Synthesis and electrochromic properties of mesoporous tungsten oxide†
Wei Chenga, Emmanuel Baudrina, Bruce Dunna and Jeffrey I. Zinkb
aUniversity of California, Los
Angeles, Department of Materials Science and Engineering, Los Angeles, CA 90095, USA
bUniversity of California, Los
Angeles, Department of Chemistry and Biochemistry, Los Angeles, CA 90095, USA
First published on 10th October 2000
Abstract
The electrochromic properties of mesoporous tungsten oxide are described and compared to standard sol–gel films of WO3. The block copolymer template used in the synthesis was removed by solvent extraction and calcination methods, leading to mesoporous tungsten oxides which range from amorphous to crystalline depending on the removal treatment. The pores are not ordered but do exhibit a fairly regular diameter with an average size of 4 to 5 nm. The mesoporous tungsten oxides exhibit a well behaved electrochromic response and their electrochemical and optical reversibilities are better than those of the analogous sol–gel film. In addition, the mesoporous films exhibit higher rates for coloration and bleaching. The high surface area of these materials is responsible for this behavior and also leads to multiple peaks in the voltammetric scans.
Introduction
Electrochromism offers a relatively simple means of producing variable light transmission by electrochemical oxidation and/or reduction. Over the last several years, electrochromism has received considerable interest for automotive applications (self-dimming rear view mirrors) and “smart” windows as well as for optical displays.1 Electrochromism has been widely studied in conducting polymer systems (e.g., polythiophene, polypyrrole) and in inorganic oxides (e.g., WO3, IrO2, NiO). Among the latter group of materials, WO3 has received the most attention.2 WO3 films have been prepared by a variety of methods including vacuum evaporation,3 anodic oxidation,4 spray pyrolysis,5 sol–gel synthesis,6,7 sputtering8 and laser ablation.9 As a result there is a reasonable understanding of how the preparation method and microstructure influence electrochromic behavior.The present paper reports the electrochromic properties of mesoporous tungsten oxide films. The ability to form mesoporous oxides based on the use of various structure-directing agents has been widely investigated over the past several years.10–12 While much of this research has emphasized SiO2, it is now well established that surfactant templating methods can be extended to a number of other metal oxides including WO3.13 There has been only limited study of the functional properties of the non-silicate mesoporous oxides and, thus, there is little understanding of how the presence of pores in the size range of 2 to 20 nm influences the electrical, optical or magnetic properties of the mesoporous material. In the case of mesoporous tungsten oxide, it is interesting to consider how the penetration of electrolyte through the pore network will affect the nature of the electrochromic response. In this paper we compare the properties of tungsten oxide films prepared by sol–gel methods with mesoporous films that use the same chemistry to form the oxide but also incorporate a block copolymer to generate the mesoporous microstructure. The results indicate that the mesoporous material exhibits better kinetics for coloration and bleaching in comparison to standard sol–gel derived tungsten oxide.
Experimental
Two types of tungsten oxide films were prepared in this study. In one case, sol–gel derived tungsten oxide films were deposited from a coating solution via the dip-coating procedure. The solutions were made by reacting 0.98 g WCl6 (Aldrich) with 10 g anhydrous ethanol as follows:14 | (1) |
The solution became blue as a small fraction of WVI reduced
to WV.15 After stirring for several
hours, the sol was filtered to make the final coating solution. ITO coated
glass (15 Ω □−1) was used
as the film substrate. The dip coating speed was 12.5 mm s−1.
The as-deposited film was left in air for one hour to allow the solvent
to evaporate and was then fully dried in air at 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C. Hydrolysis
and condensation of the film occurred from exposure to ambient moisture during
the drying treatments.6,13 To obtain
powder samples, the coating solution was dried in an open Petri dish in air
at 40°C for several days.
°C. Hydrolysis
and condensation of the film occurred from exposure to ambient moisture during
the drying treatments.6,13 To obtain
powder samples, the coating solution was dried in an open Petri dish in air
at 40°C for several days.
