCl reference electrode (EAg/AgCl) was converted to the reversible hydrogen electrode (RHE) scale according to the Nernst equation:41–43| ERHE = EAg/AgCl + 0.059pH + E0Ag/AgCl | (1) |
In the Mott–Schottky plot, the flat band potential was measured according to the following equation:44–48
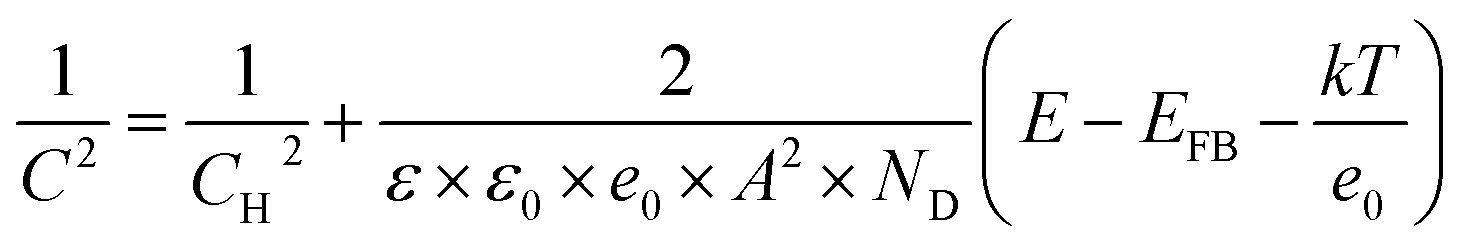 | (2) |
2.5 Theoretical calculations
The VASP computational package was used for all of the calculations in this article.50 We applied the projector-augmented-wave method with the Perdew–Burke–Ernzerhof GGA functional.51–53 The electronic convergence limit was set to be 1 × 10−5 eV. van der Waals d2 correction was taken into account.54 The optimization of atomic coordinates was considered to be converged if the Hellmann–Feynman force was smaller than 1 × 10−2 eV Å−1. A slab exposing the (001) surface was constructed. Each slab consisted of 8 phosphorus atoms. A CO2 molecule was put on the surface of the phosphorus. All the atoms were relaxed during optimization. The vacuum region was about 15 Å in height. Nudged elastic band calculations were used to study the kinetic barriers55 during CO2 reduction to CO. Kinetic barriers between optimized adsorbate geometries were calculated by the NEB method. Maximum forces of 0.03 eV A−1 were used as the optimization criteria. The Gibbs free energy was obtained by ΔG0 = ΔE0 + ΔZPE − TΔS. ΔG0 is calculated using DFT, ΔZPE is the difference in zero point energies in the reaction, ΔS is the change in entropy and T is 298 K.562.6 Femtosecond transient absorption spectroscopy
Femtosecond transient absorption spectra were recorded based on a commercial femtosecond Ti/sapphire regenerative amplifier laser system (Mai Tai, Spitfire-Pro, Spectra Physics Company) and an automated data acquisition transient absorption spectrometer (Ultrafast, Helios). The amplifier was seeded with the laser output from the oscillator (Maitai, Spectra Physics) and generated pulses of about 120 fs at 800 nm with a repetition rate of 1 kHz and an average power of around 2.6 W. For the present experiments, part of the fundamental pulse (approximately 95% of the amplified 800 nm output from the Spitfire) was used for second harmonic generation in a BBO crystal to obtain a 400 nm laser as the pump, and the probe pulse was obtained by focusing the remaining 5% of the amplified 800 nm output on a Ti/sapphire crystal to generate a white-light continuum spectrum that covers the range of 420 to 780 nm. The pump and probe beams were focused onto the sample and should maintain the temporal and spatial overlap at the sample. The signal was recorded by detecting the intensity of the probe light. A cell with a 2 mm optical path length and a stir bar was used to keep the sample even. The instrument response function of this system was determined to be 150 fs, and the data were stored as three-dimensional (3D) wavelength–time–absorbance matrices.2.7 Nanosecond transient absorption spectroscopy
Ns-TA experiments were carried out with an EOS Multichannel pump–probe transient absorption spectrometer (Ultrafast company). A Nd:YAG laser with a third harmonic process was used to produce the 365 nm pump laser. A continuous light source was generated by using a photonic crystal fiber with the range of 350–2250 nm. When the sample is excited by the pump light, the probe light will pass through the sample and the absorption signal will be detected by a fiber optics-coupled multichannel spectrometer with CMOS sensors with an intrinsic resolution of 2 nm. The normalized intensity was measured according to: | (3) |
3 Results
3.1 Structure of phosphorus photocatalysts
