Lix(Mn1 − yCoy)O2 intercalation compounds as electrodes for lithium batteries: influence of ion exchange on structure and performance†
Alastair D. Robertson, A. Robert Armstrong, Amelia J. Fowkes and Peter G. Bruce*
School of Chemistry, University
of St. Andrews, St. Andrews, Fife, UK KY16 9ST. E-mail: pgb1@st-and.ac.uk
First published on UnassignedUnassigned6th October 2000
Abstract
LixMn1 − yCoyO2
compounds were synthesised by a low temperature route involving ion exchange
from sodium precursors. Neutron diffraction confirmed that the structures
are layered (space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m). Materials
synthesised from the precursors by ion exchange using LiBr in ethanol at 80
m). Materials
synthesised from the precursors by ion exchange using LiBr in ethanol at 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C
possess vacancies on the transition metal sites which pin residual Na+
ions. Such transition metal vacancies and Na+ ions are not
observed on refluxing at 160°C in hexanol. We show that lithium
intercalation accompanies the ion exchange process. The presence of Na+
in the Li+ layered materials induces disorder perpendicular
to the layers and this has been modelled. The performance of the materials
depends on the ion exchange conditions. The y = 0.025
compound obtained in ethanol exhibits a particularly high capacity to cycle
lithium. The initial discharge capacity is 200 mA h g−1
with a fade rate of only 0.08% per charge/discharge cycle on extended
cycling. This performance is delivered despite conversion to a spinel-like
phase during cycling and is markedly superior to the cycling ability of directly
prepared spinels over a similar composition range.
°C
possess vacancies on the transition metal sites which pin residual Na+
ions. Such transition metal vacancies and Na+ ions are not
observed on refluxing at 160°C in hexanol. We show that lithium
intercalation accompanies the ion exchange process. The presence of Na+
in the Li+ layered materials induces disorder perpendicular
to the layers and this has been modelled. The performance of the materials
depends on the ion exchange conditions. The y = 0.025
compound obtained in ethanol exhibits a particularly high capacity to cycle
lithium. The initial discharge capacity is 200 mA h g−1
with a fade rate of only 0.08% per charge/discharge cycle on extended
cycling. This performance is delivered despite conversion to a spinel-like
phase during cycling and is markedly superior to the cycling ability of directly
prepared spinels over a similar composition range.
Introduction
Rechargeable lithium batteries have developed into a major technology in the last ten years. Production in 1998 reached 600 million cells worldwide and has typically doubled each year. The reason for this success is that such batteries can store up to three times the energy per unit weight and volume compared with conventional rechargeable batteries. As a result, rechargeable lithium batteries are revolutionising a variety of commercial sectors including mobile communications and mobile computing, as well as making literally lifesaving contributions in medicine by powering implantable devices. Although apparently at opposite ends of the chemistry spectrum there is a certain similarity between lithium battery technology and pharmaceuticals. In both cases the key to advancing the commercial product lies in research into new compounds. In the field of lithium batteries this means discovering and developing new positive and negative electrodes as well as new electrolytes. Once obtained, the engineering required to exploit the new materials in cells is not as formidable as in the exploitation of many advances in chemistry.The current generation of lithium-ion batteries utilise the layered intercalation compound LiCoO2 as the positive electrode.1,2 On charging such cells, lithium is extracted from LiCoO2, the ions pass across the electrolyte and are inserted between the carbon layers in the graphite negative electrode, discharge reverses this process thus reforming LiCoO2 at the positive electrode. One of the greatest challenges in developing a new generation of rechargeable lithium batteries lies in the need to replace LiCoO2 with a new lithium intercalation host that is cheaper, less toxic and safer when in the charged state than LiCoO2 (Co4+, present in LixCoO2, is not a particularly stable oxidation state of cobalt, especially when in the presence of an organic electrolyte). It is also important that any new alternative can store, and hence deliver, more charge on cycling the cells. The LiCoO2 electrode can only be cycled over a composition range corresponding to the removal and insertion of one half of the lithium ions giving rise to a specific capacity of only 130 mA h g−1.3
Manganese oxides are particularly attractive alternatives to LiCoO2
because they have the potential to offer advantages such as lower cost and
toxicity as well as improved safety.4,5
As a result, considerable effort has been expended on optimising the spinel
intercalation host LiMn2O4 as a positive electrode for
rechargeable lithium batteries despite its rather low practical capacity of
110 mA h g−1.4,6–9
Given the success of the layered structure-type, as embodied in LiCoO2,
there has been much interest worldwide in the synthesis and investigation
of layered LiMnO2. This compound has proved elusive but has, quite
recently, been prepared for the first time.10,11
The synthesis involves using Na+ to template the formation
of a layered structure, then replacement of Na in NaMnO2 by Li.