The second type of film prepared in this study is mesoporous tungsten oxide. (In
this paper, the two types of materials are referred to as sol–gel tungsten
oxide and mesoporous tungsten oxide to distinguish between the two different
types of microstructure.) The coating solution in this case consisted
of 0.98 g WCl6
(Aldrich), 10 g anhydrous
ethanol and 0.25 g tri-block copolymer (BASF Pluronic P123)
HO(CH2CH2O)20(CH2CH(CH3)O)70(CH2CH2O)20H (designated as EO20PO70EO20).
The dip-coating and drying conditions were the same as indicated for the
sol–gel tungsten oxide. Solvent extraction and calcination methods were
used to remove the copolymer. Extraction was accomplished by immersing the
films twice into anhydrous ethanol for 10 minutes. Calcination involved heating
in air to 300°C or 400°C for 2 h at a heating
rate of 1°C min−1.
The sol–gel and mesoporous tungsten oxide materials were characterized by a variety of methods. For the most part, films were characterized. However, in those instances where films were not convenient to use because of the limited amount of material, the corresponding powders were analyzed. The film thickness was measured using an Alpha-step Profilometer. The film was gently scratched to produce the necessary step. X-Ray diffraction (XRD) of the films was carried out on a Crystalogic Diffractometer using Cu-Kα radiation (λ=1.5418 Å). Transmission electron microscopy (TEM) was performed on a JEOL 2000 electron microscope operating at 200 KeV. The TEM samples were prepared by scratching the film off the substrate, dispersing the particles in ethanol, and depositing them onto a holey carbon film on a Cu grid. Powders (sol–gel and mesoporous) were used for thermogravimetric analysis (TGA 2950; TA Instruments) and nitrogen adsorption–desorption isotherm measurements (Micromeritics ASAP 2000). Chemical analysis of the powders was performed commercially (Galbraith Laboratories).
The electrochemical properties were determined using a three electrode configuration in which the working electrode consisted of the dip coated tungsten oxide film deposited on ITO coated glass. The reference and counter electrodes were a saturated calomel electrode (SCE) and a platinum grid, respectively. The electrolyte was a 0.1 M aqueous solution of sulfuric acid. The voltammetry and chronoamperometry experiments were performed using a PAR 273 potentiostat (EG&G instruments). UV-Visible absorption measurements were performed with a Shimadzu UV-260 spectrophotometer. The transmittance was normalized to 100% at the beginning of the experiment.
Results
Structural and microstructural characterization
The thickness of the tungsten oxide films was in the range of 0.4 to 0.5 µm for all the samples characterized in this work. The compositions of the sol–gel and mesoporous tungsten oxides can be generally written as: WO3 − x·nH2O and WO3 − x·nH2O·mP123 respectively. More detailed information concerning the composition was obtained by TGA and chemical analysis as described in the following paragraphs.The thermogravimetric analysis curve for sol–gel tungsten oxide (Fig. 1a) suggests a three step process:
the desorption of adsorbed water between room temperature and 200°C,
the subsequent loss of tightly bonded water molecules between 250°C
and 470°C, and a gain of oxygen at higher temperature due to the
oxidization and crystallization of WO3. The composition of the
as-made sol–gel tungsten oxide can be estimated as WO2.95·1.3H2O
which is close with the WO3·1.3H2O formula reported
by Krings et al.16 who used WOCl4
as the precursor.
 | ||
| Fig. 1 TGA curves for the (a)
sol–gel tungsten oxide and (b) mesoporous tungsten oxide.
The samples were heated in air at a rate of 5°C min−1. | ||
The TGA curve of the mesoporous tungsten oxide indicates that the co-polymer
is pyrolyzed over the temperature range 140°C to 350°C (Fig. 1b). The compositions of the mesoporous
tungsten oxides can be estimated as: WO2.94·2.8H2O·0.011P123,
WO2.94·1.3H2O·0.001P123, WO2.94·0.8H2O·0.0005P123
and WO2.97·0.17H2O for the as-made, solvent
extracted, 300°C calcined and 400°C calcined samples,
respectively. These results show that the co-polymer can be removed to
nearly the same extent by solvent extraction as by calcination at 300°C.
