
N-doped hierarchical porous carbon derived from bismuth salts decorated ZIF8 as a highly efficient electrocatalyst for CO2 reduction†
Pengfei
Yao
ab,
Tianyu
Li
 a,
Yanling
Qiu
a,
Qiong
Zheng
a,
Huamin
Zhang
a,
Jingwang
Yan
a and
Xianfeng
Li
*a
a,
Yanling
Qiu
a,
Qiong
Zheng
a,
Huamin
Zhang
a,
Jingwang
Yan
a and
Xianfeng
Li
*a
aDivision of Energy Storage, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian 116023, P. R. China. E-mail: lixianfeng@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 12th November 2020
Abstract
With the excessive consumption of fossil fuels, CO2 emission issues and the energy crisis have become more prominent. The electrocatalytic reduction of CO2 (ERC) driven by intermittent renewable electricity can not only mitigate carbon dioxide emission, but also store renewable energy. However, the poor activity, selectivity and stability of the catalysts inhibit their practical large-scale application. Herein, we report a facile synthesis method for a synthetic N-doped hierarchical porous carbon catalyst, which was obtained by directly carbonizing the ZIF8 precursor decorated with the low boiling metal salts (BiCl3). Plentiful bismuth salts can not only efficiently destroy the Zn–Nx bond and promote the aggregation and evaporation of the Zn species at a relatively low temperature, but also enhance the formation of a conductive and hierarchical porous catalyst. In addition, the tedious post-processing to remove the metal salts is avoided. Density functional theory calculations suggest that the coordinated unsaturated Zn–Nx sites tremendously inhibit the activity of the nearby pyridinic N sites, which play an important role in the catalytic performance. As a result, the catalysts achieve about 90% faradaic efficiency for CO at a very low overpotential of −0.49 V, which is much lower than the most reported N-doped carbon catalysts.
1. Introduction
With the rapid depletion of fossil fuels, CO2 emission issues and the energy crisis have become more prominent. The electrocatalytic reduction of CO2 (ERC) can not only store the intermittent renewable electricity in high value-added fuels and chemicals, but also mitigate the carbon dioxide emission.1,2 However, CO2 is very stable under ambient environments, and a very high overpotential is needed to drive the carbon dioxide reduction reaction. Additionally, the ERC involves multiple proton–electron transfer processes and accompanies the competitive hydrogen evolution reaction in the similar potential window, which makes it difficult to achieve high selectivity for the target products.3,4 Thus, it is urgent to design a series of robust catalysts to achieve high activity and selectivity for CO2 reduction products. Over the past few decades, many kinds of catalysts have been developed for ERC, such as metals, metal oxides, metal alloys and carbon materials.5–8 Among the metal-based catalysts, copper is one of the most promising materials to produce hydrocarbons.9,10 However, Cu-based catalysts are confronted with many problems, including poor stability, high overpotential and low selectivity. Precious metals, such as Ag and Au, can efficiently transfer CO2 into CO at a lower potential,11,12 but high cost limits their wide applications. Therefore, seeking the nonprecious catalysts with higher activity and selectivity is urgent. In recent years, carbon-based catalysts have attracted enormous attention due to the high conductivity, large surface area, tunable porous structure, good stability, and low cost.13–15 The hierarchical porous carbon materials are the most promising catalytic supports for the CO2 reduction reaction among the carbon-based family. The porous structure of the catalysts can not only enhance the CO2 adsorption, but also promote mass transportation, which can efficiently improve the performance for the electrochemical reduction of CO2.16,17 Additionally, doping heteroatoms into the porous carbon matrix, especially N, S and B, can further improve the property of ERC as well. This is because the heteroatoms adjacent to the carbon can adjust the spin and charge density of the carbon atoms, and can thus promote the adsorption and activation of CO2.2,4 The zeolitic imidazolate frameworks (ZIFs) are regarded as ideal precursors to produce porous carbon catalysts due to their controlled porosity, large surface area and uniform heteroatom decoration.18,19 However, ZIF-derived N-doped porous carbon materials also have many challenges, including relatively low stability, conductivity, narrow pore size distribution and high metal content, which are not conducive to realizing the high selectivity and activity for ERC. The low electronic conductivity brings a slow electron transfer to form CO2˙−. The single microporous structures of ZIFs are not beneficial to transporting protons and electrons. In addition, the high Zn–Nx content derived from ZIFs will promote competitive hydrogen evolution reactions.15,20 Thus, it is necessary to solve these problems in order to achieve the excellent performance for ERC. Many methods have been developed to build the hierarchical porous architectures by using hard and soft templating ways. However, these fabrication procedures require tedious post-treatment to remove the template. It is well known that some metal-doped carbon materials can realize high conductivity due to the graphitization of the carbon matrix induced by the doped metal atoms. Among the ZIFs family, ZIF8 is the best choice to get the metal-free N-doped carbon catalysts, because most metal (Zn) ions will evaporate during the high temperature carbonization process. Herein, we report a facile synthesis method to