Molecular access to intracrystalline tunnels of sepiolite†
Eduardo Ruiz-Hitzky
Instituto de Ciencia de Materiales de
Madrid, CSIC, Cantoblanco, 28049, Madrid, Spain. E-mail: eduardo@icmm.csic.es
First published on UnassignedUnassigned3rd October 2000
Abstract
The availability of the structural micropores in sepiolite for the access of molecular or ionic entities is an old and controversial subject, which is revised in this work in the light of the most recent experimental data obtained in our research group. In this way evidence of the accessibility of molecules, voluminous ions and polymeric species to the intracrystalline tunnels of the mineral is presented on the bases of the application of several techniques to the study of the mineral adsorption properties.
Introduction
The access of molecular entities to intracrystalline cavities of solid materials is a conventional topic which is currently the subject of increasing interest in view of novel applications based on such behaviour.1 Thus, new synthetic molecular sieves based on zeolite type topologies (zeotypes) have been developed in the last two decades, in the search for nanoporous solids containing large structural cavities which will allow the access of more voluminous molecules.2 More recently, the discovery by Beck and co-workers of a family of well organised mesoporous solids, MCM-41 silica consisting of tubular SiO2 of about 40 Å diameter, has resulted in great progress in the field of nanoporous materials.3 The controlled access of reagents to the internal cavities of these mesoporous solids is the basis for the preparation of selectively functionalised MCM-41 organic–inorganic derivatives with anisotropic topochemical behaviour.4 Among other novel porous materials, those based on pillared layered solids, e.g. smectite silicates incorporating silica for permanent layer separation5 or layered titanates containing alumina pillars,6 are being currently reported in view of their selective surface properties. However, the ability of organic species, from small molecules to certain polymers, to penetrate into crystalline solids of different topology and structural organization is well known.7 In this way, 2D matrices, such as certain layered silicates (i.e., smectites), transition-metal oxides (i.e., V2O5 xerogel), oxyhalides (i.e., FeOCl) and dichalcogenides (i.e., TaS2), as well as 3D matrices like the zeolite and ALPO families, act as host solids for organic species. Nanocomposite materials can be prepared and designed by the combination of such inorganic matrices with organic species interacting at the molecular level.8 Layered solids impose a 2D arrangement of the intercalated species, whereas solids of 3D topology induce cluster formation and, in some cases, one-dimensional alignment of the molecules remaining organised into the intracrystalline cavities.8a,9Very important consequences derive from these behaviours: see, for instance, the use of zeolites in shape selective catalysis2 or the role of more sophisticated nanocomposite materials based on the inclusion of dyes, metal clusters, etc. in the development of new devices for optical, electronic and other applications.8a,b,9c An example is, for instance, the possibility of the formation of intracrystalline fibrils of conductive polymers (i.e., polyaniline) using the MCM-41 mesoporous solids10 and other porous materials.11
Amongst the porous microcrystalline solids with similar zeolite 3D topology,
we can include lesser known natural silicates such as sepiolite which, however,
may be of enormous industrial importance as a raw material, especially owing
to its adsorbent properties.12 Sepiolite is
a microcrystalline hydrated magnesium silicate with Si12O30Mg8(OH,F)4(H2O)4·8H2O as the unit cell formula,13
showing a microfibrous morphology with particle size in the 2–10 µm
length range. Structurally it is formed by an alternation of blocks and cavities (tunnels)
that grow up in the fibre direction (c-axis)
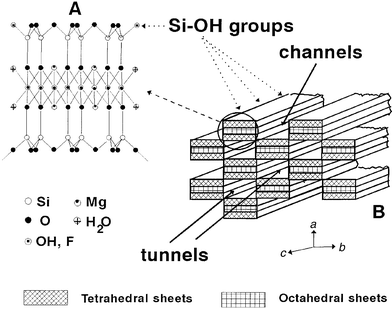
(Fig. 1). Each structural block is composed
of two tetrahedral silica sheets sandwiching a central sheet of magnesium
oxide–hydroxide. Owing to the discontinuity of the silica sheets, silanol
groups (Si–OH) are present on the “external surface”
of the silicate particles.14 These groups
are located at the edges of the channels
(i.e. those
tunnels acceding to the external surface of the silicate) and are directly
accessible to reagents allowing the preparation of organic–inorganic
materials derived from sepiolite containing different surface organic functions.15 The dimensions13a
of the cross-section of sepiolite tunnels are about 11 × 4 Å2.