Amorphous phosphorus is formed of irregular particles without any crystal structure, and no lattice can be found in the TEM images (Fig. S1†). Fibrous P is prepared via a CVD method (see the Experimental section). The fibrous P product is a dark red powder (Fig. 1a). This can be dispersed in ethanol (Fig. 1b). In the SEM image, the fibrous P is in the form of microrods with a length of 0.5–5.0 μm and a diameter of 0.2–1.5 μm (Fig. 1c). In the TEM and HRTEM images, a lattice fringe with a d-spacing of 6.39 Å is visible, which is characteristic of the (−110) facets of fibrous phase red P (Fig. 1d and e).23 The corresponding EDX spectrum displays a strong peak for P (Fig. 1f), which is consistent with the chemical composition of elemental red P. The signal for carbon should come from the substrate in the TEM characterization.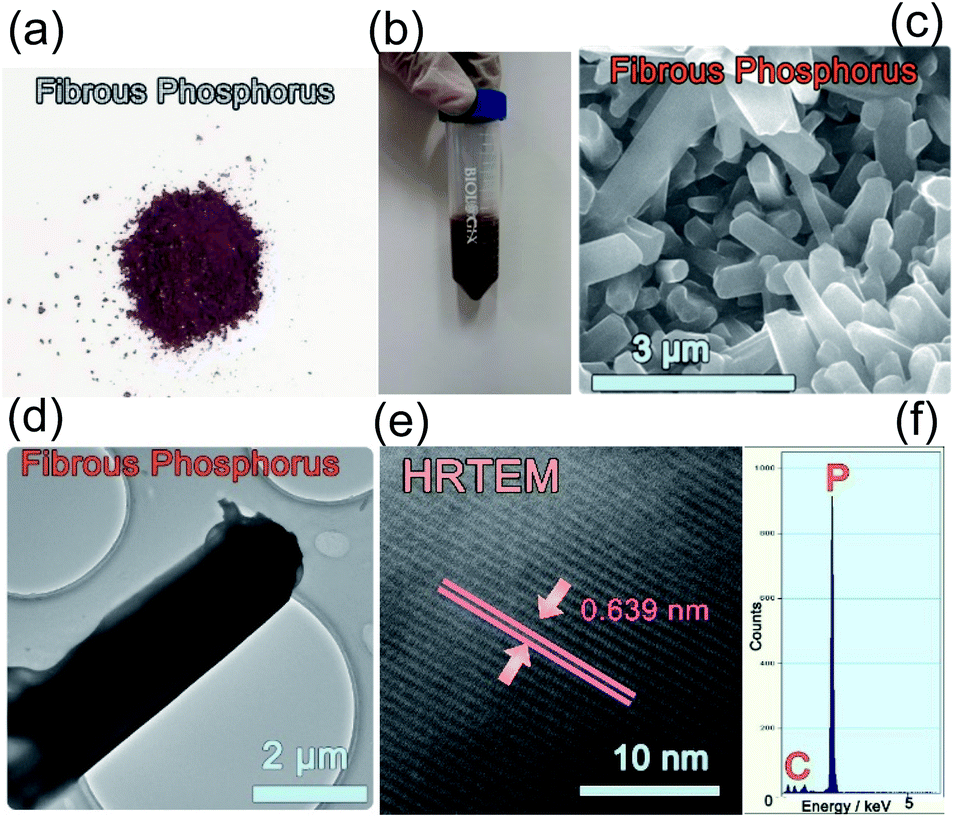 | ||
| Fig. 1 Photos of crystalline fibrous red P as (a) a powder and (b) a suspension. (c) SEM, (d) TEM, (e) HRTEM images and (f) EDX spectrum of crystalline fibrous red P. | ||
Amorphous P is found to be a linear inorganic polymer with a broad molecular weight distribution. According to a previous report,57 based on the study of scanning tunneling microscopy (STM) and gel permeation chromatography (GPC), amorphous P is a zig-zag chain structure rather than a two- or three-dimensional structure. It should adopt a zig-zag ladder structure (Fig. 2a).57 In contrast, fibrous P consists of covalently linked phosphorus pentagonal tubes that consist of alternating P8 and P9 cages linked by a P2 chelating bridge. The unit cell of fibrous P is composed of two parallel polyphosphide tubes, each of which contains 21 P atoms (Fig. 2b).14
 | ||
| Fig. 2 The molecular structures of (a) amorphous phosphorus and (b) fibrous crystalline phosphorus. (c) XRD, (d) Raman and (e) 31P solid state nuclear magnetic resonance spectra of amorphous phosphorus and fibrous crystalline phosphorus. (f) Band diagram of fibrous crystalline phosphorus obtained from theoretical calculations. | ||
To investigate their structure, XRD, Raman and FT-IR analysis were performed. The XRD pattern of amorphous P exhibits few diffraction peaks, suggesting its amorphous character (Fig. 2c). The XRD pattern of fibrous P is well aligned with the standard simulated pattern of fibrous phase red P (CSD 391323).23 39 of the main diffraction peaks in the XRD pattern of fibrous red P were compared with those of the standard fibrous phase P one by one (Fig. S2 and Table S1†). The 39 diffraction peaks are consistent with those of standard fibrous P. As Hittorf’s P exhibits a similar structure to fibrous P, we also carefully compared the XRD pattern of standard Hittorf’s P with that of our fibrous P sample. Compared with Hittorf’s P, our sample does not exhibit the typical peaks of Hittorf’s P at 9.6°, 10.4° and 11.1° (grey region in Fig. S3†) and at 13.8° and 19.3° (asterisks). The Raman spectrum (Fig. 2d) of fibrous P shows much more apparent peaks compared to that of amorphous P. The location of the Raman peaks is in good agreement with previous reports, which confirms its phase as fibrous P. The more obvious Raman peaks for fibrous P should be due to its higher crystallinity, which is beneficial to Raman vibrations.
The 31P solid state nuclear magnetic resonance spectra of amorphous P and fibrous P are exhibited in Fig. 2e. A broad wide peak can be found in the spectrum for amorphous P, while several sharp peaks are visible for fibrous P, indicating the crystalline structure of fibrous P. It is probable that more detailed nuclear magnetic resonance signals can be obtained for fibrous P with higher crystallinity, while the nuclear magnetic resonance peaks overlap for amorphous P, which results in a broad peak.
The fine P 2p XPS spectra of amorphous P and fibrous P resemble each other (Fig. S4†). They chiefly display a strong peak centered at 130.1 to 130.2 eV, which corresponds to the P0 atom.58,59 The domination of the P0 peak suggests that our purified red P is mainly in the elemental state.
