Conjugate additions of heteronucleophiles to enones and alkynoates. A ‘benign by design’ functionalization of heteroaromatics
Martín Avalos, Reyes Babiano, José L. Bravo, Pedro Cintas, José L. Jiménez, Juan C. Palacios* and María A. Silva
Departamento de Química Orgánica, Facultad de Ciencias, Universidad de Extremadura, E-06071, Badajoz, Spain.. E-mail: palacios@unex.es
First published on 17th January 2001
Abstract
A simple and convenient functionalization of pyrrole and thiophene nuclei has been accomplished by one-step clay-catalyzed conjugate addition reactions of these heteroaromatics with some enones and alkynoates under very mild conditions. The experimental protocol can be easily modified to accommodate one or two alkyl groups.
Green ContextPyrroles and thiophenes are extremely useful compounds with applicability in numerous areas including bioactive compounds, organic semiconductors and addressable genechips. Thus their functionalisation is of considerable interest though traditionally reliant on hazardous and wasteful procedures. Here novel facile functionalisation procedures are described. These are solvent-free, employ safe and reusable solid acids and subject to microwave acceleration. A variety of mono- and bis-alkylated products have been prepared using this procedure.DJM |
Introduction
In a series of recent investigations we have disclosed that homo-Diels–Alder reactions can be conducted under solvent-free conditions using inexpensive and innocuous mineral solids as catalysts.1 Furans undergo competitive cycloaddition and Michael-type reactions, and the product distribution is largely dependent on the stoichiometry and experimental conditions.2 The incessant demand for increasingly green technologies is encouraging scientists to think more on environmentally friendly chemical processes.3 In this context, important generic areas of chemistry such as acid-catalyzed reactions need to be rethought and modified. Besides furan, the π-excessive heteroaromatics pyrrole and thiophene react with electrophiles via Friedel–Crafts-type reactions to give useful precursors for natural product syntheses. However, these reactions often require acid catalysts and/or high temperatures to be achieved,4–6 and oligomeric side products are also obtained. Remarkably, polypyrrole and polythiophene films are currently being explored in applications such as biosensores, organic semiconductors, or addressable gene-chips.7,8 It may be possible to substitute the above-mentioned forcing conditions for solid catalysts (e.g. clays, zeolites) or supported reagents, thereby making unnecessary the use of solvents and separation agents.9 We now report a facile functionalization of these important heterocycles involving their solventless condensation with enones adsorbed on montmorillonite K10 to afford mono- and bis-alkylated products in moderate to good overall yields.Results and discussion