They are filled by two types of water molecules: i)
co-ordinated
water molecules which are bonded to Mg2+ ions located
at the edges of octahedral sheets, and ii)
zeoliticwater,
associated by hydrogen bonding to the former. This latter type of water molecules
is easily removed by exposure to vacuum or by thermal treatment at about 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C,
whereas the former needs more drastic conditions (>350°C,
dynamic vacuum) resulting in the complete dehydration of the silicate.
The loss of these co-ordinated water molecules causes folding of the structure
and the disappearance of the tunnels in agreement with both XRD patterns and
IR spectroscopic results.14
°C,
whereas the former needs more drastic conditions (>350°C,
dynamic vacuum) resulting in the complete dehydration of the silicate.
The loss of these co-ordinated water molecules causes folding of the structure
and the disappearance of the tunnels in agreement with both XRD patterns and
IR spectroscopic results.14
 | ||
| Fig. 1 Schematic model representing the sepiolite structure. | ||
HRTEM confirms the structural arrangement of tunnels belonging to sepiolite as illustrated in Fig. 2 (adapted from ref. 16) in good agreement with XRD data, but the adsorption properties related to these microporous cavities are as yet ambiguous.17 It is generally admitted that some small polar molecules such as ammonia, methanol, acetone and ethylene glycol can access these intracrystalline cavities replacing water molecules, as evidenced by different techniques including IR spectroscopy.18 The tunnels may also be available to several gases and vapours after elimination of the zeolitic water molecules. In this way, many years ago, Barrer and co-workers reported the selective adsorption of linear versus branched hydrocarbons, suggesting an implication of the microporous open tunnels.19 Serna and Fernández-Alvarez20 confirmed the intracrystalline sorption of hexane and hexene contrarily to benzene or cyclohexane adsorptions by sepiolite occurring in the last cases at the external surface of the silicate. Nevertheless, Inagaki and co-workers17 have deduced, from the corresponding adsorption isotherms obtained at the same temperature, that benzene molecules are able to migrate inside the internal surface of sepiolite through the tunnels. Therefore, the entrance of molecules into such structural cavities remains as controversial behaviour. In the present work such topic is revised, mainly based on our experiences related to adsorption of different species on sepiolite.
 | ||
| Fig. 2 HRTEM images of sepiolite samples. A and B: natural samples; C: anhydrous sepiolite (adapted from ref. 16: Reproduced with kind permission of the Mineralogical Society of Great Britain and Ireland). | ||
Experimental
Materials
The sepiolite used is a natural mineral from Yunclillos (Toledo, Spain) provided by TOLSA S.A. with a 99% content of pure silicate. The surface area (N2, BET) is 335 m2 g−1 and the cationic exchange capacity CEC is close to 0.15 meq g−1. The granulometric fraction used was <200 mesh. Pyridine (Py; Merck, >99%) and 2,6-dimethylpyridine (Merck, >98%) were purified by vacuum distillation after drying over 4 Å molecular sieves. The cationic dye methylene blue (MB) was supplied by Carlo Erba. The n-heptane was also supplied by Carlo Erba (RPE-ACS purity grade) and was dried over 4 Å molecular sieves (Fluka). Poly(ethylene oxide), PEO, of molecular weight = 105 Dalton was supplied by Aldrich.
Methods and equipment
BET specific surface area was determined from an N2 adsorption isotherm at 77 K on sepiolite degassed at 120°C during
12 h, using a Coulter Omnisorp 100 sorptometer. Ar adsorption on samples
of sepiolite treated with MB and sepiolite degassed at 120°C and
250°C was carried out at 87 K using the same apparatus.
Ar isotherms were also obtained on sepiolite degassed at 120°C
and 350°C (6 h) using a Varian ASAP 2000 instrument.Adsorption amounts of Py and its 2,6-dimethyl derivative were determined
from UV (Varian 2300 spectrophotometer) absorbance changes at λmax = 251 nm
before and after adsorption from n-heptane solutions, using 1 g
of sepiolite and 50 ml of solutions of different concentrations. The
system was maintained at 25°C during 7 days with intermittent
shaking.