obtain a N-doped porous carbon catalyst with relatively high stability, conductivity, hierarchical porous and low metal content, which can directly carbonize the ZIF8 precursor decorated by a low-boiling metal salt (BiCl3). The N-doped porous carbon materials (ZIF8-950-4Bi) were synthesized, as shown in Fig. 1. First, ZIF8 was fabricated according to a previous report with a slight modification.21,22 Subsequently, ZIF8 was evenly dispersed in ethanol, followed by the addition of BiCl3. Then, the N-doped porous carbon catalyst was obtained through simply pyrolyzing the mixture of ZIF8 and bismuth salts without the post-processing. The BiCl3 salt begins melting and undergoes decomposition to form the bismuth nanoparticles at relatively low temperature, which can promote the decomposition of ZIF8, especially the break of the Zn–N bond of ZIF8. Therefore, it can effectively promote the aggregation and vaporization of the Zn species at much lower temperature. In addition, the low boiling bismuth nanoparticle will evaporate at high temperature, which creates many mesoporous structures and catalyzes the graphitization of the carbon skeleton. By regulating the content of BiCl3, we can make the hierarchical porous structure, high electronic conductivity and minimal Zn content catalysts. The hierarchical porous architectures can promote the diffusion and adsorption of carbon dioxide to the active sites, and the high electronic conductivity brings the fast electron transfer to form CO2˙−. In addition, the low content of the coordinated unsaturated Zn–Nx will efficiently reduce the shielding effect on the nearby pyridinic N sites. As a result, the ZIF8-950-4Bi achieves about 90% faradaic efficiency for CO at a lower overpotential than the most reported N-doped carbon catalysts. | ||
| Fig. 1 Preparation of the N-doped hierarchical porous carbon. | ||
2. Experimental section
2.1 Synthesis of ZIF8
ZIF8 was synthesized according to a previous report with a slight modification.23 First, 2-methylimidazole (7.0 g) was added to 150 mL methanol with stirring for 10 min. Meanwhile, Zn(NO3)·6H2O (3.5 g) was added to 150 mL methanol with stirring for 10 min. Then, the 150 mL methanol containing Zn (NO3)·6H2O was poured into the 150 mL methanol containing 2-methylimidazole. After stirring for 12 h, the ZIF8 was obtained by centrifugation and washing for three times, followed by drying at 70 °C for 12 h.2.2 Synthesis of ZIF8-950-XBi
Typically, 96 mg BiCl3 was added to 40 ml ethanol with sonication for 20 min. After that, 0.2 g ZIF8 was added to the ethanol solution containing BiCl3 with sonication for 10 min. After stirring for 12 h, ZIF8-4Bi was obtained by centrifugation and washing for three times, followed by drying at 70 °C for 12 h. Subsequently, ZIF8-4Bi was calcined at 950 °C for 2 h at a rate of 3 °C min−1 in Ar atmosphere. The obtained samples are denoted as ZIF8-950-4Bi. ZIF8-950, ZIF8-950-Bi, and ZIF8-950-2Bi were obtained by the same procedure, with only changing the BiCl3 (0, 24 or 48 mg) content.2.3 Synthesis of ZIF8-T-4Bi/ZIF8-T
First, ZIF8-4Bi was obtained with the same procedure as the previous method, and then it was calcined at T for 10 min at a rate of 3 °C min−1 in Ar atmosphere (ZIF8-T-4Bi), where T is the reaction temperature. The synthesis processes of ZIF8-T were the same as ZIF8-T-4Bi, which only replaces ZIF8-4Bi with ZIF8.2.4 Electrode preparation
The catalysts were coated onto both sides of the glass carbon electrode (2 × 1.5 × 2.0 cm2) by the following procedures. 2.0 mg catalysts were dispersed in 320 μL mixed solvent, including 20 μL 5% Nafion solution and 300 μL ethanol by sonicating for 0.5 h. Then, the catalyst inks were dropped onto the glassy carbon electrode with a loading weight of 0.33 mg cm−2.2.5 Materials characterization
The scanning electron microscopy (SEM) images were used to analyze the morphology of the catalysts by a JEOL JSM-6360 with an acceleration voltage of 20 kV. A high-resolution transmission electron microscopy (TEM) pattern was obtained by a JEOL JEM-2000EX (120 kV) microscope. The X-ray diffraction (XRD) patterns were collected by a Rigaku D/Max2500PC diffractometer with Cu Kα radiation (λ = 1.5418 Å). The X-ray photoelectron spectroscopy (XPS) measurements were conducted by an ESCALAB 250Xi spectrometer equipped with a nonmonochromatized Al Kα X-ray source. The pore structures and surface area of the samples were measured by a gas adsorption analyzer (ASAP-2010), and the pore size distributions for all samples were simulated by NLDFT. The micro-Raman spectra were acquired from an InVia Raman microspectrometer (Renishaw, UK) with 532 nm radiation at a laser power of 0.4 mW. The thermogravimetric analysis-differential scanning calorimetry (TG-DSC) characterization of the ZIF8 and ZIF8-4Bi was measured by a TG meter (NETZSCH STA 449 F3) under Ar atmosphere from 25 to 800 °C. The Bi content was measured by inductively coupled plasma mass spectrometry (Perkin Elmer NexION 350X).2.6 Electrochemical measurements