XRD patterns for the sol–gel tungsten oxide and mesoporous tungsten
oxide films are compared in Fig. 2.
There are some similarities: the as-deposited materials are amorphous
while the films heat-treated at 400°C for two hours crystallize
to WO3. At 300°C, there is a subtle difference in behavior
as the sol–gel film exhibits the onset of crystallization while the
mesoporous film is still amorphous.
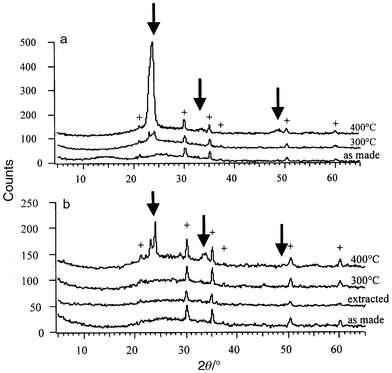 | ||
| Fig. 2 XRD patterns for (a) sol–gel and (b) mesoporous tungsten oxide thin films heated to different temperatures. The crosses (+) indicate the Bragg peaks from the ITO coating of the substrate and the arrows show the main tungsten oxide peaks. | ||
The morphology of the mesoporous tungsten oxide was characterized by TEM and gas sorption methods. TEM images for extracted and calcined films (Fig. 3a,b) indicate that the pores are not ordered although they do exhibit a fairly regular pore diameter. Pore sizes show a wider distribution (40–80 Å) for the extracted film than for the calcined one (40–60 Å). The TEM images are consistent with the 50% porosity level obtained by gas sorption (Table 1).
| Sample | Surface areaa/m2 g−1 | Average pore diameterb/Å | Porosityc |
|---|---|---|---|
| a BET (Brunauer–Emmett–Teller) method.b BJH (Barrett–Joyner–Halenda) desorption.c Estimated from pore volume determined by N2 isotherm curve at P/P0 = 0.99 (single point), and from density measured by a He gas pycnometer (AccuPyc1330, Micromeritics).13 | |||
| Solvent extracted | 155 | 50 | 0.55 |
| 300°C treated | 135 | 45 | 0.46 |
| 400°C treated | 45 | 106 | 0.49 |
 | ||
| Fig. 3 TEM images of mesoporous
tungsten oxide film after (a) ethanol extraction and (b)
calcination at 300°C. (c) Nitrogen adsorption–desorption
isotherms and BJH pore size distribution plot (inset) for mesoporous
tungsten oxide powder calcined at 300°C. | ||
Nitrogen adsorption–desorption isotherms (Fig. 3c)
exhibit a type IV curve which is characteristic of mesoporous tungsten oxide.13 The pore size for this material (calcined
at 300°C) showed a fairly narrow distribution, centered at
45 Å
(Fig. 3c inset).
The surface area (BET), average pore diameter (BJH desorption)
and porosity level are listed in Table 1
for the extracted, 300°C treated and 400°C treated
mesoporous tungsten oxide. The results for the 300°C treated sample
are similar to those reported by Yang et al.13
It would seem that as the calcination temperature increases to 400°C,
the mesopore structure collapses due to the crystallization of the inorganic
wall, leading to a decrease in surface area, and an increase in average pore
size. In comparison, the sol–gel tungsten oxide material has a smaller
surface area (∼20 m2 g−1
for the dried powder).
Voltammetry and optical measurements
The mesoporous tungsten oxide films exhibit a well behaved electrochemical response (Fig. 4b,c). The voltammograms (from −0.8 V to 0.8 V vs. SCE) are similar to those reported in prior studies of proton insertion in tungsten oxide.16 There is a well-defined anodic peak for the samples calcined at 300°C and 400°C
and good electrochemical reversibility is observed after the first cycle.