IR spectra of Py adsorbed on the vapor phase were recorded using a Perkin-Elmer 580B double beam spectrophotometer coupled to a M-3500PE data station. Sepiolite samples of about 10 mg were pressed (2000 Kg cm−2) to obtain thin wafers of 13 mm diameter which were mounted in a stainless steel holder and placed into a typical vacuum cell with CaF2 windows. The dichroic effect of vibration bands of sepiolite and Py adsorbed on sepiolite was examined by tilting the sample (wafer) with respect to the IR beam from 0 to 45°, rotating the vacuum cell and registering the IR at both angles of inclination. Dichroic studies (νOH of Si–OH and Mg–OH groups) were also investigated on oriented films of pure sepiolite of 36 mm diameter, prepared by very slow filtering of the silicate colloidal suspensions (0.5% of sepiolite in weight) through a Millipore filter. Comparable results were obtained with both types of sample preparations indicating the preferential orientation of sepiolite by using the hydraulic press to prepare the silicate in the form of wafers.
Solid samples of sepiolite–MB were characterized by IR spectroscopy in a Fourier transform IR (FTIR) Nicolet 20SXC spectrophotometer, using samples as wafers.
The enthalpy changes associated with the MB adsorption process were determined
at 25 ± 0.01°C with a LKB-2107 sorption
microcalorimeter operating in the batch mode. A suspension of 50 mg
of sepiolite and 500 µl of water was placed in the batch cell
of the microcalorimeter. Aliquots of 25 µl of an aqueous 0.05 M
solution of the dye were added one by one using a peristaltic pump, at a flow
rate of 8 ml h−1. Eight “injections”
were made consisting of MB amounts ranging between 25% and 200%
of the sepiolite CEC. The dye solution was previously placed in a thermostatic
bath at 25°C until temperature equilibration, to minimize the
difference of temperature between the cell and the solution.
Results and discussion
Different authors21 have shown the ability of nitrogen and argon to enter the intracrystalline cavities although the role of the structural micropores of sepiolite in the adsorption of gases is still controversial. The access to the intracrystalline cavities of sepiolite is clearly revealed by the Horwarth–Kawazoe22 analyses applied to the treatment of the adsorption isotherms of argon at 87 K on sepiolite previously degassed at 120°C, i.e.
until complete elimination of the intracrystalline zeolitic water
molecules. From these analyses a pore size distribution (Fig. 3)
showing an effective pore diameter in the 6–7 Å range is
obtained which is in agreement with the dimensions of the structural cavities.
In this experiment, it is shown that Ar atoms penetrate into the sepiolite
tunnels, revealing the predominance of open pores with a mean diameter of
6.7 Å. A similar experiment carried out with the same sepiolite
sample, but in this case pre-treated at 350°C under dynamic
vacuum to eliminate the co-ordinated water molecules, indicates that these
pores are fully suppressed (Fig. 3)
by effect of the thermal treatment. This observation is in agreement with
the structural changes (crystal folding)
(Fig. 2) associated with water loss that
give the so-called anhydrous sepiolite.23
So, the ability of argon atoms to penetrate into the sepiolite tunnels is
then avoided. Besides, other open pores, which are mainly attributed to defects
in the crystal growth, remain almost unchanged (Fig. 3).
From the adsorption/desorption isotherms the calculated specific surface
area (BET, Ar) and the pore volume of sepiolite degassed at 120°C
are 300 m2 g−1 and 0.14 cm3 g−1,
respectively, whereas such parameters obtained from the same sample, but pre-heated
at 350°C under dynamic vacuum, are 152 m2 g−1
and 0.07 cm3 g−1, respectively.
Inagaki and co-workers17 found comparable
values (ca. 290 m2 g−1)
for the surface area deduced from the adsorption isotherms on a Turkish sepiolite
of several small molecules such as N2, H2O, NH3
and C2H5OH, indicating their ability to access the interior
of the microporous solid.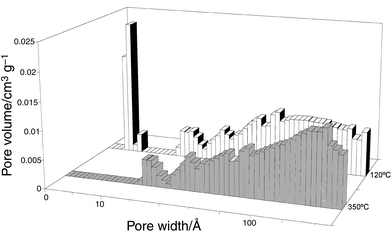 | ||
| Fig. 3 Pore size distribution
of sepiolite degassed at 120°C and 350°C (Ar,
Horwarth–Kawazoe method). | ||
Pyridine (Py) is a molecule of compatible geometry (larger
axis of about 7.5 Å) for potential insertion into sepiolite
through the tunnels. In fact, when sepiolite is placed in contact either with
Py vapours or with Py in an organic solvent such as n-heptane,
pyridine molecules are spontaneously adsorbed by the silicate even without
previous dehydration treatment. In all cases, the obtained adsorption isotherms
of Py from n-heptane at 25°C can be ascribed to a
typical H isotherm (Giles and co-workers classification)24 with a plateau at about xm = 1.8 × 10−3 mol g−1 corresponding to the maximum Py surface coverage value. Taking
into account the area calculated for one Py molecule (28.6 Å2, ref. 17), the surface
covered by this molecule in a monolayer disposition is about 310 m2 g−1
which corresponds to complete surface coverage, i.e. including the internal
surface as a result of the penetration into the tunnels of the silicate.
