Delamination and restacking of layered double hydroxides†
Fabrice Leroux*, Mariko Adachi-Pagano, Mourad Intissar, Samuel Chauvière, Claude Forano* and Jean-Pierre Besse
Laboratoire des Matériaux Inorganiques, CNRS UPRES-A no. 6002, Université Blaise
Pascal, 63177, Aubière
cédex, France. E-mail: forano@cicc.univ-bpclermont.fr; fleroux@chimtp.univ-bpclermont.fr
First published on UnassignedUnassigned6th October 2000
Abstract
Layered double hydroxides recovered after exfoliation have been characterized by solid state chemistry techniques. The nature of the recovered materials is highly dependant on the drying process; when gently dried, a well-ordered phase is obtained, either by freeze-drying or reconstruction, but the material becomes amorphous after evaporation of the solvent. Delamination was used to prepare interstratified LDHs, making this technique a new route to the formation of a wide range of tunable materials.
Introduction
It is of interest to prepare highly dispersed phases from low-dimensional solids and, to some extent, single layers. Much research has been devoted to this field, which is rapidly expanding to include a large number of materials.1,2 Exfoliation is of practical use in preparing pillared materials and is also involved in nanocomposite preparation.3,4 By taking advantage of the anisotropy in the chemical bonding, individual layers may be obtained by spontaneous exfoliation of the solids in aqueous solution. This is the case for clay minerals with lower layer charge density, such as laponite, which readily swells in water.5,6 More generally, dispersion of clays in water, so-called “liophobic colloidal systems”, has been extensively studied with regard to smectite-type materials.7,8 Added to their relatively low layer charge, these systems display characteristics conducive towards exfoliation, such as an edge to surface net charge difference combined with a small particle size. It is well established that the two properties promote the de-aggregation process, with the former considered as an initiator of exfoliation. Although delamination is much more difficult when the layers are tightly bound together, i.e. when the charge density of the layers is high, and where the particle size is large, in order to achieve exfoliation, it is necessary to diminish the attractive forces between layers via the exchangeable ions. One method consists of converting the 2D solid into its protonic form and reacting this with a suitable large basic molecule, e.g. layered protonic titanate with alkyl ammonium cations.9,10 Exfoliation is observed after stirring for seven days at room temperature. Exfoliation of zirconium hydrogen phosphate11 and oxovanadium phosphate12 requires propyl or butylamine. Delamination of layered chalcogenides was reported for MoS213,14 and TaS2.15 MoS2 disperses into single layers on reaction of LixMoS2 with water, allowing the preparation of a large variety of nanocomposites.16 In the case of layered double hydroxides, a two-step method has been used to obtain a highly stable colloidal suspension; exchange with an anionic surfactant was followed by reaction in refluent BuOH.17 In contrast to their cationic counterparts, anionic clays display a layer charge density as high as for mica associated with a large particle, making ultrasonic treatment inefficient in pushing apart the layers. Up to now, the incorporation of large guest compounds such as polyoxometallate anions, such as α-(SiW11O39)8−,18 or metalloporphyrin anions19 into an LDH matrix is only successful after pre-swelling using spacers, such as terephthalate ions,20 or by reconstruction.21 After transformation into an amorphous state (often referred to as the LDO—layered double oxide) via thermal treatment, LDH materials can be recovered. However, this process is not universal; some compositions (such as CoRFe; R is the ratio of MII to MIII) do not present such behavior due to the formation of stable phases at high temperature or due to their surface acidity.21 Alternative methods for exchange reactions with large guest compounds are: direct synthesis by adjusting the pH,22 reaction in an ethanol–water mixture,23 or in glycerol,24 or micellar organic phase extraction.25In this paper, we describe the delamination of the dodecylsulfate-exchanged LDH ZnRAl(OH)2(1 + R){CH3(CH2)11SO4}·nH2O and its reconstitution. We will show that our method can be used to prepare a wide range of materials, such as those obtained by interstratification of two LDHs with layer charge densities matching each other. Delamination–restacking represents a new pathway for the preparation of tunable materials from LDHs.
