Microstructural development of the La0.5Li0.5TiO3 lithium ion conductor processed by the laser floating zone (LFZ) method†
Alejandro Várez*a, María L. Sanjuánb, María A. Lagunab, Jose I. Peñac, Jesús Sanzd and German F. de la Fuentec
aDepartamento de Materiales, Universidad Carlos III de Madrid, Avda. Universidad, 30, 28911, Leganés, Spain. E-mail: alvar@ing.uc3m.es
bInstituto de Ciencia de Materiales de
Aragón, CSIC-Universidad de Zaragoza, 50009, Zaragoza, Spain
cInstituto de Ciencia de Materiales de
Aragón, CSIC-Universidad de Zaragoza, 50015, Zaragoza, Spain
dInstituto de Ciencia de Materiales de
Madrid, CSIC, 28049, Cantoblanco, Spain
First published on 12th October 2000
Abstract
La2/3 − xLi3xTiO3 (0 < x < 0.17) compounds have reached great prominence in the last few years due to their high lithium ion conductivity. They present a perovskite-related structure (ABO3) with A-cation deficiency. However, neither the phase diagram nor the solidification processes are well known, which makes the growth of single crystals difficult. In this vein, we have processed La0.5Li0.5TiO3 polycrystalline powder by the LFZ method and studied the processed material by means of SEM, EDS, XRD and micro-Raman techniques. Three phases have been detected: the major phase (A) is a La–Li–Ti–O perovskite with 3x < 0.5, that forms large single crystal grains. Phase B is rutile TiO2 and appears as small precipitates surrounding phase A. Finally, we observe eutectic mixtures of phase A and a third phase (C) containing Ti and O in a ratio lower than 1∶2. Phase C is identified as a Li–Ti oxide, probably Li2Ti3O7.
Introduction
Li ion-conducting materials have been widely studied in the last few years because of their potential applications as solid electrolytes in high energy batteries and other electrochemical devices. Nowadays one of the best Li ion conductors at room temperature is found in oxides with a perovskite related structure1–5 with the general formula La2/3 − xLi3xTiO3. Although these materials present a relatively wide range of Li solutions the limits are under discussion. Robertson et al.3 have published a tentative phase diagram where the minimum and the maximum solubilities of Li were established as x = 0.04 and 0.14 respectively. Fourquet et al.4 found a slightly narrower composition range (0.06 ≤ x ≤ 0.14), while Kawai and Kuwano2 expand the upper limit up to x = 0.15. These small differences could be attributed to the slightly different synthesis conditions employed. In all the cases mentioned, the structural model proposed for samples at room temperature was a perovskite doubled along the c-axis with tetragonal symmetry.Recently, we found that the compositional range could be enlarged in both
limits (0.03 ≤ x ≤ 0.167) by heating
up to 1350![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C.6,7 We have also
observed, by means of X-ray diffraction experiments, a change of symmetry
from tetragonal to orthorhombic of the doubled unit cell when the Li content
decreases below x = 0.06. These structural modifications
are associated with cation vacancies ordering along the c-axis
into two alternating A sites of the perovskite (ABO3). This ordering
disappears gradually with the lithium content and with the temperature. When
the Li-rich end member (x = 0.167) is quenched from
1300°C into liquid nitrogen, a primitive cubic cell, (ap × ap × ap), with a unique A site is detected by X-ray diffraction
experiments.6 However, the neutron diffraction
(ND) pattern of this sample was indexed in a hexagonal unit cell (space group R-3c).
Rietveld refinement of ND data indicated that La ions occupy the A site, while
Li cations are fourfold oxygen coordinated at the centre of square faces of
the cubic perovskite.8 As a consequence of
this fact the number of vacancies of perovskite is higher than that deduced
in former structural analyses where La and Li occupied A sites.