Comparable results were obtained by Inagaki and co-workers17
from the adsorption isotherms of Py in the vapour phase at the same temperature (25°C).
The IR spectra of an orientated
film of sepiolite recorded before and after exposure to Py vapours (Fig. 4) indicate some significant features.
Firstly, the broad absorption in the 3650–3000 cm−1
region, in which the bands assigned to the co-ordinated water molecules (3627
and 3550 cm−1; νOH) appear,
is deeply modified (strong decrease in intensity and enlargement of the
band shifting towards lower frequencies). The corresponding bending vibrations (1617
and 1626 cm−1, Fig. 4)
are also perturbed (shift towards higher frequencies, i.e. near
1650 cm−1) indicating the penetration of the
Py into the tunnels where such water molecules are mainly located. The adsorbed
Py molecules remain associated with the mineral surface by hydrogen bonding
between the nitrogen heteroatom of Py and the co-ordinated water molecules,
therefore there is formation of “water bridges” linking Mg2+
ions and the Py molecules inside the tunnels through the co-ordinated
H2O molecules. Thermal treatment (3 h at 250°C,
under dynamic vacuum) results in the elimination of these water molecules
and, according to the IR spectra (shift of the Py ring band at 1443 cm−1
to 1450 cm−1) direct co-ordination between
the Py nitrogen and the Mg2+ ions takes place. The second
important feature derived from the IR spectra analysis is the dichroic effect
associated with the band at 750 cm−1, which is assigned
to B2 vibrations of the Py molecule. In this way, a significant
increase in intensity of this band (Fig. 5)
is observed when the spectrum is registered with the IR beam at two angles
of incidence (0° and 45°) with respect to the oriented film
of sepiolite. In contrast, the A1 and B1 bands near
1450, 1490, 1580 and 1600 cm−1 remain unchanged when
the angle of incidence is modified. The changes of dipole moments associated
with such B2 vibrations are perpendicular to the plane of the Py
molecule in agreement with a preferential molecular arrangement of Py molecules
into the tunnels with the plane of the Py ring lying parallel to the [100]
plane of the sepiolite crystal, as represented in Fig. 6.
Besides, the stretching OH vibration bands of Si–OH and Mg–OH
groups, which appear at about 3720 and 3680 cm−1 respectively
in the IR spectrum of the untreated sepiolite, are also dichroic (Fig. 7). This clearly indicates the orientation
of the intracrystalline adsorbed pyridine relative to those hydroxys, i.e.
with the O–H axis in the same direction as the crystallographic axis a,
whereas the Py ring lies parallel to the b,c plane. On the other
hand, the strong decrease in intensity of the 3720 cm−1νOH
band (Figs. 4 and 7)
is attributed to hydrogen bonding between the N atoms of Py and the hydroxy
groups of the silicate surface producing a shift towards low frequency values.
The shifted band is not observed in the spectrum because it should be overlapped
with νOH bands of water molecules.