Our results with pyrrole and thiophene have been collectively summarized in Schemes 1 and 2, and Table 1. Condensations with N-phenylmaleimide proceed slowly, presumably due to steric reasons. Reactions can either be stirred or irradiated with focused microwaves, thereby resulting in shorter reaction times especially with thiophene derivatives (Scheme 2). These fast reaction conditions also favor the formation of monoalkylated products, although lower yields are often obtained owing to the volatility of the products (Table 1, entry 6). It should be noted that irradiated reactions can equally be conducted in a domestic oven with similar results. Nevertheless, our reactions employing a monomode reactor (see Experimental section) exhibited increased efficiency and reproducibility. In addition, and unlike domestic ovens, the reaction temperature can be controlled by power modulation within a wide range of Watts and measured with accuracy by IR detection. Furthermore, this device offers a homogeneous distribution of the electric field inside the cavity.10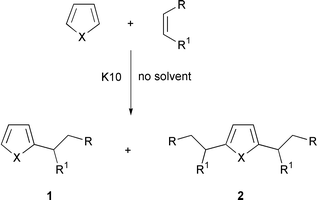 | ||
| Scheme 1 | ||
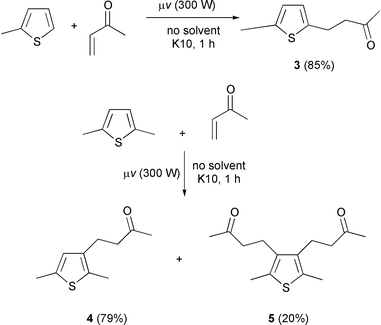 | ||
| Scheme 2 | ||
| Entry | X | R | R1 | Substrates (Molar ratio) | Conditions | t | Yield (%)a |
|---|---|---|---|---|---|---|---|
| a Isolated yields of flash-chromatographed products.b Using monomode reactor with focused microwaves (2.45 GHz, 5% relative intensity). | |||||||
| 1 | NH | COMe | H | 1∶2 | 0 °C | 45 min | 2 (100) |
| 2 | NH | COMe | H | 1∶1 | 0 °C | 1 h | 1 (15), 2 (68) |
| 3 | NH | COMe | H | 2∶1 | 0 °C | 1 h | 1 (30), 2 (50) |
| 4 | NH | –CONPhCO– | 1∶1 | 25 °C | 12 h | 1 (16) | |
| 5 | NH | –CONPhCO– | 2∶1 | 25 °C | 48 h | 1 (30) | |
| 6 | NH | –CONPhCO– | 1∶1 | μν (300 W)b | 1 h | 1 (16) | |
| 7 | S | COMe | H | 1∶1 | 25 °C | 3 d | 1 (14), 2 (64) |
The reaction of pyrrole with maleic anhydride gives rise to a mixture of 6 and the dicarboxylic derivative 7, the latter arising from ring opening of the anhydride moiety at the catalytically active acid sites of montmorillonite, and is the exclusive product after a prolonged reaction time (Scheme 3). The distinctive identification between compounds 6 and 7 could be accomplished on the basis of their IR data, as 7 exhibits characteristic bands at ca. 3000–2500 cm−1 due to OH stretching. Likewise, and unlike 6, compound 7 was insoluble in chloroform. No reaction was observed between thiophene derivatives and N-phenylmaleimide or maleic anhydride.
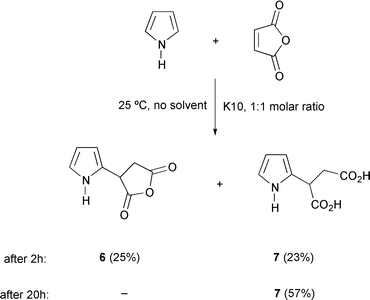 | ||
| Scheme 3 | ||
This clay-catalyzed protocol could also be extended to alkynic substrates such as dimethyl acetylenedicarboxylate providing an interesting reaction mixture of double addition products (8 and 9) accompanied by a Z/E mixture of alkenes (10, Scheme 4), which could be purified by flash chromatography and identified by spectroscopic methods. It is well known that simple pyrroles or thiophenes show little tendency to react as 4π components in cycloadditions owing to their enhanced aromaticity. In fact, Diels–Alder reaction between pyrrole and methyl vinyl ketone catalyzed by Cr(III)-exchanged montmorillonite in organic solvents has been observed, although the cycloadduct was obtained in 35% yield, but no reaction could be detected for thiophene under these Lewis-acid conditions.11 On the other hand, conjugate addition reactions with electrophilic alkynes are accelerated by high pressure or by Lewis-acid catalysts.4
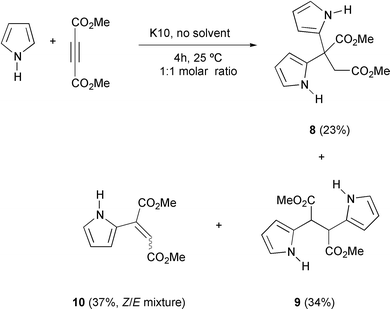 | ||
| Scheme 4 | ||
Compound 9 was obtained as a single diastereomer as evidenced by its NMR spectra which showed only one signal set. Likewise, it was difficult to establish a spectroscopic distinction between isomers 10Z and 10E, because of the similarity of their coupling pattern.