After the fifth cycle, the loss is less than 1%
(and most of this
loss results from film dissolution, vide infra). For comparison,
proton insertion in the sol–gel tungsten oxide films leads to significantly
more capacity loss, in the range of 50% for the first cycle. The voltammograms (Fig. 4a) do not exhibit well defined anodic
peaks and instead are rather featureless over the voltage range. The electrochemical
characteristics for the sol–gel and mesoporous films heated to different
temperatures are listed in Table 2. | ||
| Fig. 4 Voltammetric response
for sol–gel tungsten oxide (300°C calcination)
and mesoporous tungsten oxide (300°C and 400°C
calcination) thin films. The sweep rate was 10 mV s−1
and the electrolyte was 0.1 M H2SO4. | ||
| Samplea | Anodic peak voltage/V (vs. SCE) | Amount of charge intercalated QR/C cm−2 | Amount of charge deintercalated QO/C cm−2 | ΔQ/QR (%)b |
|---|---|---|---|---|
| a MP = mesoporous; S–G = sol–gel.b First cycle. | ||||
| 40°C polymer not removed | −0.37 | 22.35 | 12.64 | 43.5 |
| MP 40°C ethanol extracted | −0.44 | 26.45 | 19.36 | 26.8 |
| −0.07 | ||||
| +0.18 | ||||
| MP 300°C | −0.37 | 25.98 | 18.74 | 27.8 |
| MP 400°C | −0.19 | 14.27 | 9.92 | 30.5 |
| S–G 40°C | −0.48 | 35.26 | 21.85 | 38.0 |
| S–G 300°C | −0.35 | 24.97 | 10.65 | 57.3 |
| S–G 400°C | −0.10 | 27.97 | 14.08 | 49.7 |
One of the most interesting results obtained with mesoporous tungsten oxide
is the development of multiple anodic peaks. Whereas during the first cycle
there is only a spike at −0.8 V on reduction and a peak at −0.37 V
on oxidation, new features become apparent in the sample calcined at 300°C
after three cycles. There is a new feature in reduction between +0.2 V
and −0.2 V and new peaks between +0.1 V and −0.1 V
in oxidation. In addition, the original anodic peak is displaced to −0.45 V.
The evolution of these changes is shown in Fig. 5a.
Electrochemical measurements made on the mesoporous samples prepared by solvent
extraction and by calcining at 400°C provide insight into the
origin of this behavior. As shown in Table 1,
these two samples exhibit the largest and smallest surface areas, respectively.
The mesoporous tungsten oxide prepared by solvent extraction exhibits three
anodic peaks during its first cycle at −0.44 V, −0.07 V
and +0.18 V (Fig. 5b).
That is, no cycling was required to induce this behavior. The 400°C
calcined material (not shown) required electrochemical cycling in
order to achieve the evolution of multiple peaks, however, the resolution
of the peaks is far less obvious than the results shown in Fig. 5.
 | ||
| Fig. 5 (a) Evolution
of the cyclic voltammogram during cycling for a 300°C mesoporous
tungsten oxide thin film calcined at 300°C. The arrows show the
evolution in the position and intensity of the peaks with cycling. (b)
Cyclic voltammogram for solvent extracted mesoporous tungsten oxide (0.1 M
H2SO4, 10 mV s−1).
The sweep rate was 10 mV s−1 and the electrolyte
was 0.1 M H2SO4. | ||
The development of multiple peaks during repetitive insertion–deinsertion has been reported previously.17 Kim and Pyun attributed the three anodic peaks to three types of hydrogen injection sites: reversibly active, shallow trap site (reversible) and deep trap site (irreversible). It would seem that the surface area of the film is an important factor in generating this response. Increasing the surface area will necessarily increase the number of surface active sites; this is one reason that high surface area materials are widely used for catalytic application.18
The optical properties of mesoporous tungsten oxide are well behaved in
much the same manner as the electrochemical properties. The optical change
for the calcined 300°C sample during the voltammetric sweep is
completely reversible (Fig. 6a).