Electrochemical measurements were conducted by a VersaSTAT 3 potentiostat (EG&G Instrument) in a homemade H-type cell. The Pt foil was a counter electrode, and the Ag/AgCl saturated KCl electrode was used as the reference electrode. The prepared electrode was used as a working electrode. The counter electrode was positioned in the anode chamber, while the reference and working electrodes were both placed in the cathode chamber. The cathode chamber contained 160 ml of CO2-saturated 0.5 KHCO3 solution, and the CO2 gas was continually bubbled into the electrolyte at a rate of 25 ml min−1. The anode electrolyte was about 80 ml of 0.5 M KHCO3. The anode and cathode chamber were separated by a piece of Nafion 115 membrane. The potentials of the reference electrode can be converted to the reversible hydrogen electrode (RHE) with the formula ERHE = EAg/AgCl + 0.197 + 0.0591pH. The gas product was detected by gas chromatograph (GC, Shimadzu GC-2014). Nitrogen (99.999%) and argon (99.999%) were used as the carrier gases. Hydrogen gas and CO were quantified by a thermal conductivity detector (TCD) and flame ionization detector (FID), respectively. The aqueous product was quantified by ion chromatography (ICS-1100, Dionex Corporation). The linear sweep voltammetric characterization was measured between 0 V and −1.0 V (vs. RHE) with a scan rate of 10 mV s−1 with a VersaSTAT 3 potentiostat (EG&G Instrument).3. Results and discussion
3.1 Structural characterization
As shown in Fig. S1a and b,† the ZIF8 precursor was successfully synthesized, and has dodecahedral morphology with a size of around 100 nm. The diffraction peaks of the as-prepared ZIF8 are consistent with the XRD spectra of the reported ZIF8 structure.24 The ZIF8 precursor has a high surface area (1157 m2 g−1) and micropore structure with a narrow range of 1.1 nm (Fig. S1c and d†). After mixing with BiCl3, ZIF8 can retain the dodecahedral shape (Fig. S2a–d†). After high temperature pyrolysis of the mixture, the shape of the products is completely different from the precursor with increasing bismuth salts (Fig. 2a and S3†). ZIF8-950-4Bi is composed of many carbon nanoparticles derived from ZIF8, which stack with each other to form three-dimensional porous structures. The element mapping images suggest (Fig. 2b) that only C, N, O and Zn are evenly dispersed on the surface of ZIF8-950-4Bi. In addition, the ICP and XPS results demonstrated that the bismuth salts could be almost completely removed during the high temperature carbonization process (Table S1 and Fig. S5h†). Furthermore, the Zn content rapidly decreases with increasing bismuth salt content, which verifies that the majority of Zn atoms can be efficiently removed by the metal salts (Table S2†). The structure of other samples added with various content of bismuth salt is similar to ZIF8-950-4Bi. However, a more abundant mesoporous structure and ordered, curly graphitic carbon layers can be found with increasing bismuth salt content (Fig. 2c and d and S4a–f†), which can efficiently promote the mass transportation and enhance the electrical conductivity. | ||
| Fig. 2 (a) SEM images of ZIF8-950-4Bi. (b) SEM images of ZIF8-950-4Bi and the corresponding elemental mapping images for C, N, O and Zn. (c) TEM and (d) HRTEM images of ZIF8-950-4Bi. | ||
The chemical composition and electronic states of the catalysts were analysed by X-ray photoelectron spectroscopy (XPS). The XPS spectra revealed the presence of C, N, O and a few Zn on the surface of the samples. Furthermore, with the increase of bismuth salts, the content of the Zn element measured by XPS tends to be consistent with the Zn element measured by EDS (Table S2†), which indicates that the bulk content of Zn reaches the equilibrium state with the surface content of Zn for ZIF8-950-4Bi. The high-revolution N1s XPS results of ZIF8-950-4Bi can be deconvoluted into four peaks at 398.5, 399.3, 401.5 and 401.2 eV, and correspond to pyridinic N, Zn–N, pyrrolic N and graphitic N, respectively, as shown in Fig. 3a.15,25,26 The N1s and Zn2p spectra of other samples are shown in Fig. S5.† The content and ratio of the four types of nitrogen are listed in Table S3.† Although ZIF8-950-4Bi contains a relatively lower content of pyridinic N than that of the control samples, the ratio of Zn–N for ZIF8-950-4Bi is much lower than other samples, which can efficiently reduce the competitive hydrogen evolution reaction. Additionally, the ratio of the graphitic N for ZIF8-950-4Bi obviously increased, further suggesting the higher electrical conductivity. Furthermore, the content of Zn–N bond is about two times that of the Zn species from the elemental analysis of XPS, demonstrating that the coordination numbers of the Zn species in ZIF8-4Bi-950 is about two. The nitrogen adsorption–desorption isotherm of all samples were measured to explore the specific surface area and porous structure, as shown in Fig. 3b. At relatively low pressure, the adsorption curve of the catalysts rapidly increases, indicating the existence of abundant micropores. Furthermore, with the increasing content of metal salts, the obvious hysteresis loop can be found, suggesting the formation of the abundant mesoporous structure in ZIF8-950-4Bi, which is consistent with the TEM results. The results verified that the bismuth salt can efficiently create a mesoporous structure. The pore size distributions of ZIF8-950-4Bi are focused on 0.7 nm, 0.9 nm, 1.1 nm and some mesoporous structure (>2 nm) by density functional theory (Fig. 3c). The surface area and pore volume are summarized in Table S4.† The ZIF8-950-4Bi possesses a high surface area, and abundant microporous and mesoporous structures. The high surface area is conducive to exposing more active sites. The hierarchical porous architectures can promote the diffusion and adsorption of carbon dioxide to the active sites for improving the ERC activity. The Raman spectra of ZIF8-950-4Bi shows obvious D and G bands belonging to the disordered carbon and sp2-bonded graphitic carbon, respectively.27,28 The ID/IG values of ZIF8-950-4Bi are 0.95, which is lower than the control samples of 1.00 and 0.98 for ZIF8-950 and ZIF8-950-Bi, respectively, and the same as ZIF8-950-2Bi (Fig. 3d), indicating a relatively high graphitization degree in accordance with the TEM results.