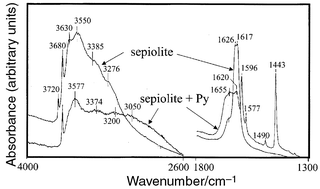 | ||
| Fig. 4 IR spectra of natural sepiolite and sepiolite treated with pyridine vapours. | ||
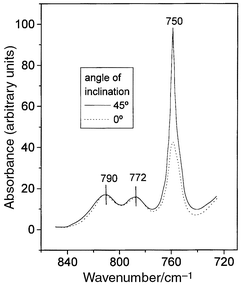 | ||
| Fig. 5 Dichroic effect in the B2 IR absorption band of pyridine adsorbed on sepiolite. | ||
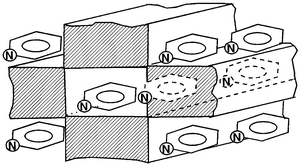 | ||
| Fig. 6 Idealised representation of the pyridine molecules arranged at the sepiolite external surface and inside the tunnels. | ||
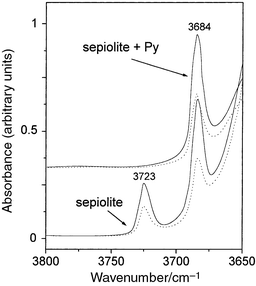 | ||
| Fig. 7 Dichroic effect of the OH stretching bands of natural sepiolite and sepiolite–pyridine samples. | ||
Molecules more voluminous than Py, such as 2,6-dimethylpyridine, are adsorbed to a lesser extent (xm = 0.6 × 10−3 mol g−1) than Py under identical experimental conditions. The corresponding surface coverage is about 140 m2 g−1 which may be related to the extension of the external surface of sepiolite (similar to “folding sepiolite”). In these cases, the IR spectra show few changes in the bands associated with water molecules, indicating that only the H2O located at the surface (channels) are accessible to this type of pyridine derivative. Thus, we can assume that the penetration of this type of molecules into the tunnels is avoided.
Methylene blue (MB) is a cationic dye with molecular dimensions
17 × 7.6 × 3.25 Å3
which may be suited to migration inside the sepiolite tunnels. The adsorption
isotherms at 25°C from water solutions25
are also H type, corresponding to processes where the solid has great affinity
towards the solute.24 However, it is well
known that MB has the ability to form aggregates26
(dimers,
trimers) in water solutions depending on the concentration of the dye.
This fact leads to a slight lowering of the amount of dye adsorbed on sepiolite
when the initial dye solution is at high concentrations in agreement with
previous results.27 Such behaviour could be
explained on the basis that at these high concentrations, the volume of the
aggregates formed does not allow the MB access to the structural micropores
of sepiolite, lowering the amount of dye that is able to be adsorbed. The
formation of such molecular aggregates has been elucidated by UV-VIS spectroscopic
studies of the sepiolite/dye system.25a
A convincing result supporting the access of MB and other cationic dyes of
comparable molecular dimensions such as thioflavine-T (TFT)28 consists of the IR modification of bands associated
with the co-ordinated water molecules mainly located inside the tunnels.
Thus, for dye adsorptions from very low concentrations (<10−3 M),
in which monomeric species predominate, the IR spectrum resembles that of
vacuum-dried sepiolite, showing the modification of bands at 1650–1600 cm−1
which are assigned to bending H–O–H vibrations and suggesting
that the dye molecules partially replace the weakly adsorbed water molecules (zeolitic
water) located within the tunnels. In this situation, MB could interact
with the co-ordinated water molecules contributing to the development
of the 1650 cm−1 band. Such spectral modifications
are not observed for MB adsorptions at higher initial concentrations or when
more voluminous dyes, such as crystal violet and methyl green, are adsorbed
on sepiolite.27 In contrast, TFT, which is
of comparable molecular size to MB, gives similar spectral changes.28
Horwarth–Kawazoe analyses22 have
been applied to data from Ar adsorption isotherms (87 K)
of: i) sepiolite degassed at 250°C under dynamic vacuum,
and ii) sepiolite containing MB after adsorption from dye solutions at
low initial concentrations. The graphical results represented in Fig. 8
show a significant decrease, similar for both samples, of the adsorption of
Ar atoms. This result indicates that MB molecules inside the tunnels are preventing
the entrance of Ar, creating an impediment comparable to that caused by the
thermal treatment that provokes the structural folding of sepiolite.
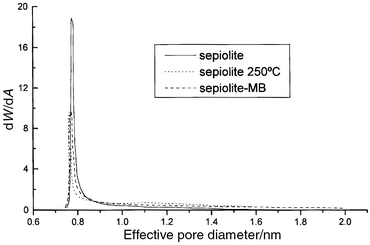 | ||
| Fig. 8 Graphical results of
the Horwarth–Kawazoe analyses (Ar adsorption, 87 K)
of sepiolite (degassed at 120°C and 250°C)
and sepiolite treated with methylene blue. | ||
The accessibility of MB to the sepiolite tunnels is also supported by the microcalorimetric experiments carried out by de Castro and co-workers.27 Thus, in the adsorption of MB on sepiolite it was observed that for low initial MB concentrations the process is endothermic, whereas adsorption from higher concentrations produces an exothermic effect. The endothermic effect could be attributed to the enthalpy balance between the heat absorbed by the water displacement and the heat evolved by the MB adsorption. Such a situation implies that the MB entering the sepiolite replaces water molecules located in the structural pores. By contrast, the adsorption of dyes of larger size, such as crystal violet, is always an exothermic process.