Experimental section
Preparation of the materials, and description of the delamination process
The starting materials (Cl)-ZnRAl (R = 2, 3 and 4) were prepared by a coprecipitation, as described by Miyata.26 A solution of ZnCl2 (Aldrich) and AlCl3 (Aldrich) in the molar ratio R was prepared. This solution was added dropwise into 250 ml decarbonated water under vigorous stirring, at a constant pH value depending on R (pH = 8.3, 8.7 and 9.3 for R = 2, 3, and 4, respectively). The addition of NaOH (1 M) was complete after 24 h. The suspension was aged in the mother liquor with stirring for 24 h. The white solid products were isolated by repeated centrifuging and washing with decarbonated water and were finally dried at room temperature.The amount of sodium dodecylsulfate salt (Prolabo, 98%) added corresponded to twice the anionic exchange capacity (AEC) of the LDHs. The solution was stirred for 72 h under a nitrogen atmosphere. The exchanged (DS)-LDH was isolated using the same technique described for the pristine material.
After drying at room temperature, 20 mg of the DS-exchanged
LDH was dispersed in 100 ml butanol by a 15 min ultrasonic treatment,
and then placed under nitrogen gas. BuOH (Acros, 99%) was
used as received. The delamination process was performed overnight under reflux
conditions at 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C. Larger amounts of surfactant derivative LDH,
up to 150 mg, delaminate in 100 ml BuOH solution over a longer
period.
°C. Larger amounts of surfactant derivative LDH,
up to 150 mg, delaminate in 100 ml BuOH solution over a longer
period.
Solid materials were recovered after drying. The solvent was removed by
evaporation at 120°C or by freeze-drying. The dried materials
were washed with decarbonated water and then dried at room temperature. Restacking
of the LDH layers in the presence of Cl− or CO32−
ions was performed by adding an aqueous solution containing the sodium salt
to the mother liquor. The materials obtained were characterized by elemental
analysis (Service Central d'Analyse, CNRS, Vernaison, France).
Characterization techniques
Powder X-ray diffraction patterns were obtained with a Siemens D500 diffractometer (Cu-Kα radiation). Patterns were recorded over the 2θ range 2 to 75° in steps of 0.04° with a count time of 2 s. FTIR spectra were recorded on a Perkin-Elmer 2000 FT spectrometer employing the KBr dilution technique. Thermogravimetric (TG) analysis were performed on a Setaram TGA 92 instrument with a linear heating rate of 5°C min−1 under air. Scanning
electron micrographs were recorded with a Cambridge Stereoscan 360 operating
at 20 kV at Techinauv, A. S. (Aubière, France).X-Ray absorption spectroscopy
Zn K-edge XAFS (X-ray absorption fine structure) studies were performed at LURE (Orsay, France) using X-ray synchrotron radiation emitted by the DCI storage ring (1.85 GeV positrons, average intensity of 250 mA) at the D44 line. Data were collected at room temperature in transmission mode at the Zn K-edge (9658.6 eV). The quantity of powdered sample was chosen to obtain edge jumps of about Δμx ≈ 1. A double-crystal Si(111) monochromator scanned the energy in 2 eV steps from 100 eV below to 900 eV above the Zn K absorption edge, three spectra were recorded for each sample. The accumulation time was 2 s per point. After extraction by standard procedures, the EXAFS spectra were evaluated by the classical plane wave single scattering approximation. Long metal to metal distance correlations P3 and P4 (see text) arising from multiple scattering phenomena were not refined. Fourier transformations of EXAFS spectra were made after multiplication of the signal by a k3 factor over a 2.8–14 Å−1 Kaiser apodization window with τ = 2.5. The resultant χ(k) signal was fitted by using the formula χ(k) = S02ΣAi(k) sin[2kri + ϕi(k)], with the amplitude Ai(k) = (Ni/kri2)F(k) exp(−2k2σi2), where ri is the interatomic distance, ϕi the total phase shift of the ith shell, Ni the effective coordination number, σi the Debye–Waller factor, and Fi(k) the backscattering amplitude. EXAFS signal treatments27 and refinements were performed using a proprietary program package at LURE.28 The residual ρ factor is defined as ρ = [Σ{k3χexp(k) − k3χtheo(k)}2/Σ{k3χexp(k)}2]1/2. To estimate the relative part of the cations contributing to the metal–metal correlation, the number of backscattering atoms was free to move during the refinement. This guarantees all possible ratios between Al3+ and Zn2+ for the second shell. The commonly accepted fitting accuracy is about 0.02 Å for the distance and 10 to 20% for the number of neighbors.Results and discussion
Characterization of ZnRAl LDHs
The molar ratio Zn/Al and the relative ion contents of the prepared materials are very close to the expected values, indicating that the reaction was complete (Table 1). As the Zn2Al sample shows the highest layer charge in the LDH series, the delamination process was studied in detail.ZnRAl materials are well crystallized, as can be seen
from the X-ray diffraction pattern (Fig. 1),
the diffraction peaks are typical of the layered double hydroxide structure.29 The reflections were indexed in a hexagonal lattice
with a R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m rhombohedral symmetry, commonly used
as a description of the LDH structure. Miller indexing is given in Fig. 1 and refined cell parameters are reported
in Fig. 2. An increase in the Zn to
Al ratio increases the unit cell parameters. As a corresponds to
the metal to metal distance, replacement of Zn2+ cations by
the smaller Al3+ cations in the octahedral sheet decreases
the a parameter. The increase in the interlamellar distance is related
to the decrease of the net charge of the layers with the gradually diminishing
Al3+ content as the cause of the expansion of the layers.