No alkene derivatives could be detected in the reaction between pyrrole and methyl propiolate, but rather products 11 and 12 resulting from successive Michael-type additions (Scheme 5). The latter results are especially attractive because they represent a facile entry to polypyrrolic fragments and porphyrin precursors,12 which are currently accessible via the classical reaction of pyrroles with aldehydes13 or the self-condensation of dipyrromethanes.14 It should also be pointed out that compound 12, as well as the symmetrical compounds 2 (X = NH), show H-3 signals as doublets (J = 2.8 Hz) as a consequence of a long range coupling between H-3 and the NH proton which could further be determined by selective spin–spin decoupling. In fact, it is well documented that the NH proton of a pyrrole or an amide is coupled to vicinal protons, even though the NH absorption may be broad as result of partial decoupling by the nitrogen electrical quadrupole moment.15
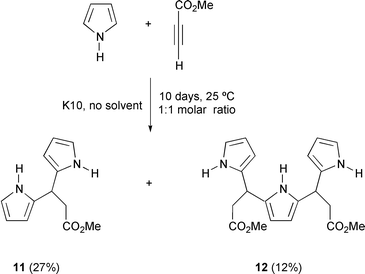 | ||
| Scheme 5 | ||
With a few exceptions, such as 1 (X = NH, R = R1 = CONPhCO), 8 and 9 which could be obtained as crystalline solids and gave satisfactory combustion analyses, most samples were homogeneous oils by TLC analysis. Finally, the mechanism associated with these processes is a subject of an ongoing investigation, since it is unclear if the conjugate addition follows a concerted16 or stepwise pathway,17 and a theoretical study is under way.
In conclusion, we have shown that a clay material drives conjugate addition reactions of heteroaromatics to enones and alkynes in absence of organic solvents. The protocol combines atom economy, efficiency and provides access to mono- and di-alkylated products under mild conditions. Although this procedure will require further exploration of its scope, it is hoped that the above results will be of benefit to the scientific community engaged in solvent-free condensations.
Experimental
All commercial reagents and solvents were used without further purification. Montmorillonite K10 was purchased from Aldrich. Column chromatography and TLC analyses were performed on Merck silica gel 60 (400–230 mesh) and GF254, respectively. NMR spectra were recorded on a 400 MHz Bruker spectrometer in CDCl3. Microwave-irradiated reactions were conducted in a Synthewave® 402 reactor from Prolabo.Typical procedure
To commercially available montmorillonite K10 (2.5 g), cooled previously at 0 °C, was added dropwise the corresponding heterocycle (5.0 mmol) and methyl vinyl ketone (5.0 mmol) with vigorous stirring, and the reaction mixture was kept at 0 °C (or 25 °C). Upon standing for an approprite reaction time, the mixture was extracted with CH2Cl2 (20 mL) and MeOH (20 mL), filtered, and concentrated to give a residue, which was purified by flash chromatography (diethyl ether–hexane or ethyl acetate–hexane eluent systems).Reaction of pyrrole and methyl vinyl ketone
2-(3′-Oxobutyl)pyrrole: δH(CDCl3) 8.51 (br s, NH), 6.62 (m, H-5), 6.06 (dd, J = 2.9, 2.3 Hz, H-4), 5.86 (br s, H-3), 2.83 (2H, t, J = 6.0 Hz, H-1′, H-1″), 2.76 (2H, t, J = 6.0 Hz, H-2′, H-2″), 2.14 (3H, s, CH3), δC(CDCl3) 209.7 (C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) O), 131.4
(C-2), 116.7 (C-5), 107.8 (C-4), 105.2 (C-3), 44.1 (C-2′), 30.0
(CH3), 21.2 (C-1′). 2,3-Bis(3′-oxobutyl)pyrrole:
δH(CDCl3) 8.47 (1H, br s, NH), 5.71
(2H, d, J = 2.8 Hz, H-3, H-4). 2.73–2.81 (8H, m, H-1′,
H-1″, H-2′, H-2″), 2.16 (6H, s, 2 ×
CH3), δC(CDCl3) 209.1
(CO), 130.4 (C-2), 104.7 (C-3), 43.9 (C-2′), 27.0
(CH3), 21.4 (C-1′).