The practical capacity of stoichiometric LiMnO2 is not significantly
higher than LiMn2O4 spinel.11–13
Doping LiMnO2 by replacing some Mn with other ions such as Co or
Al has been carried out.14–17
Lix(Mn1 − yCoy)O2,
like the parent LiMnO2, cannot be synthesised directly but may
be obtained from the analogous sodium phase by ion exchange. In earlier studies
this was achieved by refluxing at 160°C in hexanol containing
an excess of LiBr. More recently we have found that ion exchange in ethanol
at lower temperatures yields materials with superior performance as electrodes
in rechargeable lithium batteries.18 Here
we describe in more detail the synthesis of Lix(Mn1 − yCoy)O2 under different ion exchange conditions and the influence of such
conditions on the structure and performance of the materials.
The layered compounds discussed in this paper possess the O3 structure defined by an ABC stacking of close packed oxide layers (i.e. cubic close packing). Dahn's group are exploring different layered oxides based on the O2 stacking of oxide layers (i.e. ACAB).19
Experimental
Nax(Mn1 − yCoy)O2 phases were prepared by mixing Na2CO3(Aldrich, 99.5+%), Mn(CH3CO2)2·4H2O (Aldrich, 99+%) and Co(CH3CO2)2·4H2O (Aldrich, 98+%) in distilled water and in stoichiometric ratios corresponding to x = 1. Following rotary evaporation at 80°C,
the mixtures were heated in air, first at 250°C for 12 h
then at 630–720°C for 1 h, before being quenched
to room temperature. The higher temperatures were employed for the more Co
rich compositions.A range of sodium phases with different Co contents were then subjected
to ion exchange. Attempts to form the lithium phase by exchange in molten
salts such as LiNO3/LiCl proved unsuccessful in that the solid
converted to spinel. The most aggressive conditions which still preserved
the layered structure involved refluxing in hexanol at 160°C with
a 7–8 fold molar excess of LiBr. By carrying out such a reflux for 8 h
a single phase compound was obtained. Milder ion exchange conditions were
also possible but longer times were required to obtain a complete reaction.
Ion exchange was carried out in ethanol in the presence of a 7–8 fold
molar excess of LiBr involving refluxing at 80°C, and in the same
solvent at 25°C.
Chemical analyses for sodium and lithium were carried out by flame emission and for manganese and cobalt by atomic absorption spectroscopy. The average oxidation state of the transition metal ions was determined by redox titration using ferrous ammonium sulfate/KMnO4.
Manganese-rich compounds exhibit fluorescence when irradiated by copper X-ray radiation. Generally, an analysing monochromator is placed between the sample and the detector to reduce this problem. However, such an analyser precludes the use of a primary beam monochromator. Excellent X-ray data may be obtained using a curved crystal primary monochromator and a focussing geometry in transmission mode. Such an arrangement delivers high resolution and minimises preferred orientation, which can be particularly severe for layered compounds when examined using Bragg–Brentano (reflection) geometry. The problem is that Cu radiation must be replaced by a different source to eliminate the fluorescence. We employ an iron source (λ = 1.936 Å) with a curved germanium monochromator and a small angle position sensitive detector operating in transmission mode on a Stoe STADI/P diffractometer.
The structures of the materials were further characterised using neutron diffraction. Time-of-flight powder neutron diffraction data were collected on the POLARIS high-intensity, medium-resolution instrument at ISIS, Rutherford Appleton Laboratory. Since lithium and cobalt are neutron absorbers, the data were corrected for absorption. The structures were refined by the Rietveld method using the program TF12LS based on the Cambridge Crystallographic Subroutine Library (CCSL).20,21 Neutron scattering lengths of −0.19, −0.3703, 0.278 and 0.5803 (all ×10−12 cm) were assigned to Li, Mn, Co and O respectively.22 Lattice parameters were obtained from X-ray diffraction data by Rietveld refinement using GSAS.23
Surface area measurements were carried out using the BET method employing a Micromeritics Gemini 23670 instrument and yielded values of 3–6 m2 g−1.