m rhombohedral symmetry, commonly used
as a description of the LDH structure. Miller indexing is given in Fig. 1 and refined cell parameters are reported
in Fig. 2. An increase in the Zn to
Al ratio increases the unit cell parameters. As a corresponds to
the metal to metal distance, replacement of Zn2+ cations by
the smaller Al3+ cations in the octahedral sheet decreases
the a parameter. The increase in the interlamellar distance is related
to the decrease of the net charge of the layers with the gradually diminishing
Al3+ content as the cause of the expansion of the layers.
 | ||
| Fig. 1 Diffraction patterns of (Cl)-ZnRAl, (a) R = 2, (b) R = 3 and (c) R = 4. The first two reflections of the DS derivative are displayed in the inset. | ||
 | ||
| Fig. 2 Evolution of the lattice parameters a (square) and c (circle) as a function of R. The layer charges of (CO32−)-Zn2Alrecov was estimated from the (110) position (see text). | ||
DS incorporation between the layers of the ZnRAl LDHs phases is confirmed by the increase in the basal spacing. Its value is the sum of the layer thickness, estimated to be 4.8 Å for the brucitic-like layers, and the interlayer distance. A basal spacing of 25.2 Å (Fig. 1 inset) indicates that the surfactant tails are interdigitated in the interlamellar space, as the length of the whole DS molecule is 20.8 Å.30 Our attempts to prepare ZnAl LDHs with the same DS arrangment (bilayer) as in Ni4Al31 and Mg2Al LDH32 were not successful.
The thermal behaviour of DS-exchanged derivatives was studied (Fig. 3a). To assign weight losses, thermogravimetric
analysis was also conducted on sodium dodecylsulfate salt. DS-Na decomposes
in two steps, centered at 210 (denoted 3) and 255°C (denoted
3′). The total weight loss is in agreement with the formation of
sodium sulfate (observed by X-ray diffraction at 700°C)
from two surfactant molecules (Δmexp = 76.5%, Δmth = 75.4%).
The small DTA signal (Fig. 3b)
before 100°C indicates a conformational rearrangment of the molecule
before it decomposes. For (Cl)-Zn2Al, loss of intracrystalline
water (1) and dehydroxylation (2) occurs at 150 and 250°C,
respectively. For the DS derivative, the three processes (1 + 2 + 3)
below 400°C are superimposed (Fig. 3b),
making it difficult to dissociate the water loss and dehydroxylation (1 + 2)
processes from the decomposition of the organic species. Due to intercalation,
decomposition of interlayer DS does not proceed as described for the surfactant
molecule. Decomposition of the alkyl chains is the first process. The CH2
vibration bands disappear in the IR spectra of (DS)-Zn2Al
heated to 200°C, and are completely absent after 400°C (Fig. 3c). In the meantime, the SO4
polyhedra vibration is shifted by ca. 100 cm−1
to lower frequency, indicating the local symmetry changes from C3v
(in