 | ||
| Fig. 3 (a) The high-resolution N 1s spectrum of ZIF8-950-4Bi. (b) Nitrogen adsorption–desorption isotherm of all the samples. (c) Pore size distributions of all the samples. (d) Raman spectra of all of the samples. | ||
To further clarify the effect of the bismuth salts on the mechanism of structure evolution, a series of ex situ characterization techniques for ZIF8-T and ZIF8-T-4Bi were carried out to track the pyrolysis process. There are mainly two stages for the weight loss of ZIF8-T, as shown in Fig. 4a. The former weight loss is originally from the loss of crystalline water before 550 °C, and the ZIF8 can still maintain the crystal structure, as shown in Fig. 4c. When the temperature rises to over 550 °C, the obvious endothermic process and weight loss are observed from the thermogravimetric analysis-differential scanning calorimetry (TG-DSC). We speculate that the latter stage of weight loss is attributed to the decomposition and carbonization of ZIF8. Additionally, from the XRD results, the crystal structure of ZIF8 completely disappears after 550 °C, and the weak diffraction peak of ZnO appeared at about 600 °C. When the temperature is elevated to over 650 °C, there is only the existence of the C (002) and C (101) faces, indicating that the majority of Zn was evaporated over 650 °C (Fig. 4d). However, with the addition of bismuth salts, the structural evolution of ZIF8-4Bi is extremely different from that of ZIF8. There are mainly three stages for weight loss, as shown in Fig. 4b. The first stage for weight loss is originally from the loss of crystalline water before 350 °C, and ZIF8-4Bi can still maintain the crystal structure (Fig. 4e). Furthermore, the SEM and TEM studies show that ZIF8-4Bi is covered by many nanosheets of BiClO, which is produced by the hydrolysis of BiCl3 (Fig. S6a and b†). When the temperature rises to about 350 °C, the obvious endothermic process and weight loss are observed from TG-DSC. Furthermore, the diffraction peak of Bi metal is appeared. Additionally, from the results of SEM and TEM, we can find many Bi nanoparticles on the surface of ZIF8 (Fig. S6c and S7†). Thus, we speculate that BiCl3 begins melting and decomposing to form the bismuth nanoparticles at about 350 °C, which is accompanied by the seriously structural destruction of ZIF8. Unexpectedly, the diffraction peak of ZnO appeared at about 450 °C, which is much lower than that of ZIF8 without the addition of bismuth salts. Therefore, we conjecture that the Zn–N bond should be destroyed by the BiCl3 melting process, and some unconstrained element Zn reacts with element O from the ZIF8 decomposition to produce ZnO at about 450 °C. Furthermore, the obvious endothermic process is observed at about 450 °C, as shown in Fig. 4b, which may originate from the break of the Zn–N bond. In addition, the crystal structure of ZIF8 completely disappears at about 450 °C. When the temperature rises to over 550 °C, the structure evolution is similar to that of ZIF8. After the temperature rises to about 650 °C, there is only the existence of a diffraction peak of C and bismuth metal, indicating that the majority of Zn evaporated over 650 °C (Fig. 4f). The TEM shows that the Bi nanoparticles completely disappeared when the temperature increased over 750 °C (Fig. S6e†), and the XRD also verifies that the diffraction peak of the Bi nanoparticles has disappeared over 750 °C. When the temperature rises to 950 °C, the obvious mesoporous structure of ZIF8-4Bi appears because of the vaporization of the Bi nanoparticles (Fig. S6f†). Additionally, the DFT calculation further proved that the addition of bismuth can increase the bond length of Zn–N, which is conducive to destroying the Zn–N bond (Fig. S8†). As a conclusion, the bismuth salts not only efficiently destroy the Zn–N bond, and promote the aggregation and vaporization of the Zn species at the relatively low temperature, but also produce an abundant mesoporous structure.