In order to evaluate the performance of the layered materials, composite electrodes were constructed by mixing the active material, Kynar Flex 2801 (a copolymer based on PVDF) and carbon, in the weight ratios 85 ∶ 5 ∶ 10. The mixture was prepared as a slurry in THF and spread on to aluminium foil using a Doctor Blade technique. Following evaporation of the solvent and drying, electrodes were incorporated into an electrochemical cell in which the second electrode was formed from lithium metal and the electrolyte was a 1 molal solution of LiPF6 (Hashimoto) in propylene carbonate (Merck). Electrochemical measurements were carried out using a Biologic MacPile II.
Results and discussion
X-Ray diffraction and compositions
X-Ray diffraction patterns of the as-synthesised sodium compounds exhibit reflections which may be indexed on a layered rhombohedral structure, space group R3m, as expected for NaxMn1 − yCoyO2, as well as reflections associated with unreacted Na2CO3. Prolonged heating or higher temperatures results in disproportionation of the solid solutions. Following ion exchange of the sodium phases and subsequent washing and drying, X-ray diffraction patterns of the resulting lithium phases show no evidence of the carbonate phase, Fig. 1. Under all ion exchange conditions reported here, the X-ray diffraction data for the lithium phases may be indexed on a rhombohedral unit cell in space Rm, typical of phases with the α-NaFeO2
structure type, such as LiCoO2. Under the mildest conditions (25°C),
several weeks may be required to obtain products which yield the X-ray
data shown in Fig. 1. | ||
| Fig. 1 Powder X-ray diffraction
patterns for the LixMn1 − yCoyO2
phases, y = 0.1, prepared from the same sodium
phase but ion-exchanged under different conditions: (a) ethanol
25°C, (b) ethanol 80°C, (c) hexanol
160°C. | ||
The lattice parameters and c/a ratios are presented in Table 1. In all cases the a lattice parameter decreases with increasing Co content. The a lattice parameter lies in the basal plane of the layered structure and is particularly sensitive to changes in the average transition metal to oxygen bond length. Previous studies have shown that Co enters the layered lithium manganese oxide structure in the trivalent state.15 The reduction in a is consistent with replacement of the larger high spin Mn3+ ion (ionic radius = 0.645 Å) by the smaller low spin Co3+ ion (ionic radius = 0.545 Å). Comparing the a lattice parameters for the samples prepared under different ion exchange conditions, it is evident that the values of a for the compounds prepared by refluxing in hexanol are greater than those obtained by refluxing in ethanol, which are in turn slightly greater (with the exception of y = 0.3) than those for the samples prepared by ion exchange at room temperature in ethanol. The c lattice parameters show no clear trend with Co content for the sample prepared in hexanol but decrease with Co content for both sets of samples prepared in ethanol. There is an overall trend towards larger c axis values on changing the preparative conditions from hexanol to ethanol reflux and then to room temperature ion exchange.
| Co content | a/Å | c/Å | a/c |
|---|---|---|---|
| a Space group Rm. | |||
| (a) Hexanol, 160°C | |||
| 0.025 | 2.8872(4) | 14.4333(21) | 4.999 |
| 0.05 | 2.8797(3) | 14.4534(18) | 5.019 |
| 0.1 | 2.8734(3) | 14.3843(12) | 5.006 |
| 0.2 | 2.8666(2) | 14.4041(9) | 5.025 |
| 0.3 | 2.8468(2) | 14.4286(8) | 5.068 |
| (b) Ethanol, 80°C | |||
| 0.025 | 2.8678(3) | 14.6067(19) | 5.093 |
| 0.05 | 2.8667(2) | 14.5817(14) | 5.086 |
| 0.1 | 2.8549(3) | 14.557(1) | 5.099 |
| 0.2 | 2.8502(1) | 14.541(2) | 5.102 |
| 0.3 | 2.8345(2) | 14.5275(8) | 5.125 |
| (c) Ethanol, 25°C | |||
| 0.025 | 2.8638(3) | 14.6622(18) | 5.120 |
| 0.05 | 2.8626(2) | 14.6522(14) | 5.118 |
| 0.1 | 2.8527(3) | 14.6288(11) | 5.128 |
| 0.3 | 2.8368(1) | 14.5782(19) | 5.139 |
The results of detailed compositional analysis on the ethanol and hexanol refluxed samples, each at four different Co contents, are presented in Table 2. Several aspects of the results are worthy of note. The total alkali metal content is significantly less than unity in all cases and this is consistent with the presence of Na2CO3 in the X-ray diffraction patterns collected on the sodium phases prior to ion exchange. The sodium phases are deficient in their alkali metal content and this deficiency is carried through into the lithium phases on ion exchange. Some sodium remains after exchange. The amount is negligible in the case of samples prepared in hexanol but significant for those prepared in ethanol. In the case of the materials prepared in hexanol, the Co and Mn contents are close to those anticipated from the nominal composition and there are few if any vacancies on the transition metal sites. In contrast, the ethanol derived materials are somewhat transition metal deficient and as a result have significant vacancy concentrations on the transition metal sites.