°C.6,7 We have also
observed, by means of X-ray diffraction experiments, a change of symmetry
from tetragonal to orthorhombic of the doubled unit cell when the Li content
decreases below x = 0.06. These structural modifications
are associated with cation vacancies ordering along the c-axis
into two alternating A sites of the perovskite (ABO3). This ordering
disappears gradually with the lithium content and with the temperature. When
the Li-rich end member (x = 0.167) is quenched from
1300°C into liquid nitrogen, a primitive cubic cell, (ap × ap × ap), with a unique A site is detected by X-ray diffraction
experiments.6 However, the neutron diffraction
(ND) pattern of this sample was indexed in a hexagonal unit cell (space group R-3c).
Rietveld refinement of ND data indicated that La ions occupy the A site, while
Li cations are fourfold oxygen coordinated at the centre of square faces of
the cubic perovskite.8 As a consequence of
this fact the number of vacancies of perovskite is higher than that deduced
in former structural analyses where La and Li occupied A sites.
The aim of the present work has been to grow monocrystalline fibers of the fast ionic conductor La2/3 − xLi3xTiO3 (x = 0.167) to study the phase diagram under directional solidification.
Experimental
1 Crystal growth
°C for 6 hours.
In order to avoid lithium losses the heating rate used during the synthesis
of the La0.5Li0.5TiO3 powders and during
the sintering of the precursor was 1°C min−1.For subsequent characterisation and convenience in handling, the fibers were mounted in a matrix of thermosetting polymer that was cured under pressure and temperature simultaneously. Afterwards the mounted fiber was cut in a transverse and a longitudinal direction. Finally it was subjected to grinding and polishing with diamond paste.
2 Characterisation
X-Ray diffraction (XRD) patterns were recorded with CuKα radiation in an X'Pert Philips diffractometer, with (θ/2θ) Bragg–Brentano geometry, equipped with a curved graphite monochromator. The mounted fiber was located into a multipurpose stage that enables the XRD analysis of large three-dimensional samples. Data were taken with a 0.5° divergence slit, a receiving slit of 0.01° and a set of soller slits with an axial divergence of 1°. The 2θ range analysed was 10–90°, with a step scan of 0.02 and a counting time of 5 s for each step. The working conditions were 40 kV and 55 mA.A Philips XL30 scanning electron microscope equipped with a backscattered (BSE) and an energy dispersive X-ray (EDAX) detector was employed to study the microstructure and composition of the fibers. The microscope almost always operated in the backscattered mode between 15–20 kV, and all the EDAX analyses were registered at the same conditions of voltage (20 kV), take-off angle and live time. For each different phase observed on the BSE images, four independent analyses were carried out. Stoichiometric samples of Li2TiO3, Li2Ti3O7 and La2Ti2O7 were used as the standards for Li, Ti and La determination.
Raman spectroscopy experiments were performed at room temperature in a Dilor XY spectrometer with a diode array multichannel detector. Light from a coherent Ar+ laser at 514.5 nm was focused onto the sample through a ×50 or a ×100 microscope objective lens, providing a maximum spatial resolution of about 2 µm. A confocal diaphragm was sometimes used to gain in-depth resolution. The light power at the sample was 10 mW and the spectral resolution was typically 3 cm−1.