DS) to Td in SO42−
(Fig. 3c). This entire thermal process has
been proposed previously.31
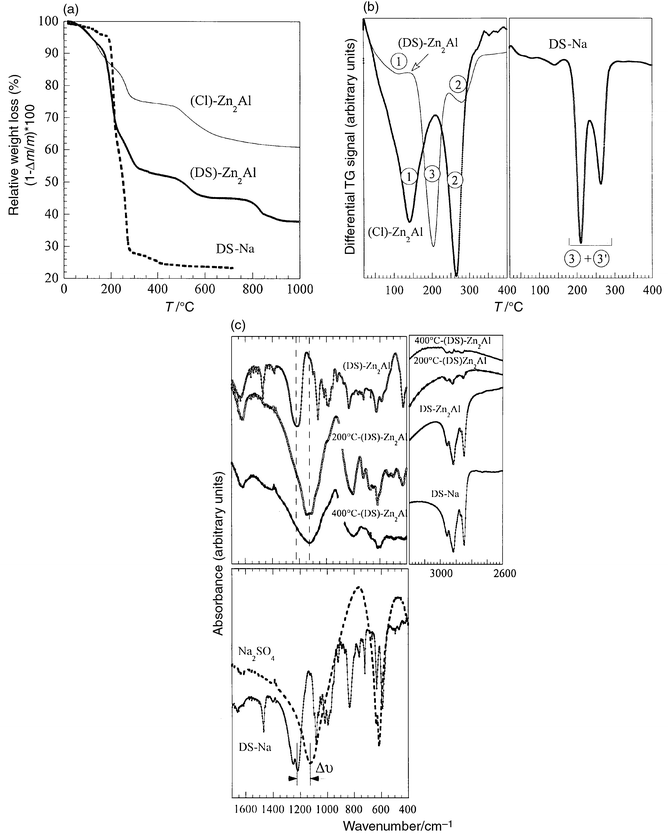 | ||
| Fig. 3 DTA and DTG diagrams
of (Cl)-ZnAl, (DS)-ZnAl and DS: (a) DTA and
(b) DTG (weight losses are indicated). (c) FTIR spectroscopy:
IR spectra of the sample heated to 200 and 400°C and of Na2SO4
are also shown. | ||
As Zn2+ and Al3+ retain the same oxidation state during the thermal treatment, the molecular mass can be calculated from the total weight loss according to the equation: MW of ZnRAl(OH)2(1 + R){CH3(CH2)11SO4}·nH2O*(1 − Δm/m) = MW of “ZnRAlOR + 3/2”, where Δm/m represents the weight loss in percent obtained from the TGA curve (Fig. 3a). Mw obtained from this equation is in agreement with the elemental analysis data [for R = 2, Δm/m = 0.63 (Δm/mtheor = 0.66); for R = 3, Δm/m = 0.58 (Δm/mtheor = 0.58)].
Delamination process
The process of delamination in BuOH is summarized in Fig. 4. Other solvents were also used. It was previously mentioned that dispersion of (DS)-Zn2Al in methanol, ethanol, propanol or hexane under reflux conditions yields unstable colloidal suspensions, whereas higher alcohols, such as pentanol and hexanol, give rise to stable translucent solutions. The optimal time for completion of the delamination process depends on the nature of the LDH material. Twelve hours reaction was required for complete delamination of Zn2Al. | ||
| Fig. 4 Pathways for LDH synthesis, DS-exchange, delamination in BuOH and recovery by drying. The morphology of the samples is indicated by SEM or TEM pictures. | ||
Before any treatment, the samples were dried at room temperature. It is well known that the water content has an important effect on the particle texture.33 Since the water content may also influence the delamination process (vide infra), (DS)-Zn2Al was dried in vacuum overnight. This treatment did not change the X-ray diffraction pattern of the material. After refluxing in BuOH, the resulting solution was cloudy/milky, indicating that the solid was mostly highly dispersed but had not been exfoliated. After one hour, a white powder settled to the bottom of the vessel and this material was dried and analyzed. The phase exhibits a strong contraction of the interlayered domain from 25.2 to 16.8 Å. This indicates a high degree of interdigitation of the alkyl chains. Considering the thickness of the brucitic layer (vide supra), the surfactant tails are tilted at ca. 57° from the perpendicular axis of the inorganic layers (along c). Also, some of the surfactant anions were removed from the interlayer space as indicated by the TG analysis (not shown). The final weight loss step, attributed to the decomposition of SO4 to SO2, remained quantitatively high, showing that SO42− anions remained in the structure, balancing the layer charge.