°C and hexanol at 160°C
| Nominal Co content | Composition | Average TM oxidation state | No. of TM vacancies (%) | Mn3+ occupancy of TM sites (%) |
|---|---|---|---|---|
| (a) Ethanol, 80°C | ||||
| 0.025 | Na0.051Li0.564Mn0.893Co0.025O2 | 3.687+ | 8.2 | 26.4 |
| 0.05 | Na0.032Li0.604Mn0.878Co0.051O2 | 3.621+ | 7.1 | 30.1 |
| 0.10 | Na0.035Li0.539Mn0.870Co0.087O2 | 3.580+ | 4.3 | 31.5 |
| 0.30 | Na0.019Li0.593Mn0.657Co0.297O2 | 3.551+ | 4.6 | 13.1 |
| (b) Hexanol, 160°C | ||||
| 0.025 | Na0.009Li0.619Mn0.960Co0.028O2 | 3.413+ | 1.2 | 55.0 |
| 0.05 | Na0.008Li0.608Mn0.949Co0.053O2 | 3.377+ | 0.0 | 57.0 |
| 0.10 | Na0.012Li0.642Mn0.901Co0.094O2 | 3.363+ | 0.0 | 54.0 |
| 0.30 | Na0.011Li0.626Mn0.692Co0.316O2 | 3.336+ | 0.0 | 35.3 |
The chemical analyses presented in Table 2 enable a more detailed interpretation of the structural data presented in Table 1. The c axis parameter of these layered compounds is particularly sensitive to the size and quantity of the guest species in the alkali metal layers. It is known that the weak van der Waal's bonding between adjacent oxide ion sheets in layered oxides leads to a general expansion of the c axis with decreasing alkali metal content. Larger guests will also lead to a c axis expansion. For the samples prepared in ethanol, the alkali metal content is lower and the quantity of Na higher and this is consistent with the longer c axis observed in the case of the ethanol samples compared with those prepared in hexanol. The presence of vacancies on the transition metal sites of the compounds prepared in ethanol leads to an increase in the average Mn oxidation state compared with the samples obtained by hexanol reflux. The lower alkali metal content for the ethanol samples also serves to increase the average Mn oxidation state, Table 2. The higher oxidation state for the samples prepared in ethanol reflects a greater proportion of Mn4+ compared with the hexanol samples. Since Mn4+ (ionic radius = 0.53 Å) is smaller than high spin Mn3+ (ionic radius = 0.645 Å) this results in a shorter average transition metal to oxygen bond length, the effect of which is to reduce the a lattice parameter for the ethanol samples, as is observed in Table 1.
None of the samples reported here exhibits a cooperative Jahn–Teller distortion. In such transition metal manganates a Jahn–Teller distortion arises when the occupancy of the octahedral transition metal sites by high spin Mn3+ is greater than 50%. The exact value will vary from system to system depending on the balance between the lower electronic energy achieved by lowering the symmetry and the energetics involved in the lattice distortion. It is evident from Table 2 that the highest Mn3+ occupancy is 57%, with most compositions exhibiting values significantly lower than this. Evidently there is insufficient occupancy of the transition metal sites by Mn3+ to promote a cooperative Jahn–Teller distortion.