Results
a X-Ray diffraction experiments
Fig. 1 shows the X-ray diffraction (XRD) patterns of the longitudinal and cross-section of the fiber as well as the starting polycrystalline material. In the case of the longitudinal section of the fiber, most of the diffraction peaks are coincident in position with those of the polycrystalline material, while the intensities are quite different. This is due to the preferential orientation of the samples during the directional solidification. This fact is more appreciable in the XRD pattern of the cross-section of the fiber, where the peaks corresponding to the (112) and (224) planes of the tetragonal perovskite are the most important. Likewise extra diffraction peaks (2θ ≈ 26.5 and 38.8) are observed that we attribute to the secondary phases detected on the BSE micrograph (see below). | ||
| Fig. 1 Experimental X-ray diffraction patterns for: (a) starting material, and (b) longitudinal and (c) cross-sections of the mounted grown fiber. The different intensities of the peaks are attributed to the anisotropy of the grown fiber. | ||
b Microstructural study
Fig. 2 shows a backscattered electron (BSE) micrograph of a longitudinal section of the grown fiber. Three different areas can be distinguished: a) the quenched or frozen zone (right), that corresponds to the last liquid part of the material just before switching off the laser, b) the fiber grown by directional solidification (left) and c) the solidification interface (liquid phase interface during the fiber growth). | ||
| Fig. 2 Scanning electron micrograph in the backscattered (BSE) mode of a longitudinal section of the mounted fiber. Three different zones can be distiguished: frozen zone (right), grown fiber (left) and the narrow interface of solidification (middle). | ||
In the case of the grown fiber area, we can see large grains (white contrast) oriented along the growth direction. Surrounding these columnar grains, dark contrast areas can be distinguished which have been assigned to secondary phases. A magnification of the interface zone (Fig. 3) shows a eutectic morphology, where white and black layers alternate. The lamellar spacing in this case is very small indicating that the solidification rate was rather high.
 | ||
| Fig. 3 Magnification of the black contrast area that appears in the interface of solidification of Fig. 2. | ||
The frozen zone presents a branch-out morphology with a very high degree of disorder where different phases (areas with black and white contrast on the BSE image) coexist indicating a phase separation. The presence of intermixed phases provides better mechanical properties. In fact, in this area no cracks, arising from the polishing process, are detected. At higher magnification (Fig. 4) we can see how the primary crystals (white contrast) grow from the liquid in a dendritic manner. Solidification starts at a nucleus in the interface area and quickly grows parallel to the fiber. Subsequently, secondary arms grow perpendicular, in some cases, and at 45°, in others, producing dendrite-type crystals. Between these white contrast branches, the residual liquid (dark contrast) with a composition different from that of the primary dendrites, has been solidified. It is worth noting that the dark contrast area is more abundant in the frozen than in the grown fiber areas.
 | ||
| Fig. 4 Magnification of a zone of the frozen part in Fig. 2, showing the microstructure of solidifying. | ||
In Fig. 5a a cross-section of the grown fiber is presented. At low magnification “equiaxial” grains (white contrast) can be observed. These correspond to the columnar grains detected on the longitudinal section. The grain boundaries present a black contrast that, at higher magnification (Fig. 5b), shows a eutectic aspect. We can also distinguish grey contrast areas appearing like a segregated phase.
 | ||
| Fig. 5 Transverse section of the fiber at low (a) and high (b) magnification. Three different contrasts, corresponding to three phases, can be observed at higher magnification: grey (segregated grains of titanium dioxide), white (La–Li–Ti–O perovskite) and black (lithium titanium oxide). The last two phases solidify in a eutectic manner. | ||
c X-Ray microanalysis
In order to know the composition of the different phases emerging during the solidification, we have performed microanalyses in the different areas by means of EDS. In Table 1, we present the elements detected and their proportions.| Area | Phase | Elements | Ratio | Remarks |
|---|---|---|---|---|
| Frozen | White | La∶Ti∶O | 0.7∶1∶3.1 | |
| Grey | Ti∶O | 1∶1.8 | ||
| Black | (Ti + Al)∶O | 1∶2.1 | 2–3 wt% Al | |
| Interface | Lamellar | La∶Ti∶O | 0.2∶1∶2.6 | Mixture of white and black phases |
| Grown fiber | White | La∶Ti∶O | 0.7∶1∶3.1 | |
| Grey | Ti∶O | 1∶1.9 | ||
| Black | (Ti + Al)∶O | 1∶2.3 | 2–3 wt% Al |
Taking into account the limitations of the technique, related to the detection of light elements and the absorption problem of La, we could deduce the following:
- The primary crystals (white contrast) correspond to the major phase of the grown fiber and also participate as a eutectic constituent. All the crystals present an excess of La in relation to the starting material.