We have above presented the ability of atoms (Ar), molecules (Py)
and cationic organic species (MB) to diffuse through the sepiolite
tunnels. Macromolecules could also be formed by condensation of monomers inside
the tunnels as reported by different authors. So, Inagaki and co-workers29 describe the polymerisation at 25°C
of isoprene previously adsorbed on sepiolite, ascribing to the protons belonging
to the co-ordinated water the Brønsted acidity responsible for
the initiation of the polymerisation reaction. Sandi and co-workers30 report the polymerisation of ethylene and polyethylene
in sepiolite adopting more drastic conditions, the resulting polyethylene
being subsequently carbonised by pyrolysis of the sepiolite nanocomposite.
However, the direct insertion into the tunnels of high molecular weight polymers
is also possible. In this way, we have succeed in intercalating poly(ethylene
oxide), PEO, of 105 Dalton, from aqueous solutions. The time
necessary to achieve such polymer insertion is much longer (e.g.
one week at 25°C) than the time required for intercalation
of molecular or ionic species. It could be assumed that the usual helical
conformations of PEO in the solid state, and in some cases when adsorbed in
layered silicates,31 became a zig-zag
conformation inside the tunnels due to steric hindrance. Supporting the entrance,
at least as a partial penetration, the IR spectrum shows the characteristic
perturbation of bands in the H–O–H region indicating the replacement
of zeolitic water and the interaction (H-bonding) between the
oxygen of the oxyethylene chains with the hydrogen of co-ordinated water
located in the channels and inside the tunnels. The specific surface area
of sepiolite drastically drops from 335 m2 g−1
to 124 m2 g−1 after treatment with
PEO, although it is enough that a small fraction of the polymer chain fills
the tunnel entrance to avoid subsequent molecular (e.g. N2)
penetration. At this moment, it is difficult to quantify the extent of inserted
PEO. Further details on these PEO–sepiolite nanocomposites will be reported
elsewhere.32
Conclusions
The possibility to induce versatile intracrystalline sorptions either of atoms or of neutral molecules and charged species, and even polymers, into the structural tunnels of sepiolite, appears as a feasible way to prepare organic–inorganic materials with predetermined behaviour. In fact, these processes lead to nanocomposite materials with 1D molecular arrangement. This property is particularly well illustrated in the adsorption of organic dyes, e.g. methylene blue. In these last cases, and imposed by the topology of the sepiolite tunnels, the resulting supported-dye materials show an unusual molecular disposition which is of great interest for the preparation of solids which may be potentially useful for non-linear optical applications. The molecular sieving could be of interest for the separation—or the catalytic transformation—of selected molecules belonging to a determined mixture: see for instance the ability to discriminate the adsorption of pyridine from its derivatives. The entrance of polymers, such as poly(ethylene oxide), into the sepiolite tunnels gives in a mild way stable nanocomposites containing sepiolite without zeolitic water, opening the way for new applications of this versatile mineral.In this manner we have presented in this article a contribution aimed at achieving a better understanding of the mechanism controlling the insertion of different kinds of organic species, showing the role of sepiolite as a molecular sieve and also as a host for polymer inclusions. This could be a basis for the preparation of nanocomposite materials useful for advanced technologies related for instance to photostabilisation, molecular separation, functional polymer–clay systems, etc.
Acknowledgements
This work has been carried out with financial support from the CICYT (Spain) and EU Projects as “Peace Campus”. Technical assistance from Jesus Merino and fruitful discussions with J.M. Serratosa, B. Casal, P. Aranda and C. de Castro are gratefully acknowledged.References
- (a) T. J. Pinnavaia and M. F. Thorpe (Editors), in Access in Nanoporous Materials, Plenum Press, New York, 1995 Search PubMed; (b) T. J. Pinnavaia, in Materials Chemistry an Emerging Discipline, eds. L. V. Interrante, L. A. Casper and A. B. Ellis, Adv. Chem. Ser. 245, Am. Chem. Soc., Washington DC, 1995, p. 283. Search PubMed.
- J. M. Thomas and W. J. Thomas, Principles and Practice of Heterogeneous Catalysis, VCH, Weinheim, 1997. Search PubMed.