Crystal structure
Turning to the structure of the phases, a neutron diffraction pattern for LixMn0.9Co0.1O2, the compound having been prepared by refluxing in ethanol, is shown in Fig. 2. Crystallographic parameters obtained by fitting a model based on the layered structure to these data and to the corresponding data for LixMn0.8Co0.2O2 are shown in Table 3. The fit in Fig. 2 is very good as are the weighted profile R-factors quoted in Table 3. The structure of the cobalt doped solid solutions obtained by refluxing in hexanol has been described previously14,15 and is essentially that of layered LiCoO2 with oxygen atoms occupying the 6c, Mn/Co the 3a and Li the 3b sites of space group Rm.
The structure of the solid solutions obtained by refluxing in ethanol is similar.
In refining the compounds obtained in ethanol, the sodium contents were fixed
at the values obtained from the chemical analyses (Table 2)
as were the vacancy concentrations on the transition metal sites. The occupancy
of the oxygen position was fixed at unity, however the occupancies of the
remaining ions were allowed to vary within their sites. The agreement between
the refined Mn/Co and Li contents obtained from the neutron data and the
chemical analyses is reasonable. The average alkali metal to oxygen bond lengths
obtained by refinement are shown in Table 4.
Comparison of the bond lengths between compounds prepared in ethanol and hexanol
indicates that these are greater in the former case, consistent with the larger c
axis for ethanol, and reflecting the lower alkali metal content and presence
of Na+ in the ethanol derived materials. | ||
| Fig. 2 Powder neutron diffraction
pattern for LixMn0.9Co0.1O2
prepared in ethanol at 80°C. Dots = experimental
data, solid line = best fit, lower curve = difference/e.s.d.
Upper tick marks indicate position of reflections in Rm. | ||
°C
| Atom | Wyckoff symbol | x/a | y/a | z/c | Biso | Occupancy |
|---|---|---|---|---|---|---|
| a Space group Rm; a = 2.8572(2) Å, c = 14.5433(6) Å; Rexp = 1.2%, RwP = 1.6%, RP=1.7%; RI = 3.7%.b Space group Rm; a = 2.8419(2) Å, c = 14.5617(7) Å; Rexp = 1.2%, RwP = 1.7%, RP = 1.8%; RI = 3.2%. | ||||||
| (a) LixMn0.9Co0.1O2a | ||||||
| Li/Na | 3b | 0.0 | 0.0 | 0.5 | 0.81(9) | 0.61(2)/0.035 |
| Mn/Co | 3a | 0.0 | 0.0 | 0.0 | 0.59(3) | 0.835/0.115(5) |
| O1 | 6c | 0.0 | 0.0 | 0.2626(1) | — | 1 |
| O1 B11 = B22 = 1.083(14), B33 = 0.45(2), B12 = 0.542(7) | ||||||
| (b) LixMn0.8Co0.2O2b | ||||||
| Li/Na | 3b | 0.0 | 0.0 | 0.5 | 0.96(10) | 0.52(2)/0.02 |
| Mn/Co | 3a | 0.0 | 0.0 | 0.0 | 0.39(3) | 0.769/0.181(4) |
| O1 | 6c | 0.0 | 0.0 | 0.2635(1) | — | 1 |
| O1 B11 = B22 = 0.821(11), B33 = 0.34(2), B12 = 0.411(6) | ||||||
| Co content | Hexanol at 80°C | Ethanol at 160°C |
|---|---|---|
| 0.1 | 2.146(2) | 2.160(2) |
| 0.2 | 2.147(2) | 2.162(2) |
| 0.3 | 2.142(2) | 2.152(2) |
| 0.5 | 2.135(2) | 2.142(2) |
Intercalation upon ion exchange
The parent sodium phase with 10% Co was carefully washed with water to remove any Na2CO3 and then subjected to chemical analysis, Table 5. In order to confirm the Na content both Na and C analyses were performed on the phase prior to washing. From the carbon content the amount of sodium due to Na2CO3 could be deducted from the total sodium content. The resulting sodium content so derived was 0.423, in good agreement with the value reported in Table 5. Ion exchange was carried out on the sodium phase by refluxing in ethanol at 80°C with a 7–8
fold excess of LiBr for 90 h. Two such experiments were carried out.
In both cases the lithium bromide was dried at 210°C for 16 h
prior to ion exchange. The difference between the two experiments was that
in one case 40 weight% of water (with respect to the LiBr)
was added resulting in the phase labelled B in Table 5.