There is absorption in the red during reduction as the transmittance at 650 nm
decreases to 45%. This change in coloration is completely reversible
as the transmittance returns to 100%
(normalized value) on
oxidation. The ethanol extracted samples show the same behavior. In contrast,
the sol–gel tungsten oxide films exhibit much worse reversibility (Fig. 6b). For the sol–gel film heated
at 300°C, re-oxidation leads to 90% transmittance,
while the coloration is irreversible (i.e., the films are permanently
blue colored) for the as-deposited sol–gel tungsten oxide and
the mesoporous sample heated at 400°C.
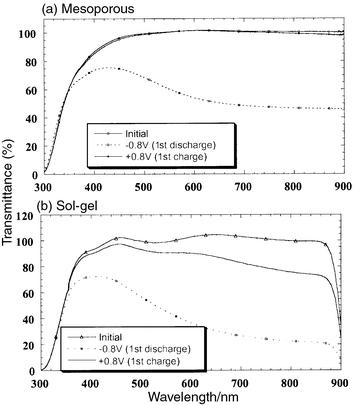 | ||
| Fig. 6 Optical transmittance
spectra for (a) mesoporous and (b) sol–gel tungsten
oxide films heated at 300°C. | ||
Coloration efficiency and chronoamperometry measurements
In order to provide more insight concerning the nature of the coloration/bleaching processes, two different types of experiments involving the transient response to a voltage step were investigated. The results in Fig. 7 represent the absorption at 650 nm in response to a voltage step applied to a mesoporous film calcined at 300°C. The step was between −0.8 V
and +0.8 V for 30 s and alternated between oxidation and
reduction. During the first reduction, the transmittance decreases from 100%
to 29% and the time to obtain 50% absorption is 9.2 s.
The following oxidation process leads to a completely transparent film in
6.9 s (the transmittance becomes somewhat greater than 100%
because of a small amount of film dissolution). That is, the tungsten
oxide is slightly soluble in the H2SO4 electrolyte.19 Subsequent cycles show quite good reversibility
of the optical properties. In contrast, coloration for sol–gel tungsten
oxide is either partially reversible or completely irreversible. | ||
| Fig. 7 Variation in the transmittance
at 650 nm as a function of time for the mesoporous tungsten oxide films
heated at 300°C and a voltage step between −0.8 V
and +0.8 V vs. SCE. The voltage changes polarity each
30 s. | ||
A parameter often used to characterize an electrochromic material is its coloration efficiency (CE) as defined by:
 | (2) |
°C).
| Samplea | Coloration timeb/s | Bleaching timec/s | CE (1st cycle)/cm2 C−1 | CE (3rd cycle)/cm2 C−1 | CE (5th cycle)/cm2 C−1 |
|---|---|---|---|---|---|
| a MP = mesoporous; S–G = sol–gel.b Time to obtain 50% change in transmission at 650 nm.c Time to obtain original transmission (100%) at 650 nm. | |||||
| 40°C polymer not removed | — | Irreversible | 16.5 | 16.3 | 24.3 |
| MP 40°C ethanol extracted | 9.7 | 7.5 | 19.5 | 21.2 | 21.6 |
| S–G 300°C | 26.2 (ΔT = 40%) | >30 | 17.1 | 24.4 | 30.6 |
| MP 300°C | 9.2 | 6.9 | 21.2 | 23 | 23.5 |
| S–G 400°C | 16.8 | >30 | 30.7 | 42.2 | 43.8 |
| MP 300°C | 26.5 (ΔT = 40%) | >30 | 22.7 | 36.7 | 35.8 |
In the second series of transient experiments, the current response to
the voltage step was determined. The responses for sol–gel and mesoporous
tungsten oxide films are shown in Fig. 8.
The kinetics during coloration (voltage held at −0.8 V for
30 seconds) are approximately the same for both films. However,
the bleaching responses (oxidation with the voltage at +0.8 V,
from 30 s to 60 s in Fig. 8)
are quite different. Whereas the sol–gel tungsten oxide exhibits a gradual
decrease in current, the mesoporous film has a two step current decay. In
addition, as the films cycle, the decay time for the sol–gel tungsten
oxide increases while the mesoporous tungsten oxide response is quite stable.