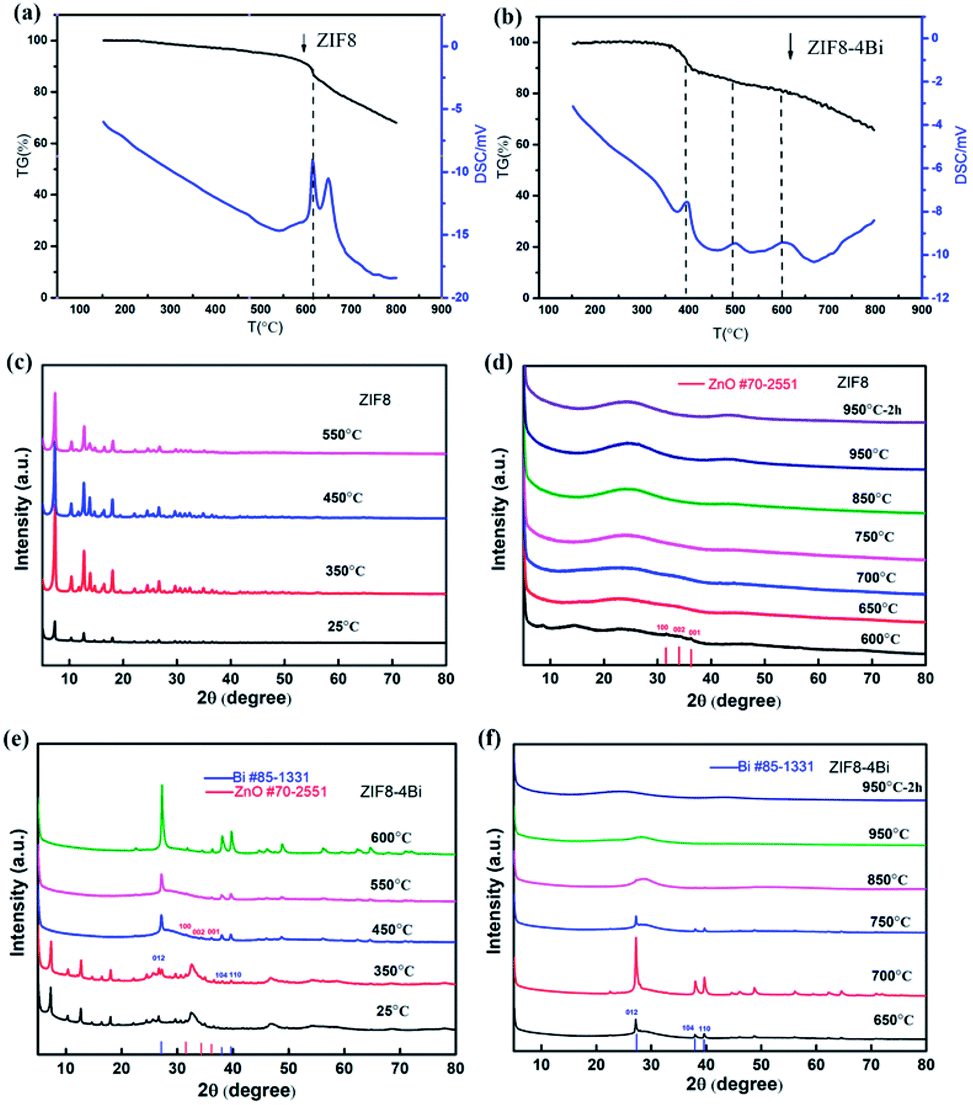 | ||
| Fig. 4 (a and b) The TG-DSC of ZIF8 and ZIF8-4Bi. (c and d) The ex situ XRD of ZIF8. (e and f) The ex situ XRD of ZIF8-4Bi in different calcining temperatures. | ||
3.2 Electrochemical characterization
The electrochemical linear sweep voltammetry (LSV) study was conducted to evaluate the catalytic performance of all samples at the potential range of 0 V to −1.0 V (vs. RHE). As shown in Fig. 5a, the onset potential of ZIF8-950-4Bi is about −0.2 V, which is more positive than that of other samples, indicating much higher electrocatalytic activities for ERC (Fig. S9a–d†). Furthermore, with the increasing content of bismuth salts, the current density rapidly rises, especially that for ZIF8-950-4Bi. The current density of ZIF8-950-4Bi is about 8 times higher than that of ZIF8-950 at −0.6 V vs. RHE, which indicates that the electrocatalytic activity can be efficiently promoted by the addition of the bismuth salts. The high electrocatalytic activity is owing to the high conductive and hierarchical porous structure of ZIF8-950-4Bi. The hierarchical porous architecture can promote the diffusion and adsorption of carbon dioxide to the active sites, and the high electronic conductivity brings the fast electron transfer to form CO2˙−, which can greatly reduce the onset potential and enhance the catalytic activity. We also normalized the current density by ECSA to evaluate the intrinsic activity of the samples (Fig. S10†). The ZIF8-950-4Bi achieved much higher jECSA than those of other samples, implying the much higher intrinsic activity. | ||
| Fig. 5 (a) Linear sweep curves of all samples in CO2-saturated 0.5 M KHCO3. (b) Faradaic efficiencies for CO production at applied potentials on all samples. (c) Tafel plots of all samples. (d) Free energy diagram at −0.11 V (vs. RHE) for the electrochemical reduction of CO2 to CO on different N defects, that is, graphene, graphitic N, pyridinic N, pyrrolic N, pyridinic N adjacent to Zn–N4, Zn–N3V and Zn–N2V2. | ||
The faradaic efficiencies of CO (FECO) for all samples were measured at the potential range from −0.3 V to −1.0 V (vs. RHE). H2 and CO are the major products, which were detected by on-line gas chromatography. As shown in Fig. 5b, with the decrease of the potential, the volcanic-like curve for FECO of all samples can be obtained. Furthermore, with the increase of the metal salts, the maximum faradaic efficiency of CO gradually rises. ZIF8-950-4Bi attains a maximum FECO of 90% at −0.6 V vs. RHE, which is almost two times higher than that of ZIF8-950. In addition, it can maintain high FECO at the wide potential range from −0.5 V to −0.7 V, which indicates that the hydrogen evolution reaction (HER) is efficiently inhibited by the addition of the bismuth salts. Based on the above results, we speculate that ZIF8-950-4Bi possesses the more abundant mesoporous structure, high electrical conductivity and low Zn–Nx content. The high electronic conductivity brings fast electron transfer to form CO2˙−. The abundant mesoporous structure is beneficial to transporting CO2. Additionally, the low Zn content will efficiently inhibit the hydrogen evolution reaction. Therefore, ZIF8-950-4Bi can achieve the superior activity and selectivity for the electrocatalytic CO2 reduction reaction.