In both cases the alkali metal content is significantly higher after the exchange
than before. In addition, the ethanol solution which was originally clear
develops a faint brown coloration during the exchange which is more intense
in the case of the dry reflux. Evidently some lithium intercalation accompanies
ion exchange under the conditions used here.
| Alkali metal content | ||
|---|---|---|
| Na | Li | |
| Na phase | 0.450 | — |
| Li phase A [ethanol, 80°C (LiBr)] | 0.022 | 0.668 |
| Li phase B [ethanol, 80°C (LiBr + H2O)] | 0.035 | 0.497 |
Disorder and hkl dependent peak broadening
Examination of the powder X-ray diffraction patterns for the samples prepared under different ion exchange conditions reveals that there is a significant variation in the full width at half maximum of the peaks as a function of their hkl values, Fig. 1. This hkl dependent peak broadening is particularly marked for the samples prepared in ethanol at room temperature and much less obvious in the case of the samples prepared in hexanol. The effect is, in particular, dependent on the l component of the reflection suggesting disorder in the c direction, possibly arising from incomplete exchange of Na+ by Li+. In a layered compound during exchange of Na+ by Li+ any given layer is likely to be almost completely occupied by Li or Na. This minimises lattice strain that would otherwise arise if ions of very different radii were to be accommodated within the same layers. Chemical analysis has shown that small amounts of Na do remain after exchange in the case of the samples prepared in ethanol for which the hkl dependent peak broadening is most prominent. hkl dependent peak broadening has been observed before in other layered systems, for example in nickel hydroxide.24In order to simulate the influence of incorporating discrete Na+ layers on the powder X-ray diffraction patterns of the lithium phases prepared in ethanol the DiFFAX code was used.25 This program calculates diffraction intensities from crystals containing coherent planar faults. It is necessary to begin with a good description of the peak shape for a reflection not affected by the disorder. Unfortunately in the case of the samples described here there is only one such peak, the (110) reflection. A pseudo-Voigt function was used to describe the peak shape, based on fitting to the (110) reflection. Since the hkl dependent peak broadening is most prominent in the samples prepared at room temperature attention was focussed on one of these, i.e. the material with 10% Co. The structure was modelled using two types of infinite layer for the compound A0.46Mn0.85Co0.096O2 where A is either Na+ or Li+. The Li+ layers were modelled with the cations octahedrally coordinated by the oxygen layers, while the coordination around the Na+ ions in the sodium layer was assumed to be trigonal prismatic in agreement with the structure of the parent sodium phase. The layer stacking was treated as recursive indicating that the diffraction intensities are to be calculated for a statistical ensemble of crystallites, each with a distinct stacking sequence, but weighted by a probability that a particular sequence will occur. The diffraction pattern was simulated for different probabilities that a sodium layer would occur. Fig. 3 shows a number of simulations corresponding to different sodium contents. By comparison of the experimental data (also shown in Fig. 3) with the simulated data the best agreement was obtained with 5% Na which is in reasonable accord with the Na content obtained by chemical analysis for the nominally 10% cobalt doped material. The agreement between the experimental and simulated X-ray data in Fig. 3(b) is reasonable although certainly not perfect. For example, although the (110) peak at 85° in 2θ is calculated to be more intense then the (018) peak at 82.75°, the intensity difference is less acute than in the observed data. Overall, there is evidently more disorder than has been taken into account in the present model.
 | ||
| Fig. 3 (a) Simulations using DiFFAX for various compositions of LixNayMn0.9Co0.1O2 (where y ranges from 0 to 0.2). (b) (i) Simulated and (ii) experimental data for the 5% Na composition. | ||
The retention of Na+ ions in materials which also contain
transition metal vacancies, such as the compounds prepared in ethanol, is
unlikely to be coincidental. The transition metal vacancies in the sodium
phase before ion exchange, which carry a high negative effective charge, may
act as traps, pinning some of the Na+ ions with relaxation
of the oxide ions around the Na+
site. As a result, such Na+
ions will be less mobile and hence may be retained during ion exchange with
Li+. Indeed such trapping is likely to enhance the staging
of Na+ and Li+ layers. The transition metal
vacancies may in fact trap Na+ ions in alkali metal layers
above and below any given O–Mn–O slab. No account has been taken
of any transition metal vacancy ordering that may occur or any clustering
of Na+ ions with the vacancies, indeed it can be difficult
to simulate such complex ordering with DiFFAX. Electron microscopy is a more
comprehensive tool for the study of these complex defects. Nevertheless, by
simulating the random incorporation of sodium layers it has been possible
to obtain diffraction profiles which are closer to the experimental data than
those assuming that the order is ideal. In the case of the materials prepared
in hexanol at 160°C it is evident that the more powerful reducing
nature of these conditions leads to elimination of the transition metal vacancies
and permits easier and hence more complete exchange of Na+
by Li+. The absence of vacancies and Na+ ions
in the lithium phases after exchange is consistent with the lack of hkl
dependent peak disordering in the X-ray data.