3.2 Band diagram of the fibrous P photocatalyst
The band structure of fibrous P is studied by theoretical calculations and electrochemical measurements. The band structure of fibrous P is theoretically calculated according to the [P21] polyphosphide tubes model (Fig. 2b).23 van der Waals correction (Grimme d2) is used to increase the accuracy because fibrous P has a layered structure.54 Fibrous P has an appreciable bandgap of 1.6 eV according to the projector-augmented-wave method with the Perdew–Burke–Ernzerhof GGA functional (Fig. 2f). This confirms its semiconductive nature. The bandgap of 1.6 eV is very close to the absorption spectra of fibrous P and amorphous P (Fig. S5a and b†). They both absorb solar energy up to the red light region, suggesting their high utilization rate of solar energy. Their bandgaps are similar, and are calculated to be about 1.65 eV according to the Kubelka–Munk plots obtained from the absorption spectra (Fig. S5c and d†).The Mott–Schottky plots of 1/C2 as a function of the applied potential are shown in Fig. S6.† The flat band position of the as-prepared samples is estimated at 1/C2 = 0 and is determined to be −0.32 V vs. RHE.8 This value can also be considered as the position of the conduction band (CB) because it is usually accepted that in many n-type semiconductors the CB is very close to the flat band potential. This value is higher than the lowest theoretical potential for CO2 reduction (CO2 to CO, pH = 7, −0.106 V vs. RHE),60 indicating its capability to reduce CO2. As the bandgap is about 1.6 eV, the valence band of the fibrous P is calculated to be about 1.33 V vs. RHE.
3.3 Evaluation of photocatalytic CO2 reduction on phosphorus
CO2 photoreduction is carried out in a 100 mL homemade reactor (for details see the Experimental section and Fig. S7†) under the illumination of a 300 W Xe lamp. Red P powders are loaded on a glass substrate in an atmosphere of CO2, and the reaction is performed in a solid–gas system. The reaction products were examined by a gas chromatograph equipped with a Thermal Conductivity Detector (TCD) and a flame ionization detector (FID). To study the pristine activity of red P, no cocatalysts were added. We discovered that both amorphous P and fibrous P exhibit very high selectivity for CO2 reduction. Only CO is produced and no other products are detected, including CH4, HCOOH or CH3OH. Fibrous red P is also dispersed in water and illuminated for 4 hours and no peaks corresponding to formic acid, methanol or ethanol can be found (Fig. S8†). The yields of CO for pure amorphous P and fibrous P catalysts are presented in Fig. 3a. The generation rate of CO on amorphous P is about 2.1 μmol g−1 h−1, while that of the fibrous P remarkably increases to 22.0 μmol g−1 h−1, which is 10.5 times higher than that of amorphous P. This indicates that crystalline phosphorus is much more beneficial for CO2 reduction. Commercial CdS, a typical photocatalyst, is used as a reference. Its activity is only 0.60 μmol g−1 h−1. The activities of amorphous P and fibrous P are 3.50 and 36.7 times higher than that of CdS respectively (Fig. 3b). Our result is compared with other results in the literature (Fig. 3b). 30 recent results published between 2018 and 2020 are summarized in Table S2.† The mainstream photocatalysts for CO2 conversion to CO are C3N4, Fe2O3, ZnO, TiO2, BiVO4 and their composites. Our result of 22 μmol h−1 g−1 for fibrous phase red P is superior to most of the mainstream photocatalysts. It is also better than black P and its composites, especially considering that black P often needs to be coupled with other photocatalysts and needs to be controlled at small sizes. Although the activity is still lower than that of the heterojunctions of CsPbBr3 QDs/C3N4,65 Au-25@ZIF-8@TiO2 (ref. 66) and mesoporous TiO2/3D graphene/layered MoS2,67 as a single phase material and a new discovery, red P still has the potential to achieve a higher activity by building composites or loading with cocatalysts in the future. | ||
| Fig. 3 (a) Time profile showing the yield of CO from the photocatalytic reduction of CO2 on amorphous P (AP) and fibrous P (FP). (b) Comparison of the activity of different photocatalysts: (1) amorphous P, (2) fibrous P, (3) traditional CdS, (4) ZnO, (5) Au/TiO2, (6) BiVO4/Bi4Ti3O12, (7) black P, (8) black P nanosheets, (9) black P/C3N4, (10) black P/covalent triazine framework.35,37,61–64 | ||
In our control experiments, CO cannot be detected when the atmosphere of CO2 is replaced by Ar. Similarly, no CO is formed when the Xe lamp is turned off. This suggests that CO should originate from CO2 reduction. The stability of fibrous P is evaluated by its cycling stability. It is found that fibrous P maintains 99% of its initial activity after 3 cycles (Fig. S9†).
In addition to the rate of product formation by the total mass of the catalyst, the rate over the surface area of the catalyst is also calculated to evaluate the geometric effects. The BET specific surface area of amorphous P and fibrous P is 23.4 cm2 g−1 and 4.17 cm2 g−1, respectively. Then, the rate of product formation is calculated to be 0.090 μmol h−1 cm−2 and 5.276 μmol h−1 cm−2. In this regard, the activity of fibrous P is 58.8 times higher than that of amorphous P, which is about 5 times higher than the enhancement (10.5 times) calculated by total mass. This indicates that the surface of fibrous P is more favorable for CO2 reduction.