O), 131.4
(C-2), 116.7 (C-5), 107.8 (C-4), 105.2 (C-3), 44.1 (C-2′), 30.0
(CH3), 21.2 (C-1′). 2,3-Bis(3′-oxobutyl)pyrrole:
δH(CDCl3) 8.47 (1H, br s, NH), 5.71
(2H, d, J = 2.8 Hz, H-3, H-4). 2.73–2.81 (8H, m, H-1′,
H-1″, H-2′, H-2″), 2.16 (6H, s, 2 ×
CH3), δC(CDCl3) 209.1
(CO), 130.4 (C-2), 104.7 (C-3), 43.9 (C-2′), 27.0
(CH3), 21.4 (C-1′).Reaction of pyrrole and N-phenylmaleimide
3-(2′-Pyrroyl)-N-phenylsuccinimide: Mp 176.5 °C. δH(CDCl3) 9.11 (br s, NH), 7.48 (2H, t, J = 7.2 Hz, Ar), 7.40 (1H, t, J = 7.2 Hz, Ar), 7.26 (2H, d, J = 7.2 Hz, Ar), 6.82 (br s, H-5′), 6.20 (dd, J = 3.2, 2.8 Hz, H-4′), 6.00 (m, H-3′). 4.24 (dd, J = 9.2, 5.6 Hz, H-3), 3.35 (dd, J = 18.0, 9.2 Hz, H-4b), 3.20 (dd, J = 18.0, 5.6 Hz, H-4a). δC(CDCl3) 176.7, 174.7, (CO),
131.6, 129.2, 128.8, 126.4 (Ar), 125.2 (C-2′), 119.0 (C-5′),
108.5 (C-4′), 105.7 (C-3′), 38.5 (C-3), 34.2 (C-4). Anal. Calc.
for C14H12N2O2: C, 69.99; H,
5.03; N, 11.66. Found: C, 69.45; H, 4.84; N, 11.60%.Reaction of thiophene and methyl vinyl ketone
2-(3′-Oxobutyl)thiophene: δH(CDCl3) 7.11 (dd, J = 5.2, 1.2 Hz, H-5), 6.90 (dd, J = 5.2, 3.6 Hz, H-4), 6.79 (dd, J = 3.6, 1.2 Hz, H-3), 3.11 (2H, t, J = 7.6 Hz, H-1′, H-1″), 2.82 (2H, t, J = 7.6 Hz, H-2′, H-2″), 2.16 (3H, s, CH3). δC(CDCl3) 207.2 (CO), 143.5
(C-2), 126.8 (C-5), 124.5 (C-4), 123.3 (C-3), 45.2 (C-1′), 30.0
(CH3), 23.8 (C-2′). 2,5-Bis(3′-oxobutyl)thiophene:
δH(CDCl3) 6.56 (2H, s, H-3, H-4), 3.01
(4H, t, J = 7.3 Hz, H-1′, H-1″), 2.78 (4H, t,
J = 7.3 Hz, H-2′, H-2″), 2.15 (6H, s, 2 ×
CH3). δC(CDCl3) 207.0
(CO), 141.4 (C-2), 123.9 (C-3), 44.8 (C-1′), 29.8
(CH3), 23.8 (C-2′).Reaction of 2-methyl thiophene and methyl vinyl ketone