Once again, the mesoporous film exhibits better electrochemical stability
than the sol–gel film. Both the ethanol extracted and the 400°C
calcined mesoporous films exhibit the same type of response as that shown
in Fig. 8 for the 300°C
mesoporous film.
 | ||
| Fig. 8 Chronoamperometry measurements
for voltage steps between −0.8 V and +0.8 V (vs.
SCE) in 0.1 M H2SO4 electrolyte; (a)
sol–gel tungsten oxide heated at 300°C and (b)
mesoporous tungsten oxide film heated at 300°C. | ||
Discussion
The studies presented here were directed at establishing the influence of mesoporosity on the electrochemical behavior of tungsten oxide. Accordingly, tungsten oxide materials were prepared by the same synthetic route with the one distinctive difference being that one group of materials was made mesoporous by incorporation of a co-polymer. In general, the mesoporous tungsten oxide films exhibit a well behaved electrochromic response. It is interesting to note that their electrochemical and optical reversibilities are better than those of the analogous sol–gel film and there is evidence that the rate of bleaching is higher. In the following discussion, we review the means by which two microstructural features of the mesoporous film, surface area and porosity, influence the electrochemical behavior.The evolution of multiple peaks in the voltammetry experiments is a characteristic observed only with the mesoporous samples (Fig. 5). Kim and Pyun,17 who prepared porous films by magnetron sputtering, proposed that the peaks were associated with proton injection sites. The results shown here correlate with surface area; the film with the greatest surface area (via ethanol extraction) exhibited multiple peaks on the first cycle while the calcined samples required some cycling to generate the same response. Since the highest surface area is also expected to possess the greatest number of active sites, the mechanism proposed by Kim and Pyun may be involved with our films as well. A contributing factor here may be surface chemistry. The ethanol extracted films are likely to be rich in hydroxyl groups which may serve as the source of proton injection sites. The calcined materials are expected to have substantially fewer of these groups.
Another characteristic observed for the mesoporous films is the enhanced rates for the coloration and bleaching processes (Table 3). The coloration of tungsten oxide is mainly limited by a barrier at the proton-injecting contact (electrolyte–WO3 interface).20 Thus the observed decrease in coloration time (Table 3) can be related to the higher surface area (and increased current) of the mesoporous films as compared to the sol–gel films. Fig. 9a, a log–log plot of the current density (J) as a function of time (t) indicates that the mechanism of coloration is the same for the two types of materials. In Fig. 9a, a slope of −1/2 would correspond to a diffusion limited process. The fact that no such behavior is observed is consistent with previous work where coloration was shown to be controlled by charge transfer.20
 | ||
| Fig. 9 Log–log plot
of current density as a function of time for mesoporous tungsten oxide calcined
at 300°C and sol–gel tungsten oxide heated at 300°C; (a)
coloration (−0.8 V), (b) bleaching (+0.8 V). | ||
On the other hand, the bleaching kinetics show an interesting difference for the two types of films. Prior work by Faughnan et al.21 showed that the bleaching current for WO3 films in H2SO4 electrolyte decayed at a rate of t−3/4:
 | (3) |
Conclusion
The use of amphiphilic block copolymers as structure-directing agents in systems other than SiO2 offers the opportunity to create mesoporous transition metal oxide materials with interesting electrochemical and optical properties. This study has shown that mesoporous tungsten oxide exhibits the well known electrochromic properties of this oxide and that the unique microstructure leads to better electrochemical and optical reversibilities with higher rates of coloration/bleaching as compared to standard sol–gel derived tungsten oxide. The improved access of electrolyte to the oxide film via the mesoporous texture coupled with the high surface area of the film are factors which contribute to this behavior. Cyclic voltammetry and chronoamperometry measurements suggest that the mesoporous films possess proton trapping sites, a feature which is also expected from the increased surface area. The interesting prospect of increasing the kinetics of electrochromism through the presence of controlled porosity may be of substantial importance for device applications. The nature of the injected ion may also be important. Experiments in progress indicate that lithium insertion and the use of organic electrolytes result in still better reversibility (<10% capacity loss). These results will be the subject of a future report.Acknowledgements
This material is based upon work supported in part by the U.S. Army Research Office (DAAD 19-99-1-0316) and by the National Science Foundation (DMR-9729186).References
- (a) C. G. Granqvist, Handbook of Inorganic Electrochromics Materials, Elsevier, Amsterdam, 1995 Search PubMed; (b) P. M. S. Monk, R. J. Mortimer and D. R. Rosseinsky, Electrochromism: Fundamentals and applications, VCH, Weinheim, 1995. Search PubMed.