- The grey contrast phase presents a stoichiometry close to that of titanium dioxide.
- The composition of the black contrast area is similar to that of the grey contrast phase, however the amount of oxygen is slightly higher and always 2–3 wt% of Al, coming from the crucible, is present.
d Micro-Raman spectroscopy experiments
We have measured the Raman spectra of the different phases present in the fiber. The experiments were performed in parallel to scanning electron microscopy analysis and led to the following observations.In Fig. 6A Raman spectra corresponding to the major phase (white contrast) of the processed fiber are presented. A comparison with those spectra of the La2/3 − xLi3xTiO3 series (Fig. 6B) shows that the lithium content of the main phase is 0.1 < 3x < 0.3. much smaller than the starting value 3x = 0.5. On the other hand, it must be noted that, within a single grain, the spectrum changed on going from the middle to the outside, denoting a possible increase of lithium content in the perovskite when approaching the single crystal boundary (note the resemblance of spectra labelled (c) in Figs. 6A and 6B).
 | ||
| Fig. 6 (A) Raman spectra taken along a single grain of the white contrast phase, in the transverse section of the fiber. The experimental conditions were exactly the same in all three cases but from (a) to (c) spectra were taken increasingly close to the dark contrast inclusions. They are interpreted as due to La2/3 − xLi3xTiO3 perovskite with increasing lithium content. (B) Room temparature Raman spectra of La2/3 − xLi3xTiO3 ceramics with varying lithium content: (a) 3x = 0.12; (b) 3x = 0.18; (c) 3x = 0.5. All three samples were prepared under similar experimental conditions. | ||
Each individual grain proved to be a single crystal with clear polarisation properties in the Raman spectra. Within a given grain of the cross-section, the orientational dependence of the spectrum is the one expected for rotation in the (112) plane. This is consistent with the XRD result that in this section the majority of grains present a (112) plane orientation.
At least three different spectra were found in the dark contrast inclusions appearing between the major phase grains. The grey contrast phase, with a Ti∶O proportion close to 2, was unambiguously identified as rutile TiO211 (Fig. 7a). In the quenched zone the spectrum of TiO2 anatase was also found12 (Fig. 7b). The black contrast phase gave a weakly active broad spectrum, frequently superposed to peaks of the La–Li–Ti–O phase or of TiO2. With the use of a confocal diaphragm we have been able to isolate the spectrum of the pure dark contrast phase (see Fig. 8A) and identify it as that of a Li–Ti oxide. A comparison with reference spectra shown in Fig. 8B suggests that it may be Li2Ti3O7 with ramsdellite structure. However, the appearance of weak reflections in the XRD patterns close to those of the high temperature phase of Li2TiO313 adds the possibility of having this Raman inactive or weakly active phase in the fiber.
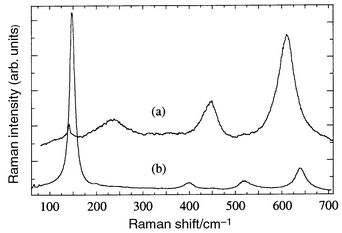 | ||
| Fig. 7 Spectra of TiO2 rutile (a) and anatase (b). Rutile was found either in the quenched or grown sections of the fiber and corresponds to the grey contrast phase of microanalysis; anatase was found only in the quenched region. | ||
 | ||
| Fig. 8 (A) Spectra of the dark contrast phase taken at two different points of the transverse section. Spectra (a) and (b) correspond to parallel and crossed polarisation measurements in an isolated dark inclusion and (c) was taken in a parallel configuration in the dark contrast area at the left of the inclusion seen in Fig. 5b. (B) Raman spectra of ceramic samples of slowly cooled (a,c,d) and quenched (b) Li2Ti3O7. Spectra (c) and (d) were taken in crossed and parallel configurations, respectively, on the same point of the sample, while (a) was taken on a different point. Quenched Li2Ti3O7 gave spectra similar to (b) throughout the whole sample. | ||
In the eutectic regions formed by white and black contrast phases the spectrum was a superposition of those of the white and black contrast phases, La–Li–Ti–O and Li–Ti–O (see Fig. 9). A comparison of this spectrum with those of ceramic powders of the same system (see Fig. 6B) allows us to conclude that the La–Li–Ti oxide in the eutectic precipitates presents a higher degree of disorder and lithium content than the same phase forming the large white contrast grains.