Thus, it appears that vacuum treatment impedes delamination of Zn2Al. The water content plays a major role in the delamination process and we believe that the replacement of the water molecules by the solvent molecules is the key process in exfoliation. The intercrystalline water could also influence the delamination process. Several LDH samples were kept for three to four weeks at various humidities (20, 40, 60 and 80% R.H.). Comparatively, LDHs kept at low RH did exfoliate, although at an R.H. as high as 80% a cloudy BuOH suspension remained. In order to prevent particle aggregation due to the effect of water, freshly prepared (DS)-LDH materials were washed with ethanol, then butanol and directly suspended in BuOH for delamination. The material exfoliated, as was evidenced by the formation of a translucent solution.
Reconstruction
Highly 2D-oriented material was recovered after freeze-drying (Fig. 4) and the X-ray diffraction pattern obtained is displayed in Fig. 5a. Initially, it cannot be said for certain that the recovered solids are LDH-like materials, rather just lamellar materials. No diffraction peak was observed for the sample recovered by evaporation. The interlamellar distance of the freeze-dried material, 29.3 Å, was larger than that of the pristine DS derivative. This may indicate a rearrangment of the DS molecules or the incorporation of further guest molecules into the interlayer space. An additional weight loss is observed at 600°C for both samples, which is more pronounced
after evaporation (Fig. 5b).
The SO4 vibration band was much larger in the latter case, showing
that the DS heads are not the sole cause of this peak (Fig. 5c).
In all probability, this is related to sulfate groups produced by decomposition
of DS. Vibration bands from the inorganic lattice between 400 and 700 cm−1
were unchanged compared to the precusor.34
The general features of the spectra are mostly unchanged after the freeze-drying
process, although bands attributed to (O–M) vibrations were
broadened after evaporation, due to the loss of stacking cohesion. This shows
that the recovered solids are highly sensitive to the drying process, giving
either ordered or disordered phases at an atomic scale, as confirmed by XAS
studies (vide infra). | ||
| Fig. 5 Comparison of recovered materials after freeze-drying and evaporation: (a) X-ray diffraction, (b) thermal analysis and (c) FTIR spectroscopy. | ||
To test the LDH nature of the solids obtained by BuOH treatment, the samples were reacted with an aqueous K2CO3 solution. The Zn/Al ratio was slightly decreased after the delamination–restacking process (Zn/Alrecov of 1.94 and 2.81, initial 2.01 and 2.98). The X-ray diffraction pattern of the product exhibits well-defined peaks (Fig. 6), which are characteristics of the LDH structure. The unit cell parameter a can be employed as an appropriate estimation of the metal ratio.35 The cell parameter a exhibits a linear correlation with the Zn/Al ratio. The position of the (110) peak of the (CO32−)-Zn2Alrecov material corresponds to a layer charge of 0.345 per metal atom, leading to a Zn/Al ratio of approximately 1.9, close to the value found by chemical analysis. The same trend was found for the (CO32−)-Zn3Alrecov sample. These results show that refluxing in BuOH induces a slight dissolution of the layers.
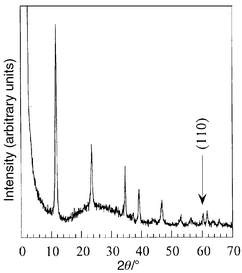 | ||
| Fig. 6 X-Ray diffraction pattern of the recovered Zn2Al sample after addition of an aqueous solution of K2CO3. | ||
Local order study