The preparation of 3, 2-methyl-5-(3′-oxobutyl)thiophene, constitutes a representative example: An equimolar mixture of 2-methylthiophene (10 mmol) and methyl vinyl ketone (10 mmol) was adsorbed on montmorillonite K10 (5.0 g) with stirring for 5 min. The homogenized mixture was then irradiated in a microwave reactor with focused electromagnetic radiation (2.45 GHz, 300 W, 5% relative intensity) for 1 h. Extraction with CH2Cl2, followed by filtration and evaporation gave 3 as a yellowish oil in 85% yield. δH(CDCl3) 6.53 (m, 2H, H-2, H-3), 3.01 (t, J = 7.3 Hz, 2H, CH2), 2.77 (t, J = 7.3 Hz, 2H, CH2), 2.41 (s, 3H, CH3CO), 2.12 (s, 3H, CH3); δC(CDCl3) 207.2 (CO), 141.2 (C-2), 137.6 (C-5), 124.6 (C-3), 124.1 (C-4), 45.1
(CH2), 29.9 (CH3CO), 23.9 (CH2),
15.1 (CH3).Reaction of 2,5-dimethyl thiophene and methyl vinyl ketone
2,5-Dimethyl-3-(3′-oxobutyl)thiophene 4: δH(CDCl3) 6.42 (1H, s, H-4), 2.65 (4H, m, H-1′, H-1″, H-2′, H-2″), 2.36 [3H, s, CH3(C-5)], 2.28 [3H, s, CH3(C-2)], 2.13 (3H, s, COCH3). δC(CDCl3) 208.12 (CO), 135.8 (C-5), 135.3 (C-2), 130.6 (C-3), 126.4 (C-4), 44.1
(C-1′), 30.0 [(CO)CH3], 22.2 (C-2′), 12.7
[CH3(C-2)], 15.1 [CH3(C-5)].
2,5-Dimethyl-3,4-bis(3′-oxobutyl)thiophene 5:
δH(CDCl3) 2.67 (4H, t, J =
7.3 Hz, H-1′, H-1″), 2.54 (4H, t, J = 7.3 Hz,
H-2′, H-2″), 2.27 (6H, s, 2 × CH3), 2.16 [6H,
s, 2 × CH3(CO)].
δC(CDCl3) 207.9 (CO), 135.2
(C-2), 129.8 (C-3), 43.8 (C-1′), 29.9 (CH3), 20.9
(C-2′), 12.9 [CH3(CO)].Reaction of pyrrole and maleic anhydride
2-(2′-Pyrroyl)succinic anhydride 6: δH(CDCl3) 8.82 (br s, NH), 6.84 (m, H-5′), 6.19 (dd, J = 2.9, 2.7 Hz, H-4′), 6.05 (br s, H-3′), 4.39 (dd, J = 9.6, 7.2 Hz, H-2), 3.43 (dd, J = 18.8, 9.6 Hz, H-3b), 3.30 (dd, J = 18.8, 7.2 Hz, H-3a). δC(CDCl3) 171.6, 168.9 (CO),
122.5 (C-2′), 119.6 (C-5′), 108.9 (C-4′), 106.5
(C-3′), 39.5 (C-2), 34.2 (C-3). 2-(2′-Pyrroyl)etanodioic acid
7: mp 126.5 °C.
δH(DMSO-d6) 10.72
(s, NH), 6.61 (m, H-5′), 5.90 (dd, J = 2.8, 2.4 Hz,
H-4′), 5.82 (br s, H-3′), 3.88 (dd, J = 10.4, 4.8 Hz,
H-2), 2.92 (dd, J = 17.2, 10.4 Hz, H-3b), 2.54 (dd, J =
17.2, 4.8 Hz, H-3a).