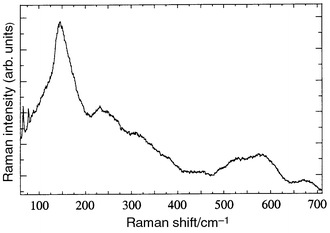 | ||
| Fig. 9 Spectrum taken in the eutectic region at the centre of the inclusion seen in Fig. 5b, close to the point where spectrum (c) of Fig. 8 was measured. It can be explained as a superposition of the spectra from the white and dark contrast phases. | ||
Discussion
Scanning electron microscopy experiments show that monocrystalline fibers can be grown by the LFZ technique. Fibers are formed by relatively large rod-like single crystals (0.5–1 mm in length × 0.2–0.5 mm in diameter) of the major perovskite phase aligned parallel to the growth direction. The lithium content of these crystals is lower than the one of the precursor and they have been grown from a liquid phase.During the processing of the fiber, TiO2 is segregated from the liquid, appearing as a proeutectic precipitate, and some Li2O is probably lost by volatilisation. At the same time, the lithium and titanium content of the liquid increases up to the eutectic composition. This liquid then freezes surrounding the single crystals of the major phase to form a eutectic mixture. From X-ray microanalysis and Raman spectroscopy results, described above, we identified that a Li–Ti oxide, probably Li2Ti3O7 (black contrast in BSE images) and the major phase lithium perovskite (white contrast) form the eutectic microstructure. In this sense we can propose the following solidification sequence:
 | (1) |
 | (2) |
The suggested solidification process, shown in Fig. 10,
is partially in consonance with the results of Robertson et al.3 These authors studied the Li2TiO3–La2/3TiO3
pseudo-binary phase diagram, from samples prepared by solid-state reactions
and pointed out the presence of a eutectic (A + Li2TiO3)
mixture, where A corresponds to a cubic phase with a composition close to
La0.525Li0.425TiO3. In our results, Raman
spectra arising from the white phase of the eutectic microstructure are quite
similar to those of La–Li–Ti–O ceramic powders with high
lithium content. However, the eutectic temperatures predicted by those authors
(1275 ± 25°C) are too low. In fact, we have
never found any sign of a liquid phase during the sintering of the precursor
at 1350°C, and we have increased the temperature up to 1700 ± 50°C
in order to melt the rod-like precursor and grow the fiber. The reason
is probably that the eutectic composition is considerably shifted toward the
lithium rich end, midway between Li2TiO3 and La0.5Li0.5TiO3,
as can be deduced from the BSE images.
 | ||
| Fig. 10 Schematic representation of the developed microstructure before and after fiber growth. | ||
As we have mentioned before the lithium content of the liquid phase increases during the processing of the fiber, as can be deduced from the fact that the black contrast area (lanthanum free), detected in the BSE micrographs, is more abundant in the frozen area than in the grown fiber. The presence of a high amount of lithium in the liquid reduces the solidification temperature operating as a flux.
On the other hand, the presence of TiO2 anatase in the frozen area indicates that this phase is stabilised at higher temperature and the rapid cooling retains that structure.
Another important aspect is the anisotropy of the fibers. This fact can be deduced from the big difference between the transverse and longitudinal XRD patterns of the fibers. As we have mentioned before, in the first case, the most intense peaks correspond to the (112) and (224) planes of the tetragonal cell and the (111) and (222) of the cubic perovskite. These planes correspond to the AO3 close packed layers of the perovskite. This fact allows us to explain the morphology of the grain boundary in Fig. 5a, where angles of 120° are mostly found.