In order to get a better insight into the delamination process, Zn K-edge spectra were analyzed. The moduli of the Fourier transformations, which correspond to the pseudo-radial distribution function (pseudo-RDF), of the EXAFS spectra of pristine (Cl)-ZnAl and its DS derivatives (with 16 and 25 Å basal spacing) are presented in Fig. 7a. The identical distribution curves show that the local order around the Zn atoms remains unchanged after the intercalation of surfactant anions in both the 16 and 25 Å phases. Furthermore, the long range order is also identical for these two samples, as exemplified by the presence of P3 and P4 peaks, arising from Zn–Me at distances of √3a and 2a, respectively.36 As the cations differ in their “electronic weight” (difference in backscattering phase and amplitude functions), the nature of the backscattering atoms composing the second shell of coordination can be distinguished. The refinements of the first two shells, which contain only single scattering contributions, are reported in Table 2. The results for the two samples (16 and 25 Å) are fairly similar, given to the identical distribution functions. The first shell corresponds to the presence of oxygen atoms, the result is in agreement with an octahedral environment of OH groups around the Zn2+. The Zn–O distance is expected to have a value of 2.11 Å in an Oh environment, from bond-valence calculations.37 The second shell is related to the presence of metal, i.e. Al3+ and Zn2+ cations. For a composition Zn2Al, intraplanar cationic order comprises a shell around MII composed of 3 Al3+ and 3 Zn2+ cations, whereas 6 Zn2+ ions surround the MIII centers.38,39 The results are close to the idealized sharing of ca. 50% between the two cations in the second shell around Zn2+. The total number of cations was slightly overestimated by the kχ(k) refinement, but the number of neighboring atoms was accurate, within the error limits.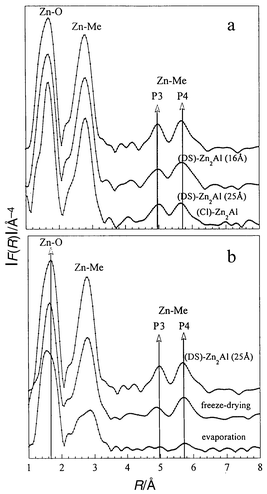 | ||
| Fig. 7 Fourier transform spectra (Zn
K-edge): (a)
(Cl)-Zn2Al, 25 Å-(DS)-Zn2Al,
16 Å-(DS)-Zn2Al, (b) delaminated
pristine 25 Å-(DS)-Zn2Al, and the solids recovered
by freeze-drying or by evaporation at 120°C. Distances are
given without phase shift corrections. | ||
| Sample | Bond | Ni | Ri/Å | σi2/Å2 | ρ (%) |
|---|---|---|---|---|---|
| (Cl)-ZnAl | Zn–O | 6.0 | 2.09 | 0.0086 | 0.4 |
| Zn–Zn | 4.2 | 3.12 | 0.0144 | ||
| Zn–Al | 4.0 | 3.00 | 0.0174 | ||
| 25 Å-(DS)-ZnAl | Zn–O | 5.8 | 2.08 | 0.0076 | 0.5 |
| Zn–Zn | 5.0 | 3.10 | 0.0119 | ||
| Zn–Al | 4.0 | 2.99 | 0.0130 | ||
| 16 Å-(DS)-ZnAl | Zn–O | 6.0 | 2.08 | 0.0079 | 0.6 |
| Zn–Zn | 4.0 | 3.11 | 0.0119 | ||
| Zn–Al | 3.5 | 3.01 | 0.0119 | ||
| (DS)-ZnAlrecov (freeze-dried) | Zn–O | 5.7 | 2.05 | 0.0085 | 0.5 |
| Zn–Zn | 3.0 | 3.11 | 0.0117 | ||
| Zn–Al | 2.9 | 3.01 | 0.0134 | ||
| (DS)-ZnAlrecov (evaporation) | Zn–O | 5.6 | 2.03 | 0.0100 | 0.3 |
| Zn–Zn | 2.5 | 3.11 | 0.0161 | ||
| Zn–Al | 2.4 | 3.01 | 0.0139 |
The effect of the delamination process is shown in Fig. 7b by a comparison of the moduli of the Fourier transform spectra of the solids reconstituted by evaporation of the solvent or freeze-drying. At a first glance, the Zn K-edge spectra appear to be similar, retaining most of the expected features for both samples, that is to say Zn–O and Zn–metal correlations. However, the intensity of the P3 and P4 peaks is much reduced (or even absent) for the solid recovered after evaporation. This suggests a random disorder or a strong distortion of the framework, both of which would cancel out the focussing effect which is enhanced by the linear Me–Me situation.40 Given the data obtained from IR spectroscopy and X-ray diffraction, the first case has to be considered; the material is highly disordered and decomposed, and appears almost amorphous. Zn2+ seems to retain a six-fold coordination sphere of oxygen atoms, although the Zn–O distance is shortened from 2.09 to 2.03 Å. Refinement curves are presented in Fig. 8. The recovered samples display a proportion of Zn2+ and Al3+ cations in the second coordination shell of roughly 50% each. In spite of its amorphous nature, the material recovered after evaporation shows the atomic arrangment characteristic of the local order of Zn2Al LDH, ruling out the possibility of phase segregation.
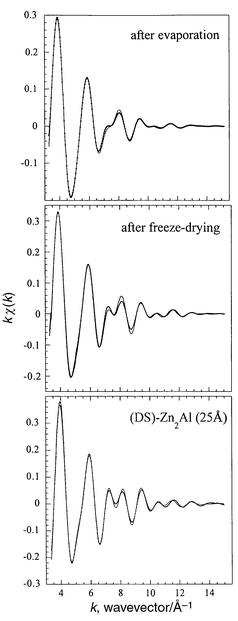 | ||
| Fig. 8 kχ(k)
Refinement of the first three shells for 25 Å-(DS)-Zn2Al,
and the solids recovered by freeze-drying or evaporation at 120°C.