To investigate the relationship between the number of active sites and the activity of the P photocatalysts, the turnover frequency is calculated. As discussed before, the TOF for heterogeneous catalysts is mostly calculated over the number of active sites or the number of moles of the catalysts. Herein, we calculated both of them.68
When the number of moles of the catalyst is used, the turnover frequency (TOF) is calculated according to:68,69
| TOF = mol of product/(mol of catalyst × time) | (4) |
The TOF for amorphous P and fibrous P is calculated to be 0.67 × 10−4 h−1 and 7.04 × 10−4 h−1 respectively.
When the number of active sites is used, we evaluate the number of active sites by measuring the quantity adsorption (QA) of CO2 (Fig. S10†). The value for amorphous P (3.0 cm3 g−1) is about 2.5 times larger than that of fibrous P (1.2 cm3 g−1). The small value for fibrous P indicates that it adsorbs less CO2. However, the BET specific surface area of amorphous P (23.4 cm2 g−1) is 5.61 times larger than that of fibrous P (4.17 cm2 g−1). Therefore, the quantity adsorption of CO2 on amorphous P is 2.24 times smaller than that of fibrous P per unit of specific surface area. This indicates that the surface of fibrous P has higher capability to adsorb CO2.
The TOF is calculated according to
 | (5) |
The TOF for fibrous P is about 20 times higher than that of amorphous P, suggesting that the activity of fibrous P is much higher than that for amorphous P.
To sum up, although the quantity adsorption for fibrous P is smaller than that of amorphous P, the TOF for fibrous P is much higher. The higher activity for fibrous P should be due to the higher reaction frequency on an individual active site.
Finally, the quantum yield (QY) is calculated according to the literature:68,70
 | (6) |
Specifically in our case
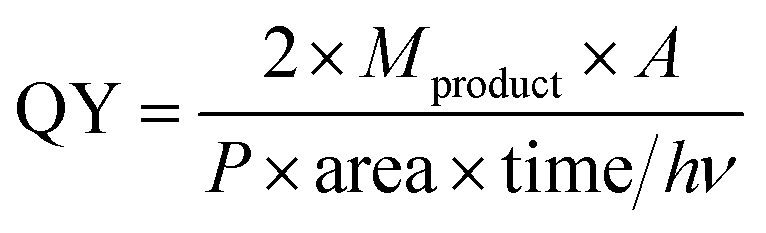 | (7) |
The capability of amorphous P and fibrous P to reduce CO2 is further investigated by examining their electrochemical behavior. The measurements are carried out in a standard three-electrode cell by linear sweep voltammetry (LSV) in an Ar or CO2-saturated 1 M KHCO3 electrolyte. As shown in Fig. 4a, amorphous P and fibrous P exhibit very small cathodic current densities in the Ar-saturated electrolyte at a bias voltage smaller than the onset potential of −0.83 V vs. RHE. Then, the cathodic current increases as a result of hydrogen evolution.9 In the CO2-saturated electrolyte, the current density of amorphous P increases slightly earlier and faster. Its onset potential shifts positively by about 0.05 V and its current density (2.0 mA cm−2) is about twice as large as that in the Ar-saturated electrolyte (1.0 mA cm−2) at −1.0 V vs. RHE. This suggests its capability to reduce CO2.
 | ||
| Fig. 4 (a) Linear sweep voltammetric curves of amorphous P and fibrous P photocatalysts in an Ar or CO2-saturated 1 M KHCO3 electrolyte in the dark with a scanning rate of 50 mV s−1. (b) Photocurrent density of AP and FP on FTO conducting glass in 1 M KHCO3 at 1.2 V vs. RHE under intermittent AM 1.5G illumination. Femtosecond transient absorption spectra of (c) amorphous red phosphorus and (d) fibrous P. (e) Femtosecond normalized single decay profile of transient absorption spectra at 630 nm and (f) nanosecond normalized single decay profile of transient absorption spectra at 530 nm. | ||
With regards to fibrous P, its onset potential shifts by about 0.12 V, and occurs much earlier than that of amorphous P. Its current density (4.6 mA cm−2) is also much higher than that of amorphous P at −1.0 V vs. RHE, demonstrating a stronger ability for CO2 reduction. In the presence of illumination, the electrodes covered with phosphorus photocatalysts are able to generate a photocurrent. Higher photocurrent density can be achieved by fibrous P (Fig. 4b). Its photocurrent density (3.7 μA cm−2) is 9.25 times higher than that of AP (0.4 μA cm−2), indicating stronger photocatalytic activity. Therefore, these results confirm that fibrous P is superior to amorphous P for CO2 reduction.