- C. G. Granqvist, Solar Energy Mater. Solar Cells, 2000, 60, 201 Search PubMed.
- O. Bohnke, M. Rezrazi, B. Vuillemin, C. Bohnke, P.A. Gillet and C. Rousselot, Solar Energy Mater. Solar Cells, 1992, 25, 361 Search PubMed.
- A. Dipaola, F. Diquarto and C. Sunseri, J. Electrochem. Soc., 1978, 125, 1344 CAS.
- R. Hurdich, Electron. Lett., 1975, 11, 142.
- P. Judeinstein and J. Livage, J. Mater. Chem., 1991, 1, 621 RSC.
- O. Lev, Z. Wu, S. Bharathi, V. Glezer, A. Modestov, J. Gun, L. Rabinovich and S. Sampath, Chem. Mater., 1997, 9, 2354 CrossRef CAS.
- B. W. Faughnan, R. S. Crandall and P. M. Heyman, RCA Rev., 1975, 36, 177 Search PubMed.
- A. Rougier, F. Portemer, A. Quédé and M. El Marssi, Appl. Surf. Sci., 1999, 153, 1 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710 CrossRef CAS.
- D. Zhao, P. Yang, Q. Huo, B. F. Chmelka and G. D. Stucky, Curr. Opin. Solid State Mater. Sci., 1998, 3, 111 CrossRef CAS.
- N. K. Raman, M. T. Anderson and C. J. Brinker, Chem. Mater., 1996, 8, 1682 CrossRef CAS.
- (a) P. Yang, D. Zhao, D. I. Margolese, B. F. Chmelka and G. D. Stucky, Chem Mater., 1999, 11, 2813 CrossRef CAS; (b) P. Yang, D. Zhao, D. I. Margolese, B. F. Chmelka and G. D. Stucky, Nature, 1998, 396, 152 CrossRef CAS.
- R. A. Walton, Prog. Inorg. Chem., 1972, 16, 1 Search PubMed.
- O. J. Klejnot, Inorg. Chem., 1965, 4, 1668 CrossRef CAS.
- L. H. M. Krings and W. Talen, Solar Energy Mater. Solar Cells, 1998, 54, 27 Search PubMed.
- D.-J. Kim and S.-I. Pyun, Solid State Ionics, 1997, 99, 185 Search PubMed.
- M. Schneider and A. Baiker, Catal. Rev. Sci. Eng., 1995, 37, 515 Search PubMed.
- The films are much more soluble in phosphoric acid..
- (a) B. W. Faughnan and R. S. Crandall, in Display Devices, ed. J. I. Pankove, Springer, Berlin, Heidelberg, 1980, Topics in Applied Physics Vol. 40, p. 181 Search PubMed; (b) P. M. S. Monk, Crit. Rev. Solid State Mater. Sci., 1999, 24(3), 193 Search PubMed.
- B. W. Faughnan, R. S. Crandall and M. A. Lampert, Appl. Phys. Lett., 1975, 27, 275 CrossRef CAS.
Footnote |
| † Basis of a presentation given at Materials Discussion No. 3, 26–29 September, 2000, University of Cambridge, UK. |
| This journal is © The Royal Society of Chemistry 2001 |