Dots are experimental data. | ||
Delamination was used to synthesize composite LDH materials by interstratification
during recovery. Nanocomposites with alternating layers have previously been
prepared by reaction of MoS2 single layers with Co2+41 or intercalation of LiAl2(OH)6+
into montmorillonite.42 Some minerals, such
as tochilinite43 or yushikinite,44
also present alternating layers; in these two cases, MgAl LDH-like layers
alternate with incommensurable layers of FexS or VS2,
respectively. LDH materials were chosen so that their layer charge densities
matched (same MII/MIII ratio). Solutions
of delaminated (DS)-Zn2Al and (DS)-Zn2Cr
were mixed, BuOH treatment was carried out for an additional 12 h and
the solution was allowed to evaporate at 120°C. The solid obtained
remained amorphous. Several lattice vibrations are clearly visible in the
IR spectrum of the product (Fig. 9).
The general cation composition can be written as (Zn2Cr)p(Zn2Al)1 − p
(p ≈ 0.5). The characterization
of this composite material and interstratification of other LDHs are presently
being studied.
 | ||
| Fig. 9 FTIR spectrum of an interstratified LDH of average layer composition “(Zn2Al)1−p(Zn2Cr)p(OH)2”. | ||
Conclusion
The dodecylsulfate of Zn2Al LDH was exfoliated in BuOH solution. The delamination process was sensitive to the preparation mode, and especially the drying process. We also succeeded in delaminating (DS)-ZnRAl materials.After recovery, the ratio of surfactant to LDH was found to be altered,
particularly after evaporation of BuOH at 120°C, leading to an
amorphous-like material. However, small structural units are visible by
X-ray absorption spectroscopy.
Acknowledgements
The authors are grateful to Dr V. Briois for her assistance with the XAS experiments and to Dr H. Roussel for having compared the XAS refinements using FeFF6. We acknowledge the experimental opportunities at LURE (Orsay, France).References
- J. Jacobson, in Comprehensive Supramolecular Chemistry: Colloidal Dispersion of Compounds with Layer and Chain Structures, ed. G. Alberti and T. Bein, Elsevier, Oxford, 1996, vol. 7, p. 315. Search PubMed.
- R. Schöllhorn, Chem. Mater., 1996, 8, 1747 CrossRef.
- P. B. Messersmith and E. P. Giannelis, Chem. Mater., 1994, 6, 1719 CrossRef CAS.
- Z. Wang and T. J. Pinnavaia, Chem. Mater., 1998, 10, 3769 CrossRef CAS.
- J. D. F. Ramsay, J. Colloid. Interface Sci., 1986, 109, 441 CrossRef CAS.
- P. A. Brahimi, P. Labbe and G. Reverdy, Langmuir, 1992, 8, 1908 CrossRef.
- S. Sonin, J. Mater. Chem., 1998, 8, 2557 RSC.
- H. van Olphen, in Chemistry of Clays and Clay Minerals, ed. A. C. Newman, Wiley, New York, 1987, p. 203. Search PubMed.
- T. Sasaki and M. Watanabe, J. Am. Chem. Soc., 1998, 120, 4682 CrossRef CAS.
- H. Rebbah, M.M. Borel and B. Raveau, Mater. Res. Bull., 1980, 15, 317 CrossRef CAS.
- E. W. Stein, Sr., C. Bhardwaj, C. Y. Ortiz-Avila, A. Clearfield and M. A. Subramanian, Mater. Sci. Forum, 1994, 115, 152 Search PubMed.
- T. Nakato, Y. Furumi, N. Terao and T. Okuhara, J. Mater. Chem., 2000, 10, 737 RSC.
- P. Joensen, E. D. Crozier, N. Alberding and R. F. Frindt, J. Phys. C: Solid State Phys., 1987, 20, 4043 Search PubMed.
- J. Heising and M. G. Kanatzidis, J. Am. Chem. Soc., 1999, 121, 638 CrossRef CAS.
- L. F. Nazar and A. J. Jacobson, J. Mater. Chem., 1994, 4, 1419 RSC.