3.4 Ultrafast transient absorption spectroscopy investigation
The higher photocatalytic CO2 reduction activity on fibrous P can be attributed to its crystal structure, which is favorable for the transfer of photoexcited charge carriers. Femtosecond transient absorption spectra (TAS) were recorded to investigate the charge carrier dynamics of amorphous P and fibrous P. The spectra were measured using a 400 nm pump with a laser pulse of ca. 50 fs duration (3.1 eV, which is a larger energy than the red P band gap of 1.69 eV). A white light continuum of 450–750 nm was used as a probe.After excitation, the pump laser causes instantaneous generation and direct excitation of charge carriers from the ground state (GS) to the emission state (EM). Plenty of photoexcited electrons are generated in the EM. As reflected in the TAS curve (Fig. 4c), in the beginning (1–10 ps), the red P nanosheets exhibit an apparent positive peak at 635 nm, which corresponds to the signal in the EM.71,72 As time goes on (10–1000 ps), this peak decays due to the loss of photoexcited electrons.73 The electrons will drop to the GS to recombine with holes or drop to the intermediate states and are trapped.73 After 1000 ps, the positive peak becomes flat and new negative peaks at 460 nm and 530 nm appear. These peaks are attributed to hole bleaching, suggesting that the signal of photoexcited holes dominates at this time scale.71 Compared with amorphous P, the positive peak for fibrous P exhibits a red shift from 635 nm to 640 nm. This suggests that the processes occur at lower energies (Fig. 4d).74
The normalized single wavelength decay profile at 630 nm is recorded to study the kinetics of the photoexcited electrons in red P (Fig. 4e). Normalization is achieved by using the equation:
| Normalized intensity = (measured intensity − lowest intensity)/(highest intensity − lowest intensity) |
The initial rise indicates the instantaneous generation and direct excitation of charge carriers from the GS to the EM. The following drop is due to the recombination of electrons and holes. The fibrous P decays slower than the amorphous P, corresponding to slower charge recombination and faster charge transfer rates in the fibrous P.
The dynamics of the photoexcited charges were investigated in a long (ns–μs) time scale by using nanosecond TA (ns-TA) spectroscopy with 400 nm excitation (Fig. S11†). The signal in this time scale should be due to the charge carriers in the trapping states of phosphorus, which should stem from the defects in phosphorus.75,76 The decay observed in both samples can be attributed to the recombination of photoexcited electrons and holes. The decay of fibrous P is much slower than that of its amorphous counterpart, implying a longer lifetime of charge carriers in fibrous P at the nanosecond scale (Fig. 4f and S12†). Therefore, fibrous P is superior to amorphous P in its charge transfer efficiency. This indicates that more photoexcited electrons can avoid being recombined and reach the surface and contribute to the CO2 reduction, making fibrous P remarkably favorable for CO2 reduction.
3.5 Mechanism for CO2 reduction on fibrous P and density functional calculations
 is a crucial step.77
is a crucial step.77
According to the literature, the bandgap of phosphorus becomes larger when it is oxidized, and the bandgap increases (larger than 2.21 eV) when more oxygen is introduced.78,79 This is larger than that of elemental phosphorus. Therefore, it is more difficult to produce photoexcited carriers in the phosphorus oxide under solar illumination. Also, with a large bandgap, it is difficult for the photoexcited electrons in elemental phosphorus to migrate to the phosphorus oxide. The electrons should be blocked by the energy barrier and concentrate in the elemental phosphorus. As a result, the oxidized surface can no longer be the active site for photoexcited electrons and CO2 reduction, and the active site should be the surface in the elemental state. This is further supported by our density functional calculations as discussed below.
| Ead = Etot − Esubstrate − ECO2 | (8) |
 | ||
| Fig. 5 (a) Structure of the CO2-FP system after structure relaxation from theoretical calculations. (b) Localized electron density map of the FP and CO2-FP systems. (c) and (d) Magnified images of (b). Two-dimensional slice showing the localized electron density map of (e) FP and (f) CO2-FP system. | ||
When a CO2 molecule is adsorbed on the surface of the fibrous P photocatalyst, the length of the C–O bonds (1.160 Å) in the original CO2 molecule is elongated to an average value of 1.193 Å. In addition, the O–C–O angle changes from 180° to 178.4°, indicating a bending of the CO2 molecule. This elongation and bending reflect the interaction between the P substrate and the CO2 molecule.
The charge in the fibrous P substrate will also be affected by the adsorbed CO2 molecule due to the interaction. Usually, some electrons will be localized in the vicinity of an atom due to chemical bonding or the periodical electric field in the crystal. As shown in Fig. 5b and c, the yellow region is the localized region. When a CO2 molecule approaches, the localized electron becomes less localized (Fig. 5d). The comparison can also be seen in the two dimensional slices shown in Fig. 5e and f. Therefore, the electrons of the fibrous P substrate near the CO2 are more transferable, which is beneficial for charge transfer from the fibrous P substrate to the CO2 molecules for their activation.
As for the oxide surface, the C–O bonds do not elongate after structure relaxation (Fig. S16†). The average length of the C–O bonds (1.163 Å) is very close to that of the original CO2 molecule. Besides, the adsorption energy of CO2 is very small (0.007 eV), which is much smaller than that of elemental phosphorus (−0.1 eV). This indicates that CO2 is not easily activated on phosphorus oxide. Furthermore, in the analysis of the localized electron density map, when the surface is covered with a layer of oxygen, the localized electron density of the substrate is not influenced by the CO2 molecules. This is different from the pure phosphorus substrate. Thus, the active site of CO2 reduction should still be elemental phosphorus. Therefore, we will discuss in detail the mechanism for CO2 reduction on an elemental phosphorus surface.