The aluminium detected in the analysis of the dark contrast phase is attributed to the crucible used in the synthesis of the precursor. The presence of this element in perovskite single-crystals is not discarded and might explain some minor frequency shifts observed when comparing the present Raman spectra with those of the La2/3 − xLi3x + yTi1 − yAlyO3 ceramics. However, the high atomic weight of lanthanum prevents the detection of aluminium in the above phase by the EDS technique. We have never detected Al in rutile TiO2, probably due to the lack of solubility of this cation in the rutile network, while Al is easily soluble in the Li2TiO3 and Li2Ti3O7 structures.14
Conclusions
The laser floating zone (LFZ) technique has been successfully employed to grow fibers of the La0.5Li0.5TiO3 fast ionic conductor. The microstructure of these processed materials presents relatively large single crystals of a lithium deficient Li–La–Ti–O perovskite. In the boundary of these large crystals, signs of a liquid phase have been observed where a eutectic reaction takes place. The eutectic components have been identified as lithium rich perovskite and a Li–Ti oxide, probably Li2Ti3O7.The obtained fibers present a strong anisotropy since they grow parallel to the [111] direction of the single-cubic perovskite [close packed layer (LaO3) stacking direction]. Taking into account that the growth of single crystals of these materials has not been successfully developed, this technology opens new possibilities for the characterisation of this kind of compound where the mechanism of the high lithium ion conductivity has not yet been established.
References
- Y. Inaguma, C. Liquan, M. Itoh, T. Nakamura, T. Uchida, H. Ikuta and M. Wakihara, Solid State Commun., 1003, 86, 689 Search PubMed.
- H. Kawai and J. Kuwano, J. Electrochem. Soc., 1994, 141, L78 CAS.
- A. D. Robertson, S. García-Martín, A. Coats and A. R. West, J. Mater. Chem., 1995, 5, 1405 RSC.
- J. L. Fourquet, H. Duroy and M. P. Crosnier-López, J. Solid State Chem., 1996, 127, 283 CrossRef CAS.
- C. Leon, M. L. Lucia, J. Santamaría, M. A. París, J. Sanz and A. Várez, Phys. Rev. B, 1996, 54, 183.
- J. Ibarra, A. Várez, C. León, J. Santamaría, L. M. Torres-Martínez and J. Sanz, Solid State Ionics, 2000, in press. Search PubMed.
- M. A. París, J. Sanz, C. León, J. Santamaría, J. Ibarra and A. Várez, Chem. Mater, 2000, 12, 1694 CrossRef CAS.
- J. A. Alonso, J. Sanz, J. Santamaría, C. León, A. Várez and M. T. Fernández-Díaz, Angew. Chem., Int. Ed., 2000, 39, 619 CrossRef CAS.
- A. Várez, F. García-Alvarado, E. Morán and M. A. Alario-Franco, J. Solid State Chem., 1995, 118, 78 CrossRef CAS.
- G. F. de la Fuente, J. C. Diez, L. A. Angurel, J. I. Peña, A. Sotelo and R. Navarro, Adv. Mater., 1995, 8, 853.
- G. A. Samara and P. S. Peercy, Phys. Rev. B, 1973, 7, 1131 CrossRef CAS.
- T. Ohsaka, F. Izumi and Y. Fujiki, J. Raman Spectrosc., 1978, 7, 321.
- E. Kordes, Fortschr. Mineral., 1933, 18, 27 Search PubMed.
- K. H. Kim and F. A. Hummel, J. Am. Ceram. Soc., 1960, 43, 611 Search PubMed.
Footnote |
| † Basis of a presentation given at Materials Discussion No. 3, 26–29 September, 2000, University of Cambridge, UK. |
| This journal is © The Royal Society of Chemistry 2001 |