- M. G. Kanatzidis, R. Bissessur, D. C. de Groot, J. L. Schindler and C. R. Kannewurf, Chem. Mater., 1993, 5, 595 CrossRef CAS , and references therein.
- M. Adachi-Pagano, C. Forano and J.-P. Besse, Chem. Commun., 2000, 91 RSC.
- E. Narita, P. D. Kaviratna and T. J. Pinnavaia, J. Chem. Soc., Chem. Commun., 1993, 60 RSC.
- S. Bonnet, C. Forano, A. de Roy, J.-P. Besse, P. Maillard and M. Momenteau, Chem. Mater., 1996, 8, 1962 CrossRef CAS.
- M. A. Drezdon, Inorg. Chem., 1988, 27, 4628 CrossRef CAS.
- K. Chibwe and W. Jones, J. Chem. Soc., Chem. Commun., 1989, 926 RSC.
- F. Kooli and W. Jones, Inorg. Chem., 1995, 34, 6238.
- T. Hibino and A. Tsunashima, Chem. Mater., 1997, 9, 2082 CrossRef CAS.
- E. D. Dimotakis and T. J. Pinnavaia, Inorg. Chem., 1990, 29, 2394.
- E. L. Crepaldi, P. C. Pavan and J. B. Valim, Chem. Commun., 1999, 155 RSC.
- S. Miyata, Clays Clay Miner., 1983, 31, 305 Search PubMed.
- G. McKale, B. W. Veal, A. P. Paulikas, S.-K. Chan and J. Knapp, J. Am. Chem. Soc., 1988, 110, 3763 CrossRef.
- A. Michalowicz, Round Midnight, EXAFS Signal Treatment and Refinement Programs, LURE, Orsay, France. Programs available on the LURE Web site; http://www.LURE.fr..
- M. Bellotto, B. Rebours, O. Clause, J. Lynch, D. Bazin and E. Elkaïm, J. Phys. Chem., 1996, 100, 8527 CrossRef CAS.
- S. Sundell, Acta Chem. Scand. A, 1971, 31, 799 Search PubMed.
- A. Clearfield, M. Kieke, J. Kwan, J. L. Colon and R.-C. Wang, J. Inclusion Phenom. Mol. Recognit. Chem., 1991, 11, 361 CAS.
- T. Kanoh, T. Shichi and K. Takagi, Chem. Lett., 1999, 117 CrossRef CAS.
- S. K. Yun and T. J. Pinnavaia, Chem. Mater., 1995, 7, 348 CrossRef CAS.
- J. T. Kloprogge and R. L. Frost, J. Solid State Chem., 1999, 146, 506 CrossRef CAS.
- E. Lopez-Salinas, M. Garcia-Sanchez, J. A. Montoya, D. R. Acosta, J. A. Abasolo and I. Schifter, Langmuir, 1997, 13, 4748 CrossRef CAS.
- H. Roussel, V. Briois, E. Elkaïm, A. de Roy and J.-P. Besse, J. Phys. Chem., 2000, 104, 5915 CrossRef CAS.
- N. E. Brese and M. O'Keefe, Acta Crystallogr., Sect. B, 1991, 47, 192 CrossRef.
- W. Hofmeister and H. von Platen, Cryst. Rev., 1992, 3, 3 Search PubMed.
- M. Vucelic, W. Jones and G. D. Moggridge, Clay Clay Miner., 1997, 45, 803 Search PubMed.
- N. Alberding and E. D. Crozier, Phys. Rev. B, 1983, 27, 3374 CrossRef CAS.
- S. Golub, C. Payen, G. A. Protzenko, Yu. N. Novikov and M. Danot, Solid State Commun., 1997, 102, 419 CrossRef.
- Q. Feng, C. Honbu, K. Yanagisawa, N. Yamasaki and S. Komarneni, J. Mater. Chem., 2000, 10, 483 RSC.
- G. A. Kakos, T. W. Turney and T. B. Williams, J. Solid State Chem., 1994, 108, 102 CrossRef CAS.
- Y. Moëlo, O. Rouer, L. Cario and B. Cervelle, Miner. Mag., 1999, 63, 879 Search PubMed.
Footnote |
| † Basis of a presentation given at Materials Discussion No. 3, 26–29 September, 2000, University of Cambridge, UK. |
| This journal is © The Royal Society of Chemistry 2001 |