- (a) J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowitcz, C. T. Kresge, K. D. Schmitt, C. T. W Chu, D. H. Olson, E. W. Sheppard and S. B McMullen, J. Am. Chem. Soc., 1992, 114, 10834 CrossRef CAS; (b) C. T. Kresge, M. E. Leonowitcz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710 CrossRef CAS; (c) Q. Huo, R. Leon, P. M. Petroff and G. D. Stucky, Science, 1995, 268, 1324 CrossRef CAS.
- F. de Juan and E. Ruiz-Hitzky, Adv. Mater., 2000, 12, 430 CrossRef.
- A. Galarneau, A. Baradawalla and T. J. Pinnavaia, Nature, 1995, 374, 529 CrossRef CAS.
- F. Kooli, T. Sasaki and M. Watanabe, Chem. Commun., 1999, 211 RSC.
- (a) M. S. Wittingham and A. J. Jacobson (editors), Intercalation Chemistry, Academic Press, New York, 1982 Search PubMed; (b) D. W. Bruce and D. O'Hare (editors), Inorganic Materials, 2nd edn., John Wiley & Sons, Chichester, 1996. Search PubMed.
- (a) G. A. Ozin, A. Kuperman and A. Stein, Angew. Chem., Int. Ed. Engl., 1989, 28, 359 CrossRef; (b) G. A. Ozin, Adv. Mater., 1992, 4, 612 CrossRef CAS; (c) E. Ruiz-Hitzky, Adv. Mater, 1993, 5, 334 CrossRef CAS; (d) M. G. Kanatzidis, L. M. Tonge, T. J. Marks, M. O. Marcy and C. R. Kannewurf, J. Am. Chem. Soc., 1987, 109, 3797 CrossRef CAS; (e) M. G. Kanatzidis, C.-G. Wu, M. O. Marcy and C. R. Kannewurf, J. Am. Chem. Soc., 1989, 111, 4139 CrossRef CAS; (f) P. Judeinstein and C. Sanchez, J. Mater. Chem., 1996, 6, 511 RSC.
- (a) P. Enzel and T. Bein, J. Phys. Chem., 1989, 93, 6270 CrossRef CAS; (b) T. Bein and P. Enzel, Angew. Chem., Int. Ed. Engl., 1989, 28, 1992 CrossRef; (c) D. W. Wöhrle and G. Schulz-Ekloff, Adv. Mater., 1994, 6, 875 CrossRef; (d) I. Braun, G. Ihlein, F. Laeri, J. U. Nöckel; G. Schulz-Ekloff, F. Schüth, U. Vietze, Ö. Weiss and D. Wöhrle, Appl. Phys. B, 2000, 70, 335 CrossRef CAS.
- (a) C.-G. Wu and T. Bein, Science, 1994, 264, 1757 CrossRef CAS; (b) C.-G. Wu and T. Bein, Science, 1994, 264, 1013.
- (a) E. Ruiz-Hitzky and P. Aranda, An. Quim. Int. Ed., 1997, 93, 197 Search PubMed; (b) E. Ruiz-Hitzky and P. Aranda, in Polymer Clay Nanocomposites, ed. T. J. Pinnavaia and G. Beall, John Wiley & Sons, in press. Search PubMed.
- (a) G. P. C. Chambers, Silicates Industriels, 1959, 24, 181 Search PubMed; (b) A. Alvarez, “Sepiolite: Properties and Uses”, in Palygorskite-Sepiolite. Occurrences, Genesis and Uses. Developments in Sedimentology, vol. 37, ed. A. Singer and E. Galan, Elsevier, Amsterdam, 1984, p. 253. Search PubMed.
- (a) K. Brauner and A. Preisinger, Miner. Petr. Mitt., 1956, 6, 120 Search PubMed; (b) J. Santarén, J. Sanz and E. Ruiz-Hitzky, Clay Miner., 1990, 38, 63 Search PubMed.
- J. L. Ahlrichs, J. C. Serna and J. M. Serratosa, Clays Clay Miner., 1975, 23, 119 Search PubMed.
- (a) E. Ruiz-Hitzky and J. J. Fripiat, Clays Clay Miner., 1976, 24, 25 Search PubMed; (b) M. N. Fernandez-Hernandez and E. Ruiz-Hitzky, Clay Miner., 1979, 14, 295 Search PubMed; (c) A. Van Meerbeek and E. Ruiz-Hitzky, Colloid Polym. Sci., 1979, 257, 178 CAS.