Electrode performance
Despite the fact that the differences in the synthesis conditions between reflux in ethanol and hexanol appear to be relatively small, there is no doubt that these differences are very significant in terms of the performance of the compounds as intercalation electrodes in rechargeable lithium batteries. In Fig. 4 we present the discharge capacity versus cycle number for the layered lithium manganese cobalt oxides prepared by refluxing in hexanol and ethanol. Cycling was carried out at 25 mA g−1, which corresponds to a practical rate of C/7. The potential range used was 2.4 to 4.6 V. For applications, it is desirable to have the highest possible discharge capacity with the lowest possible fade rate on cycling. It is evident that excellent performance is obtained from the material prepared in ethanol and with the lowest cobalt content of 2.5%. The initial discharge capacity for this compound is 200 mA h g−1 and this fades by only 0.08% per charge/discharge cycle. Higher cobalt contents prepared in ethanol show greater capacity fade and although it is possible with hexanol to obtain a slightly higher initial discharge capacity the fade is significantly greater. Given the performance of the material prepared in ethanol with 2.5% Co, coupled with the significant cost advantage of a minimal cobalt content, this material represents an interesting intercalation compound in the context of new positive electrodes for rechargeable lithium batteries. | ||
| Fig. 4 Discharge capacity
as a function of the number of cycles for some layered compounds, rate = 25 mA g−1,
potential limits = 2.4–4.6 V. ●, y = 0.025 (ethanol
80°C); △, y = 0.025 (hexanol
160°C); ×, y = 0.1 (ethanol
80°C); □, y = 0.1 (hexanol
160°C). | ||
Inspection of Fig. 4 indicates that for LixMn0.975Co0.025O2 prepared in ethanol the capacity exhibits an unusual variation in the first 10–15 cycles. The origin of this becomes clear on examining X-ray diffraction data as a function of cycle number. Such data are reported in Fig. 5. It is evident that the (018) and (110) reflections at 82.76 and 84.94° in 2θ merge on cycling and this is characteristic of a transformation from a layered to a spinel-like structure. Conversion to a spinel-like structure occurs when one quarter of the transition metal ions migrate from octahedral sites in the transition metal layers into octahedral sites in the lithium layers, this involves migration through intervening face-sharing tetrahedral sites. The lithium ions are then displaced into the vacant tetrahedral sites. It is also evident from Fig. 5 that once formed the structure remains as a spinel on subsequent cycling. This behaviour is also observed for other Co contents and for ion exchange in hexanol.15 It is interesting that the excellent performance of spinels formed in situ on cycling, e.g. LixMn0.975Co0.025O2, Fig. 4, contrasts markedly with lithium manganese oxide based spinels prepared directly.26,27 The latter are known to cycle well over a restricted voltage range around 4 V but not over the wider voltage and hence composition range employed here. Instead they exhibit severe capacity fade. Despite the apparent similarity of these two classes of material they are clearly significantly different, a matter we shall return to in more detail in a subsequent paper.
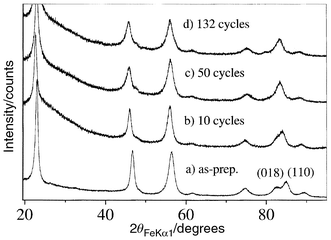 | ||
| Fig. 5 X-Ray diffraction
data as a function of cycle number for a Co content of 0.025, prepared in
ethanol at 80°C. | ||
Acknowledgements
The authors are indebted to the EPSRC for financial support and to ISIS, Rutherford Appleton Laboratory, Oxfordshire, for the provision of neutron diffraction facilities.References
- T. Naguara, Extended Abstracts, Third International Rechargeable Battery Seminar, Deerfield Beach, FL, USA, 5–7 March 1990..