The structure and energy changes are shown in Fig. 6a and b, respectively. Initially, a COOH* species is adsorbed onto the P substrate. Then, the C–O bond is elongated (state no. 2) and the free energy rises. The key step for the reduction of CO2 to CO is the breaking of the C–O bond (state no. 3) and the free energy climbs to the maximum. This step is the key step for CO2 reduction. Subsequently, the OH leaves the CO molecule and combines with another H atom to form H2O. The free energy goes downhill during this process, suggesting that this process is thermodynamically favorable. Finally, a CO molecule and a H2O molecule are formed. The free energy at the final state is about 1 eV lower than that at the initial state. However, the total process needs to overcome a barrier and the breaking of the C–O bond is the key step.
 | ||
| Fig. 6 (a) Structure change and (b) energy change for the proton-involving process during CO2 reduction to CO in the NEB calculations. (c) Structure change and (d) energy change for the proton-free process during CO2 reduction to CO in the NEB calculations. | ||
With respect to the proton-free process, optimized adsorbates of CO2 and CO + O are obtained after full relaxation as the initial and final states, respectively. In the beginning, CO2 is adsorbed and the CO2 molecules are bent and elongated as discussed above. Then, one atom becomes closer and closer to the phosphorus substrate (Fig. 6c, no. 1 to no. 3) and the energy slowly rises (Fig. 6d). At state no. 4, the C–O bond breaks and the energy greatly rises, indicating that this step is important for CO2 reduction to CO. Subsequently, the adsorbed oxygen is linked more closely with the substrate and it is easy for it to bond with more P atoms in the P substrate. At the same time, the remaining CO leaves the adsorbed O. The energy rises to its peak at this state. Therefore, this step is crucial for CO2 reduction to CO. Then, the energy quickly drops and finally the C–O bond in CO2 is broken.
Finally, the free energy for the final state is about 0.7 eV higher than that for the initial state. This is inferior to the proton-involving process where the final state is energetically downhill with respect to the initial state. Consequently, although the two processes both need to overcome an energy barrier with a similar value, the free energy of the final state shows that the proton-involving process is a downhill process. The proton-involving process should be preferred. This demonstrates that the introduction of a proton, or water, is important for CO2 reduction on P photocatalysts.
4 Conclusion
With wide solar spectrum absorption and a high conduction band, red P is found to be an elemental photocatalyst for CO2 reduction. It can selectively reduce CO2 to CO. However, due to the boundaries between particles inside amorphous P, the charge transfer limits the enhancement of CO2 reduction. In this work, crystalline fibrous phase red P is synthesized. Fibrous P shows a great improvement in CO2 reduction. Its activity is about 10.5 times higher than that of commercial amorphous red P. Its charge transfer efficiency is remarkably improved, which is confirmed by transient absorption spectra and electrochemical measurements. This study will broaden the application of red P materials and deepen our understanding of their nature as photocatalysts. Besides, this work shows that elemental photocatalysts can serve as a new kind of promising material for CO2 reduction. It will help to promote the development of new elemental photocatalysts or elemental-based composite photocatalysts for CO2 reduction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant No. 51902357), the Natural Science Foundation of Guangdong Province, China (2019A1515012143), the Start-up Funds for High-Level Talents of Sun Yat-sen University (38000-18841209), the Fundamental Research Funds for the Central Universities (19lgpy153) and the Guangdong Basic and Applied Basic Research Foundation (2019B1515120058). The theoretical calculations are supported by the National Supercomputer Center in GuangZhou and the National Supercomputing Center in Shenzhen (Shenzhen Cloud Computing Center).Notes and references
- K. Wang, Q. Li, B. S. Liu, B. Cheng, W. K. Ho and J. G. Yu, Appl. Catal., B, 2015, 176–177, 44–52 CAS.
- J. N. Qin, S. B. Wang, H. Ren, Y. D. Hou and X. C. Wang, Appl. Catal., B, 2015, 179, 1–8 CrossRef CAS.
- J. G. Yu, J. X. Low, W. Xiao, P. Zhou and M. Jaroniec, J. Am. Chem. Soc., 2014, 136, 8839–8842 CrossRef CAS.
- J. G. Yu, J. Jin, B. Cheng and M. Jaroniec, J. Mater. Chem. A, 2014, 2, 3407–3416 RSC.
- S. B. Wang, W. S. Yao, J. L. Lin, Z. X. Ding and X. C. Wang, Angew. Chem., Int. Ed., 2014, 53, 1034–1038 CrossRef CAS.
- S. B. Wang and X. C. Wang, Angew. Chem., Int. Ed., 2016, 55, 2308–2320 CrossRef CAS.
- E. E. Barton, D. M. Rampulla and A. B. Bocarsly, J. Am. Chem. Soc., 2008, 130, 6342–6344 CrossRef CAS.
- X. He, N. Zheng, R. Hu, Z. Hu and J. C. Yu, Adv. Funct. Mater., 2020, 2006505 CrossRef.
- Z. Hu, J. Gong, Z. Ye, Y. Liu, X. Xiao and J. C. Yu, J. Catal., 2020, 384, 88–95 CrossRef CAS.
- Y. C. Lan, Y. Z. Xie, J. X. Chen, Z. F. Hu and D. H. Cui, Chem. Commun., 2019, 55, 8068–8071 RSC.
- F. Xu, K. Meng, B. Cheng, S. Wang, J. Xu and J. Yu, Nat. Commun., 2020, 11, 4613 CrossRef CAS.
- Y. Lan, G. Niu, F. Wang, D. Cui and Z. Hu, ACS Appl. Mater. Interfaces, 2020, 12, 36128–36136 CrossRef CAS.
- Y. Xia and J. Yu, Chem, 2020, 6, 1039–1040 CAS.
- J. Fu, K. Jiang, X. Qiu, J. Yu and M. Liu, Mater. Today, 2020, 32, 222–243 CrossRef CAS.
- W. G. Tu, Y. Zhou, Q. Liu, S. C. Yan, S. S. Bao, X. Y. Wang, M. Xiao and Z. G. Zou, Adv. Funct. Mater., 2013, 23, 1743–1749 CrossRef CAS.
- J. Mao, T. Y. Peng, X. H. Zhang, K. Li, L. Q. Ye and L. Zan, Catal. Sci. Technol., 2013, 3, 1253–1260 RSC.