In order to further understand the electrodynamics of the CO formation, the Tafel plot of all samples is measured. The Tafel plot for ZIF8-950-4Bi is 82 mV dec−1, which is much lower than that of ZIF8-950, ZIF8-950-Bi and ZIF8-950-2Bi with the Tafel slope of 137, 88 and 90 mV dec−1, respectively (Fig. 5c). These results reveal that ZIF8-950-4Bi exhibits the fastest kinetics for the electrocatalytic CO2 reduction reaction. In addition, the Tafel slope of ZIF8-950-4Bi is close to 59 mV dec−1, indicating the fast single-electron transfer to produce CO2˙−. Therefore, ZIF8-950-4Bi can strongly accelerate the CO2˙− production with a low formation barrier, which can efficiently increase CO formation at a lower overpotential. However, without the addition of bismuth salt, the Tafel slope of ZIF8-950 is close to 118 mV dec−1, indicating that the single-electron transfer is the rate-determining step. Thus, with the decoration of bismuth salt, the activities for the catalysts are rapidly improved.
Bao et al.15 and Zhang et al.20 reported that Zn–Nx/C derived from ZIF8 leads to sluggish activity and selectivity for the electrocatalytic CO2 reduction reaction. Additionally, we prepared N-doped hierarchical porous carbon (ZIF8-950) derived from decorated ZIF8, which also showed low activity and selectivity for the electrocatalytic CO2 reduction reaction (Fig. 5b). Thus, we speculate that the Zn–Nx/C derived from ZIF8 does lead to sluggish activity and selectivity for ERC. Previous research revealed that pyridinic nitrogen is the catalytic active site, which can adsorb and activate carbon dioxide, and the positively charged carbon atoms adjacent to the pyridinic nitrogen can efficiently stabilize CO2˙− intermediates.17,28 Thus, the pyridinic nitrogen can reduce the free energy barriers for *COOH formation.29 DFT calculations based on the computational hydrogen electrode (CHE) model were used to calculate the thermodynamic limiting potential on various N-G catalysts (Fig. S11†). As shown in Fig. 5d, we speculated on the reaction mechanism of electrocatalytic CO2 to CO through two intermediates of *COOH and *CO. The pristine graphene has the highest free energy barrier (>2.0 eV) for the first proton–electron transfer due to the weak bonding of COOH. The free energy barrier for forming *COOH drops dramatically after N-doped graphene, especially the pyridinic N and pyridinic N adjacent to Zn–N4 due to the strong bonding with COOH. However, we find that the pyridinic N adjacent to Zn–N3V and Zn–N2V2 has a much higher energy barrier for forming *COOH than the pyridinic N, which indicates that the coordinated unsaturated Zn–Nx sites tremendously inhibit the activity of the nearby pyridinic N sites. Interestingly, the free energy of the *COOH adsorption at the pyrrolic N site is exergonic. However, the release of *CO from the pyrrolic-N defect needs a high energy barrier of ∼1.1 eV, which is a nonelectrochemical desorption step, irrelevant to applied voltage.28
In this work, although ZIF8-950-4Bi contains a lower content of pyridinic N than that of other control samples, ZIF8-950-4Bi contains a much lower content of coordinated unsaturated Zn–Nx than that of the control samples, which efficiently reduce the shielding effect on the sites near the pyridinic N. Furthermore, the experimental investigations also reveal that the maximum FE and electrocatalytic activities for CO decreases as the Zn–Nx content increases, which further verify that Zn–Nx derived from ZIF8 leads to sluggish activity and selectivity for the electrocatalytic CO2 reduction reaction. Additionally, ZIF8-950-4Bi possesses higher mesoporous volume (Table S4†) and electrical conductivity than those of other samples, which are beneficial to promote the mass transportation and electron transfer. Therefore, ZIF8-950-4Bi achieves lower onset potential and higher selectivity than those of other samples.