- M. Rautureau and A. Mifsud, Clay Miner., 1977, 12, 309 Search PubMed.
- S. Inagaki, Y. Fukushima, H. Dot and O. Kamigaito, Clay Miner., 1990, 25, 99 Search PubMed.
- (a) A. J. Dandy, J. Chem. Soc. A, 1971, 2383 RSC; (b) P. Fenoll Hach-Alí and J. L. Martín-Vivaldi, R. Soc. Esp. Fis. Quím., 1970, 64(B), 77 Search PubMed; (c) C. J. Serna, PhD Thesis, 1974, University of Madrid, UCM, Madrid; (d) J. M. Serratosa, in Int. Clay Conf. 1978, ed. M. M. Mortland and V. C. Farmer, Elsevier, Amsterdam, 1979. Search PubMed.
- R. M. Barrer, N. MacKenzie and D. M. MacLeod, J. Phys. Chem., 1954, 56, 568 CrossRef.
- C. Serna and T. Fernández-Alvarez, Anal. Quim., 1974, 70, 760 Search PubMed.
- (a) T. Fernández-Alvarez, in Proc. Reunión Hispano-Belga de Minerales de la Arcilla, ed. J. M. Serratosa, CSIC, Madrid, 1970, p. 202 Search PubMed; (b) A. Jiménez-López, J. D. López-González, A. Ramírez-Saenz, F. Rodríguez-Reinoso, C. Valenzuela-Calahorro and L. Zurita-Herrero, Clay Miner., 1978, 13, 375 Search PubMed; (c) Y. Grillet, J. M. Cases, M. François, J. Rouquerol and J. E. Poirier, Clays Clay Miner., 1988, 36, 233 Search PubMed; (d) L. Michot, M. François and J. M. Cases, Langmuir, 1990, 6, 677 CrossRef CAS; (e) F. Villiéras, J. M. Cases, M. François, L. J. Michot and F. Thomas, Langmuir, 1992, 8, 1789 CrossRef CAS.
- G. Horwarth and K. Kawazoe, J. Chem. Eng. Jpn., 1983, 16, 470 Search PubMed.
- (a) A. Preisinger, Clays Clay Miner., 1963, 10, 365 Search PubMed; (b) M. Nagata, S. Shimoda and T. Sudo, Clays Clay Miner., 1974, 22, 285 Search PubMed; (c) M. Rautureau and A. Mifsud, Clay Miner., 1977, 12, 309 Search PubMed.
- C. H. Giles, T. H. McEwan, S. N. Nakhama and D. Smith, J. Chem. Soc., 1960, 3973 RSC.
- (a) A. J. Aznar, B. Casal, E. Ruiz-Hitzky, I. Lopez-Arbeloa, F. Lopez-Arbeloa, J. Santaren and A. Alvarez, Clay Miner., 1992, 27, 101 Search PubMed; (b) G. Rytwo, S. Nir, L. Margulies, B. Casal, J. Merino, E. Ruiz-Hitzky and J. M. Serratosa, Clays Clay Miner., 1998, 46, 340 Search PubMed.
- (a) K. Bergman and O. Konski, J. Phys. Chem., 1963, 67, 2169; (b) S. L. Fornili, G. Scroi and V. Izzo, J. Chem. Soc., Faraday Trans 1, 1981, 77, 3049 RSC.
- C. de Castro, B. Casal, J. Merino and E. Ruiz-Hitzky, in preparation..
- B. Casal, J. Merino, J. M. Serratosa and E. Ruiz-Hitzky, Appl. Clay Sci., in press. Search PubMed.
- S. Inagaki, Y. Fukushima and M. Miyata, Res. Chem. Intermed., 1995, 21, 167 Search PubMed.
- G. Sandi, K. A. Carrado, R. E. Winans, C. S. Johnson and R. Csencsits, J. Electrochem. Soc., 1999, 146, 106.
- (a) P. Aranda and E. Ruiz-Hitzky, Chem. Mater., 1992, 4, 1395 CrossRef CAS; (b) P. Aranda and E. Ruiz-Hitzky, Acta Polym., 1994, 45, 59 CrossRef CAS.
- E. Ruiz-Hitzky and P. Aranda, in preparation..
Footnote |
| † Basis of a presentation given at Materials Discussion No. 3, 24–26 September 2000, University of Cambridge, UK. |
| This journal is © The Royal Society of Chemistry 2001 |