- Y. Fujita, H. O. Venterink, P. M. van Bodegom, J. C. Douma, G. W. Heil, N. Holzel, E. Jablonska, W. Kotowski, T. Okruszko, P. Pawlikowski, P. C. de Ruiter and M. J. Wassen, Nature, 2014, 505, 82–86 CrossRef.
- B. C. Koo, Y. H. Lee, D. S. Moon, S. C. Yoon and J. C. Raymond, Science, 2013, 342, 1346–1348 CrossRef CAS.
- T. Low, R. Roldan, H. Wang, F. N. Xia, P. Avouris, L. M. Moreno and F. Guinea, Phys. Rev. Lett., 2014, 113, 5 CrossRef.
- A. Velian and C. C. Cummins, Science, 2015, 348, 1001–1004 CrossRef CAS.
- M. Scheer, G. Balázs and A. Seitz, Chem. Rev., 2010, 110, 4236–4256 CrossRef CAS.
- F. Bachhuber, J. von Appen, R. Dronskowski, P. Schmidt, T. Nilges, A. Pfitzner and R. Weihrich, Angew. Chem., Int. Ed., 2014, 53, 11629–11633 CrossRef CAS.
- M. Ruck, D. Hoppe, B. Wahl, P. Simon, Y. Wang and G. Seifert, Angew. Chem., Int. Ed., 2005, 44, 7616–7619 CrossRef CAS.
- H. Thurn and H. Krebs, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1969, 25, 125–135 CrossRef CAS.
- A. Pfitzner, M. F. Bräu, J. Zweck, G. Brunklaus and H. Eckert, Angew. Chem., Int. Ed., 2004, 43, 4228–4231 CrossRef CAS.
- M. H. Möller and W. Jeitschko, J. Solid State Chem., 1986, 65, 178–189 CrossRef.
- A. Pfitzner and E. Freudenthaler, Angew. Chem., Int. Ed. Engl., 1995, 34, 1647–1649 CrossRef CAS.
- L. Li, Y. Yu, G. J. Ye, Q. Ge, X. Ou, H. Wu, D. Feng, X. H. Chen and Y. Zhang, Nat. Nanotechnol., 2014, 9, 372–377 CrossRef CAS.
- H. Liu, A. T. Neal, Z. Zhu, Z. Luo, X. F. Xu, D. Tomanek and P. D. D. Ye, ACS Nano, 2014, 8, 4033–4041 CrossRef CAS.
- Z. Hu, L. Yuan, Z. Liu, Z. Shen and J. C. Yu, Angew. Chem., Int. Ed., 2016, 55, 9580–9585 CrossRef CAS.
- Y. Liu, Z. Hu and J. C. Yu, Appl. Catal., B, 2019, 247, 100–106 CrossRef CAS.
- Z. Shen, Z. Hu, W. Wang, S.-F. Lee, D. K. L. Chan, Y. Li, T. Gu and J. C. Yu, Nanoscale, 2014, 6, 14163–14167 RSC.
- M. Haeser, J. Am. Chem. Soc., 1994, 116, 6925–6926 CrossRef CAS.
- X. Y. Ji, Y. Kang, T. J. Fan, Q. Q. Xiong, S. P. Zhang, W. Tao and H. Zhang, J. Mater. Chem. A, 2020, 8, 323–333 RSC.
- J. Li, P. Liu, H. L. Huang, Y. Li, Y. Z. Tang, D. H. Mei and C. L. Zhong, ACS Sustainable Chem. Eng., 2020, 8, 5175–5183 CrossRef CAS.
- W. Gao, X. Bai, Y. Gao, J. Liu, H. He, Y. Yang, Q. Han, X. Wang, X. Wu, J. Wang, F. Fan, Y. Zhou, C. Li and Z. Zou, Chem. Commun., 2020, 56, 7777–7780 RSC.
- X. Wang, J. He, J. Li, G. Lu, F. Dong, T. Majima and M. Zhu, Appl. Catal., B, 2020, 277, 119230 CrossRef CAS.
- M. S. Zhu, S. Kim, L. Mao, M. Fujitsuka, J. Y. Zhang, X. C. Wang and T. Majima, J. Am. Chem. Soc., 2017, 139, 13234–13242 CrossRef CAS.
- X. Chen, J. S. Ponraj, D. Fan and H. Zhang, Nanoscale, 2020, 12, 3513–3534 RSC.
- G. Zhang, A. Chaves, S. Huang, F. Wang, Q. Xing, T. Low and H. Yan, Sci. Adv., 2018, 4, eaap9977 CrossRef.
- M. Xu, P. M. Da, H. Y. Wu, D. Y. Zhao and G. F. Zheng, Nano Lett., 2012, 12, 1503–1508 CrossRef CAS.
- S. Hoang, S. W. Guo, N. T. Hahn, A. J. Bard and C. B. Mullins, Nano Lett., 2012, 12, 26–32 CrossRef CAS.
- L. Li, Z. Hu and J. C. Yu, Angew. Chem., Int. Ed., 2020, 20719–20725 CrossRef.
- G. M. Wang, Y. C. Ling, D. A. Wheeler, K. E. N. George, K. Horsley, C. Heske, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 3503–3509 CrossRef CAS.
- X. Y. Yang, A. Wolcott, G. M. Wang, A. Sobo, R. C. Fitzmorris, F. Qian, J. Z. Zhang and Y. Li, Nano Lett., 2009, 9, 2331–2336 CrossRef CAS.
- C. Baumanis and D. W. Bahnemann, J. Phys. Chem. C, 2008, 112, 19097–19101 CrossRef CAS.