The stability of the catalyst also plays an important role in the electrocatalytic CO2 reduction. The durability was first tested at −0.6 V (vs. RHE) for 12.5 h (Fig. S12c†). The current density and faradaic efficiency of the ZIF8-950-4Bi gradually decrease during the test period. After washing the electrode with massive water, we continue to measure the durability of the ZIF8-950-4Bi electrode without changing the other conditions. We unexpectedly discovered that the catalyst could excellently recover the original electrocatalytic property. We speculate that some active sites on the surface of the catalyst may be covered or poisoned by impurity metal ions or reaction intermediates during the long test period, which is consistent with the previous report.30 In our system, the catalyst can be recovered by simple water treatment. Furthermore, the catalyst can maintain the original structure after the long-time test (Fig. S12a and b†), indicating the robust structural stability of the ZIF8-950-4Bi electrode. Additionally, we measured ZIF8-950 with acid etching, which achieves lower selectivity and activity for ERC than that of ZIF8-950-4Bi (Fig. S13†). We speculate that the acid treatment can generally remove some metal particles, while the Zn–Nx coordination aspect is very stable. Thus, it cannot efficiently reduce the shielding effect on the sites near pyridinic N.
4. Conclusions
In summary, we report a facile synthesis method to obtain a highly active and selective N-doped porous carbon catalyst, which directly carbonizes the ZIF8 precursor decorated by the low boiling metal salts (BiCl3). The bismuth salts can efficiently adjust the conductivity, porous structure, surface area and Zn content for ZIF8, which not only achieve the hierarchical porous structure and high surface area, but also promote the aggregation and vaporization of the Zn species, and enhance the electronic conductivity. The high surface area is conducive to exposing more active sites. The hierarchical porous architectures can promote the diffusion and adsorption of carbon dioxide to the active sites, and the high electronic conductivity brings the fast electron transferring to form CO2˙−. Additionally, the low content of Zn–Nx will efficiently inhibit the hydrogen evolution reaction. Therefore, ZIF8-950-4Bi exhibits the fastest kinetics and high selectivity for the electrocatalytic CO2 reduction reaction, which reached much higher faradaic efficiency (90%) and lower onset potential for CO than that of other control samples. Furthermore, density functional theory calculations suggest that the coordinated unsaturated Zn–Nx sites tremendously inhibit the activity of the nearby pyridinic N sites, which play an important role in the catalytic performance. Our results pave a facile way to develop high performance catalysts with a tunable porous structure, surface area, conductive and N species for the electrocatalytic CO2 reduction fields.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the financial grants from the National Natural Science Foundation of China (No. 21577141).Notes and references
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478–487 CrossRef CAS.
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Adv. Mater., 2016, 28, 3423–3452 CrossRef CAS.
- W. Ju, F. Jiang, H. Ma, Z. Pan, Y. B. Zhao, F. Pagani, D. Rentsch, J. Wang and C. Battaglia, Adv. Energy Mater., 2019, 9, 1901514 CrossRef.
- Z.-L. Wang, C. Li and Y. Yamauchi, Nano Today, 2016, 11, 373–391 CrossRef CAS.
- Z. Chang, S. Huo, W. Zhang, J. Fang and H. Wang, J. Phys. Chem. C, 2017, 121, 11368–11379 CrossRef CAS.
- C. W. Lee, J. S. Hong, K. D. Yang, K. Jin, J. H. Lee, H.-Y. Ahn, H. Seo, N.-E. Sung and K. T. Nam, ACS Catal., 2018, 8, 931–937 CrossRef CAS.
- F. Li, L. Chen, G. P. Knowles, D. R. MacFarlane and J. Zhang, Angew. Chem., Int. Ed. Engl., 2017, 56, 505–509 CrossRef CAS.
- B. Zhang, J. Zhang, J. Shi, D. Tan, L. Liu, F. Zhang, C. Lu, Z. Su, X. Tan, X. Cheng, B. Han, L. Zheng and J. Zhang, Nat. Commun., 2019, 10, 2980 CrossRef.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- C. S. Le Duff, M. J. Lawrence and P. Rodriguez, Angew. Chem., Int. Ed. Engl., 2017, 56, 12919–12924 CrossRef CAS.