δC(DMSO-d6) 173.7,
173.0 (CO), 128.1 (C-2′), 117.4 (C-5′), 107.5
(C-4′), 105.2 (C-3′), 40.5 (C-2), 36.5 (C-3).Reaction of pyrrole and dimethyl acetylenedicarboxylate
Dimethyl 2,2-bis(2′-pyrroyl)butanodioate 8: mp 98 °C. δH(CDCl3) 8.79 (2H, br s, NH), 6.71 (2H, m, H-5′), 6.12 (2H, m, H-4′), 5.92 (2H, m, H-3′), 3.78 (3H, s, CH3), 3.64 (3H, s, CH3), 3.43 (2H, s, H-3). δC(CDCl3) 173.0, 172.0 (CO), 130.3 (C-2′), 118.0 (C-5′), 108.1 (C-3′),
106.6 (C-4′), 53.0 (CH3), 52.0 (CH3), 48.5
(C-2), 43.1 (C-3). Anal. Calc. for
C14H16N2O4: C, 60.86; H, 5.84;
N, 10.14. Found: C, 61.16; H, 6.04; N, 9.68%. Dimethyl
2,3-bis(2′-pyrroyl)butanodioate 9: mp 216 °C.
δH(CDCl3): 8.62 (2H, br s, NH), 6.72
(2H, m, H-5′), 6.09 (2H, m, H-4′), 6.04 (2H, m, H-3′),
4.30 (2H, s, H-2), 3.56 (6H, s, 2 × CH3).
δC(CDCl3) 172.2 (CO), 124.8
(C-2′), 118.4 (C-5′), 108.4 (C-3′), 108.2 (C-4′),
52.4 (CH3). 48.8 (C-2). Anal. Calc. for
C14H16N2O4: C, 60.86; H, 5.84;
N, 10.14. Found: C, 60.20; H, 5.66; N, 9.98%. trans-Dimethyl
2-(2′-pyrroyl)butenodioate E-10:
δH(CDCl3) 9.05 (br s, NH), 6.91 (dd,
J = 2.4, 1.2 Hz, H-5′), 6.48 (dd, J =
3.6, 2.4 Hz, H-4′), 6.25 (dd, J = 3.6, 1.2 Hz, H-3′),
6.00 (s, H-3), 3.93 (3H, s, CH3), 3.72 (3H, s, CH3).
δC(CDCl3) 168.2, 166.2 (CO),
140.0 (C-2), 123.5 (C-3), 125.9 (C-2′), 113.7 (C-5′), 111.2
(C-3′), 108.8 (C-4′), 52.9 (CH3), 51.9
(CH3). cis-Dimethyl 2-(2′-pyrroyl)butenodioate
Z-10: δH(CDCl3) 8.78
(br s, NH), 7.05 (dd, J = 2.4, 1.2 Hz, H-5′),
6.73 (dd, J = 3.6, 2.4 Hz, H-4′), 6.30 (dd, J =
3.6, 1.2 Hz, H-3′), 5.96 (1H, s, H-3), 3.90 (3H, s, CH3),
3.80 (3H, s, CH3).
δC(CDCl3) 168.9, 168.1 (CO),
138.7 (C-2), 125.8 (C-2′), 123.7 (C-3), 118.1 (C-5′), 110.5
(C-3′), 109.7 (C-4′), 52.8 (CH3), 52.1
(CH3).Reaction of pyrrole and methyl propiolate
Methyl 3,3-bis(2′-pyrroyl)propanoate 11: δH(CDCl3) 8.24 (2H, br s, NH), 6.67 (2H, m, H-5′), 6.13 (2H, m, H-4′), 5.98 (2H, m, H-3′), 4.58 (1H, t, J = 7.2 Hz, H-3), 3.67 (3H, s, CH3), 3.01 (2H, d, J = 7.2 Hz, H-2). δC(CDCl3) 173.1 (CO), 131.7
(C-2′), 117.2 (C-5′), 108.1 (C-4′), 105.3 (C-3′),
51.9 (CH3), 40.0 (C-2), 33.8 (C-3).
2,5-Bis(1′-(2-pyrroyl)-2′-carbomethoxyethyl)pyrrole
12: δH(CDCl3) 8.27, 8.19 (3H,
m, NH), 6.64 (2H, m, H-5″), 6.08 (2H, m, H-4″), 5.91 (2H, br s,
H-3″), 5.85 (2H, d, J = 2.8 Hz, H-3), 4.48 (2H, t,
J = 7.2 Hz, H-1′), 3.64 (6H, s, 2 × CH3),
2.92 (4H, d, J = 7.2 Hz, H-2′).