- T. V. Nguyen and O. B. Yang, Catal. Today, 2003, 87, 69–75 CrossRef CAS.
- N. Zheng, X. He, W. Guo and Z. Hu, Chin. Chem. Lett., 2020 DOI:10.1016/j.cclet.2020.10.039.
- K. F. Young and H. P. R. Frederikse, J. Phys. Chem. Ref. Data, 1973, 2, 313–410 CrossRef.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- F. Bachhuber, J. von Appen, R. Dronskowski, P. Schmidt, T. Nilges, A. Pfitzner and R. Weihrich, Z. Kristallogr. - Cryst. Mater., 2015, 230, 107–115 CAS.
- J. H. Montoya, A. A. Peterson and J. K. Norskov, ChemCatChem, 2013, 5, 737–742 CrossRef CAS.
- J. Rossmeisl, Z. W. Qu, H. Zhu, G. J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- S. Zhang, H.-j. Qian, Z. Liu, H. Ju, Z.-y. Lu, H. Zhang, L. Chi and S. Cui, Angew. Chem., Int. Ed., 2019, 58, 1659–1663 CrossRef CAS.
- Z. Shen, S. Sun, W. Wang, J. Liu, Z. Liu and J. C. Yu, J. Mater. Chem. A, 2015, 3, 3285–3288 RSC.
- U. Braun and B. Schartel, Macromol. Chem. Phys., 2004, 205, 2185–2196 CrossRef CAS.
- Z. Shulin, L. Sheng, G. Tao, Z. Shuaishuai, W. Jing, W. Yuping and C. Yuhui, Nano-Micro Lett., 2019, 11, 62 CrossRef.
- C. Q. Han, J. Li, Z. Y. Ma, H. Q. Xie, G. I. N. Waterhouse, L. Q. Ye and T. R. Zhang, Sci. China Mater., 2018, 61, 1159–1166 CrossRef CAS.
- A. Pougin, G. Dodekatos, M. Dilla, H. Tuysuz and J. Strunk, Chem.–Eur. J., 2018, 24, 12416–12425 CrossRef CAS.
- X. D. Liu, L. Q. Ye, S. S. Liu, Y. P. Li and X. X. Ji, Sci. Rep., 2016, 6, 9 CrossRef.
- X. Y. Wang, Y. S. Wang, M. C. Gao, J. N. Shen, X. P. Pu, Z. Z. Zhang, H. X. Lin and X. X. Wang, Appl. Catal., B, 2020, 270, 9 Search PubMed.
- M. Ou, W. G. Tu, S. M. Yin, W. N. Xing, S. Y. Wu, H. J. Wang, S. P. Wan, Q. Zhong and R. Xu, Angew. Chem., Int. Ed., 2018, 57, 13570–13574 CrossRef CAS.
- L. Y. Tian, Y. C. Luo, K. L. Chu, D. J. Wu, J. Y. Shi and Z. X. Liang, Chem. Commun., 2019, 55, 12976–12979 RSC.
- H. Jung, K. M. Cho, K. H. Kim, H. W. Yoo, A. Al-Saggaf, I. Gereige and H. T. Jung, ACS Sustainable Chem. Eng., 2018, 6, 5718–5724 CrossRef CAS.
- M. Melchionna and P. Fornasiero, ACS Catal., 2020, 5493–5501 CrossRef CAS.
- B. L. Mash, A. Raghavan and T. Ren, Eur. J. Inorg. Chem., 2019, 2065–2070 CrossRef CAS.
- Y. Xia, K. Xiao, B. Cheng, J. Yu, L. Jiang, M. Antonietti and S. Cao, ChemSusChem, 2020, 13, 1730–1734 CrossRef CAS.
- Y. Yang, M. Forster, Y. Ling, G. Wang, T. Zhai, Y. Tong, A. J. Cowan and Y. Li, Angew. Chem., 2016, 128, 3464–3468 CrossRef.
- L. Jing, R. X. Zhu, D. L. Phillips and J. C. Yu, Adv. Funct. Mater., 2017, 27, 9 Search PubMed.
- B. Pattengale, J. Ludwig and J. Huang, J. Phys. Chem. C, 2016, 120, 1421–1427 CrossRef CAS.
- S. Shen, C. X. Kronawitter, D. A. Wheeler, P. Guo, S. A. Lindley, J. Jiang, J. Z. Zhang, L. Guo and S. S. Mao, J. Mater. Chem. A, 2013, 1, 14498–14506 RSC.
- Y. Liu, Z. F. Hu and J. C. Yu, Appl. Catal., B, 2019, 247, 100–106 CrossRef CAS.
- L. Jing, R. Zhu, D. L. Phillips and J. C. Yu, Adv. Funct. Mater., 2017, 27, 201703484 Search PubMed.
- X. G. Jianzhi Huang, Y. Wei, Q. Hu, X. Yu and L. Wang, J. CO2 Util., 2019, 33, 166–170 CrossRef.
- I. V. Abarenkov, I. I. Tupitsyn, V. G. Kuznetsov and M. C. Payne, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 7881–7885 CrossRef CAS.
- A. Ziletti, A. Carvalho, P. E. Trevisanutto, D. Campbell, D. Coker and A. Neto, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91 Search PubMed.
- X. Bai, W. Chen, C. Zhao, S. Li, Y. Song, R. Ge, W. Wei and Y. Sun, Angew. Chem., Int. Ed., 2017, 56, 12219–12223 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta09177d |
| This journal is © The Royal Society of Chemistry 2021 |