- Y. Chen, C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 19969–19972 CrossRef CAS.
- Y.-C. Hsieh, S. D. Senanayake, Y. Zhang, W. Xu and D. E. Polyansky, ACS Catal., 2015, 5, 5349–5356 CrossRef CAS.
- Y. Cheng, S. Zhao, H. Li, S. He, J.-P. Veder, B. Johannessen, J. Xiao, S. Lu, J. Pan, M. F. Chisholm, S.-Z. Yang, C. Liu, J. G. Chen and S. P. Jiang, Appl. Catal., B, 2019, 243, 294–303 CrossRef CAS.
- X.-M. Hu, H. H. Hval, E. T. Bjerglund, K. J. Dalgaard, M. R. Madsen, M.-M. Pohl, E. Welter, P. Lamagni, K. B. Buhl, M. Bremholm, M. Beller, S. U. Pedersen, T. Skrydstrup and K. Daasbjerg, ACS Catal., 2018, 8, 6255–6264 CrossRef CAS.
- C. Yan, H. Li, Y. Ye, H. Wu, F. Cai, R. Si, J. Xiao, S. Miao, S. Xie, F. Yang, Y. Li, G. Wang and X. Bao, Energy Environ. Sci., 2018, 11, 1204–1210 RSC.
- W. Li, N. Fechler and T. J. Bandosz, Appl. Catal., B, 2018, 234, 1–9 CrossRef CAS.
- P. Yao, Y. Qiu, T. Zhang, P. Su, X. Li and H. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 5249–5255 CrossRef CAS.
- N. Cheng, L. Ren, X. Xu, Y. Du and S. X. Dou, Adv. Energy Mater., 2018, 8, 1801275 Search PubMed.
- H. Yang, S. J. Bradley, A. Chan, G. I. Waterhouse, T. Nann, P. E. Kruger and S. G. Telfer, J. Am. Chem. Soc., 2016, 138, 11872–11881 CrossRef CAS.
- P. Lu, Y. Yang, J. Yao, M. Wang, S. Dipazir, M. Yuan, J. Zhang, X. Wang, Z. Xie and G. Zhang, Appl. Catal., B, 2019, 241, 113–119 CrossRef CAS.
- J. Gu, C. S. Hsu, L. C. Bai, H. M. Chen and X. L. Hu, Science, 2019, 364, 1091–1094 CrossRef CAS.
- J. Wang, W. Liu, G. Luo, Z. Li, C. Zhao, H. Zhang, M. Zhu, Q. Xu, X. Wang, C. Zhao, Y. Qu, Z. Yang, T. Yao, Y. Li, Y. Lin, Y. Wu and Y. Li, Energy Environ. Sci., 2018, 11, 3375–3379 RSC.
- E. Luo, H. Zhang, X. Wang, L. Gao, L. Gong, T. Zhao, Z. Jin, J. Ge, Z. Jiang, C. Liu and W. Xing, Angew. Chem., Int. Ed. Engl., 2019, 58, 12469–12475 CrossRef CAS.
- J. Wang, X. Luo, C. Young, J. Kim, Y. V. Kaneti, J. You, Y.-M. Kang, Y. Yamauchi and K. C. W. Wu, Chem. Mater., 2018, 30, 4401–4408 CrossRef CAS.
- Q. Lai, L. Zheng, Y. Liang, J. He, J. Zhao and J. Chen, ACS Catal., 2017, 7, 1655–1663 CrossRef CAS.
- N. R. Sahraie, U. I. Kramm, J. Steinberg, Y. Zhang, A. Thomas, T. Reier, J. P. Paraknowitsch and P. Strasser, Nat. Commun., 2015, 6, 8618 CrossRef CAS.
- Q. Lai, J. Zheng, Z. Tang, D. Bi, J. Zhao and Y. Liang, Angew. Chem., Int. Ed. Engl., 2020, 59, 11999–12006 CrossRef CAS.
- J. Wu, M. Liu, P. P. Sharma, R. M. Yadav, L. Ma, Y. Yang, X. Zou, X. D. Zhou, R. Vajtai, B. I. Yakobson, J. Lou and P. M. Ajayan, Nano Lett., 2016, 16, 466–470 CrossRef CAS.
- P. P. Sharma, J. Wu, R. M. Yadav, M. Liu, C. J. Wright, C. S. Tiwary, B. I. Yakobson, J. Lou, P. M. Ajayan and X. D. Zhou, Angew. Chem., Int. Ed. Engl., 2015, 54, 13701–13705 CrossRef CAS.
- Y.-L. Qiu, H.-X. Zhong, T.-T. Zhang, W.-B. Xu, X.-F. Li and H.-M. Zhang, ACS Catal., 2017, 7, 6302–6310 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S13 and Tables S1–S4. See DOI: 10.1039/d0ta09293b |
| This journal is © The Royal Society of Chemistry 2021 |