δC(CDCl3) 173.1 (CO), 132.1
(C-2), 131.7 (C-2″), 117.2 (C-5″), 108.2 (C-4″), 105.4
(C-3), 105.3 (C-3″), 51.9 (CH3), 40.0 (C-2′), 33.7
(C-1′).Acknowledgements
Financial support from the Spanish Ministry of Science and Technology (DGICYT, PB98-0997 and AMB99-0591) and the Junta de Extremadura-Fondo Social Europeo (IPR98-A065) is gratefully acknowledged.References
- M. Avalos, R. Babiano, J. L. Bravo, P. Cintas, J. L. Jiménez, J. C. Palacios and B. C. Ranu, Tetrahedron Lett., 1998, 39, 2013 CrossRef CAS.
- M. Avalos, R. Babiano, J. L. Bravo, P. Cintas, J. L. Jiménez and J. C. Palacios, Tetrahedron Lett., 1998, 39, 9301 CrossRef CAS.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, Oxford, 1998 Search PubMed; P. T. Anastas and T. C. Williamson, Green Chemistry: Frontiers in Benign Chemical Synthesis and Processes, Oxford University Press, Oxford, 1998. Search PubMed.
- J. A. Joule, K. Mills and G. F. Smith, Heterocyclic Chemistry, Chapman & Hall, London, 1995, pp. 231–242. Search PubMed.
- K. Hayakawa, M. Yodo, S. Ohsuki and K. Kanematsu, J. Am. Chem. Soc., 1984, 106, 6735 CrossRef CAS.
- Y. Kamitori, M. Hojo, R. Masuda, T. Izumi and S. Tsukamoto, J. Org. Chem., 1984, 49, 4161 CrossRef CAS.
- V. R. Shastri and M. V. Pishko, in Electrical and Optical Polymer Systems, ed. D. L. Wise, G. E. Wnek, D. J. Trantolo, T. M. Cooper and J. D. Gresser, Marcel Dekker, New York, 1998. Search PubMed.
- J. Roncali, Chem. Rev., 1992, 92, 711 CrossRef CAS; G. Schopf and G. Kossmehl, Polythiophenes—Electrically Conductive Polymers, Springer, Berlin, 1997 R. D. McCullough, Adv. Mater., 1998, 10, 93. Search PubMed.
- J. H. Clark and C. N. Rhodes, Clean Synthesis Using Porous Inorganic Solid Catalysts and Supported Reagents, Royal Society of Chemistry, Cambridge, 2000. Search PubMed.
- A. Loupy, A. Petit, J. Hamelin, F. Texier-Boullet, P. Jacqualt and D. Mathé, Synthesis, 1998, 1213 CrossRef CAS.
- J. M. Adams, S. Dyer, K. Martin, W. A. Matear and R. W. McCabe, J. Chem. Soc., Perkin Trans. 1, 1994, 761 RSC.
- J. L. Sessler and S. J. Weghorn, Expanded, Contracted and Isomeric Porphyrins, Elsevier, Oxford, 1997. Search PubMed.
- M. Onaka, T. Shinoda, Y. Izumi and E. Nolen, Tetrahedron Lett., 1993, 34, 2625 CrossRef CAS.
- C. Pichon and A. I. Scott, Tetrahedron Lett., 1994, 35, 4497 CrossRef CAS.
- R. M. Silverstein, G. C. Bassler and T. C. Morrill, Spectrometric Identification of Organic Compounds, Wiley, New York, 1974, p. 192. Search PubMed.
- D. Niu and K. Zhao, J. Am. Chem. Soc., 1999, 121, 2456 CrossRef CAS.
- L. R. Domingo, M. T. Picher and R. J. Zaragoza, J. Org. Chem., 1998, 63, 9183 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2001 |