- K. Mizushima, P. C. Jones and J. B. Goodenough, Mater. Res. Bull., 1980, 15, 783 CrossRef CAS.
- R. Koksbang, J. Barker, H. Shi and M. Y. Saidi, Solid State Ionics, 1996, 84, 1 Search PubMed.
- M. M. Thackeray, J. Electrochem. Soc., 1995, 142, 2558 CAS and references therein..
- (a) L. Croguennec, P. Deniard, R. Brec and A. Lecerf, J. Mater. Chem., 1997, 7, 511 RSC; (b) T. Ohzuku, A. Ueda and T. Hirai, Chem. Express, 1992, 7, 193 Search PubMed; (c) J. N. Reimers, E. W. Fuller, E. Rossen and J. R. Dahn, J. Electrochem. Soc., 1993, 140, 3396 CAS.
- J. M. Tarascon, W. R. McKinnon, F. Coowar, T. N. Bowmer, G. Amatucci and D. Guyomard, J. Electrochem. Soc., 1994, 141, 1421 CAS.
- Y. Gao and J. R. Dahn, J. Electrochem. Soc., 1996, 143, 100 CAS.
- P. G. Bruce, Chem. Commun., 1997, 1817 RSC and references therein..
- H. T. Huang, C. A. Vincent and P. G. Bruce, J. Electrochem. Soc., 1999, 146, 2404 CrossRef CAS.
- A. R. Armstrong and P. G. Bruce, Nature, 1996, 381, 499 CrossRef CAS.
- F. Capitaine, P. Gravereau and C. Delmas, Solid State Ionics, 1996, 89, 197 Search PubMed.
- P. G. Bruce, A. R. Armstrong and R. L. Gitzendanner, J. Mater. Chem., 1999, 9, 193 RSC.
- G. Vitins and K. West, J. Electrochem. Soc., 1997, 144, 2587 CAS.
- A. R. Armstrong, R. Gitzendanner, A. D. Robertson and P. G. Bruce, Chem. Commun., 1998, 1833 RSC.
- A. R. Armstrong, A. D. Robertson, R. Gitzendanner and P. G. Bruce, J. Solid State Chem., 1999, 145, 549 CrossRef CAS.
- A. R. Armstrong, A. D. Robertson and P. G. Bruce, Electrochim. Acta, 1999, 45, 285 CrossRef CAS.
- Y.-I. Jang, B. Huang, H. Wang, Y.-M. Chiang and D. R. Sadoway, Electrochem. Solid State Lett., 1998, 1, 13 CrossRef CAS.
- A. D. Robertson, A. R. Armstrong and P. G. Bruce, Chem. Commun., submitted. Search PubMed.
- J. M. Paulsen and J. R. Dahn, Solid State Ionics, 1999, 126, 3 Search PubMed.
- J. C. Matthewman, P. Thompson and P. J. Brown, J. Appl. Crystallogr., 1982, 15, 167 CrossRef CAS.
- P. J. Brown and J. C. Matthewman, Rutherford Appleton Laboratory Report, RAL-87-010, 1987..
- V. F. Sears, Neutron News, 1992, 3(3), 26 Search PubMed.
- A. C. Larson and R. B. von Dreele, General Structure Analysis System, Los Alamos National Laboratory, 1995..
- C. Tessier, P. H. Haumesser and C. Delmas, J. Electrochem. Soc., 1999, 146(6), 2059 CrossRef CAS.
- M. M. Treacy, J. M. Newsam and M. W. Deem, Proc. R. Soc. London Ser. A, 1991, 433, 499 Search PubMed.
- P. Barboux, J. M. Tarascon and F. K. Shokoohi, J. Solid State Chem., 1991, 94, 185 CrossRef CAS.
- J. M. Tarascon, E. Wang, F. K. Shokoohi, W. R. McKinnon and S. Colson, J. Electrochem. Soc., 1991, 138, 2859 CAS.
Footnote |
| † Basis of a presentation given at Materials Discussion No. 3, 26–29 September, 2000, University of Cambridge, UK. |
| This journal is © The Royal Society of Chemistry 2001 |