The application of ICP-SMS, GF-AAS and HG-AFS to the analysis of water and sediment samples from a temperate stratified estuary
Ashley T.
Townsend
*a,
Jeanette
O'Sullivan
b,
Alison M.
Featherstone
c,
Edward C. V.
Butler
bd and
Denis J.
Mackey
b
aCentral Science Laboratory, University of Tasmania, GPO Box 252-74, Hobart, Tasmania 7001, Australia
bCSIRO Division of Marine Research, GPO Box 1538, Hobart, Tasmania 7001, Australia
cSchool of Chemistry, University of Tasmania, GPO Box 252-75, Hobart, Tasmania 7001, Australia
dCooperative Research Centre for Coastal Zone, Estuary and Waterway Management, Natural Sciences Precinct, QCCA Building, 80 Meiers Road, Indooroopilly, Queensland 4068, Australia
First published on 11th January 2001
Abstract
Three atomic spectrometry techniques, namely sector field inductively coupled plasma mass spectroscopy, graphite furnace atomic absorption spectrometry and hydride generation atomic fluorescence spectroscopy (ICP-SMS, GF-AAS and HG-AFS, respectively), housed at separate independent laboratories, were used to analyse water and sediment samples collected from the Huon River Estuary, SE Tasmania (Australia) in the Austral spring 1998. A dithiocarbamate-chelation/back-extraction technique was used to separate and preconcentrate Co, Ni, Cu, Zn, Cd and Pb from eight collected water samples prior to analysis by ICP-SMS and GF-AAS. A number of other elements in the waters were analysed directly (Mn, Fe and Zn by GF-AAS; As by HG-AFS), or following sample dilution (1 + 19; V, Mn, Fe, As, Mo, Ba and U by ICP-SMS). Where possible, previously corroborated GF-AAS and HG-AFS techniques were used to verify obtained ICP-SMS results. From the analysis of four reference waters (SLEW-1 and -2, SLRS-3 and NASS-5), good agreement, to within ±10–20%, was typically found between certified (or information only values) and measured results (irrespective of analytical technique). Exceptions included Zn (and sometimes Fe) that could not be quantified by ICP-SMS due to elevated blank signals, and As which was found to lie below ICP-SMS detection limits. For Huon Estuary water samples, inter-method agreement was within ±10–20% (for those elements amenable to analysis by more than one technique). Nitric acid extracts of two certified reference materials (Buffalo River Sediment and BCSS-1) and six Huon Estuary sediments were analysed by ICP-SMS (for Al, Sc, V, Cr, Mn, Fe, Co, Ni, Cu, Zn, As, Cd and Pb) and HG-AFS (for As). Results from the certified reference materials indicated extraction efficiencies of 60–70% (for most elements). A close correlation between ICP-SMS and HG-AFS was obtained for leachable As in the sediments. In terms of potential inorganic contaminants, the Huon Estuary was found to be a relatively ‘clean’ water system. The elemental concentrations measured in water and sediment samples from this region were found to lie within current Australian guidelines for estuaries. In general, no one analytical technique was able to accurately determine all elements in all samples from this relatively pristine estuarine environment. A combination of all three analytical techniques was necessary for the successful analysis of the elements considered in this study.
Introduction
Reliable and accurate analytical data are a necessary requirement when evaluating the environmental condition of natural waters (groundwaters, rivers, estuaries, open ocean samples, etc.). A comparison between a number of analytical techniques lends support to the delivery of credible environmental information, while certified reference materials (CRMs) can be used to help determine the relative success and suitability of an analytical technique or method. The critical assessment and validation of new analytical techniques against established and previously corroborated methods are crucial as new approaches evolve.In 1998, a preliminary survey of the contaminants in the Huon River Estuary (Tasmania, Australia) was carried out by scientists from CSIRO Division of Marine Research and the University of Tasmania.1 As part of this project, an inter-laboratory, multiple-method approach was used for the analysis of trace metals in water and sediment samples. Earlier studies by some of the authors looking at water samples from other regional river and estuary systems typically employed graphite furnace atomic absorption spectrometry (GF-AAS) under ultra-clean conditions for trace element analysis.2,3 In recent years, inductively coupled plasma mass spectroscopy (ICP-MS) has emerged as a valuable multi-element tool for the rapid analysis of trace metals in a variety of sample types.4 More recently, sector field ICP-MS instruments (ICP-SMS), offering the analyst increased spectral resolution and sensitivity over conventional quadrupole ICP-MS instruments, have become available.5–7 The capacity to separate many spectral interferences from isotopes of interest at relatively low atomic masses (m/z < 80) has proved advantageous in a wide range of research areas.8–10 However, the application of ICP-SMS instruments to environmental studies, including trace metal analysis in marine samples, has generally been limited.11–16
The aims of this study were: (i) to evaluate ICP-SMS for the analysis of environmental samples from relatively uncontaminated systems; (ii) to verify the ICP-SMS results against established atomic spectrometric methods for a number of elements [GF-AAS for Mn, Fe, Co, Cu, Zn and Cd; hydride generation atomic fluorescence spectroscopy (HG-AFS) for As] using certified standards and Huon Estuary samples; and (iii) to apply ICP-SMS to the analysis of water and sediment samples from the Huon River Estuary, providing additional information on the relative health of the water body. This study draws on the experience of local researchers familiar with trace metal analyses of open ocean samples (and with access to clean room facilities).
Experimental
The Huon River survey site
The Huon River catchment is located in southern Tasmania, and has a total area of 3130 km2. Much of the catchment (∼1600 km2) lies within a World Heritage Area. A detailed map of the lower Huon River and Estuary is shown in Fig. 1. The Huon Estuary is an important waterway for emerging aquaculture and tourist industries, with aquaculture the main primary industry in the region. It is a largely unmodified system, and thus samples from the estuary can be considered as typical of a substantially natural estuarine environment. Activities that can degrade water quality in the estuary include sewage treatment, forestry, quarrying, fish and fruit processing, agriculture and aquaculture. Currently, there is no secondary industry in the region, although a pulp mill operated at Port Huon between 1962 and 1991, with a brief closure between December 1982 and June 1986. Agriculture in the Huon Valley consists mainly of horticulture, which in the past has had a history of intensive pesticide use. A number of small municipalities lie on the banks of the Huon River, with a total population in the catchment of approximately 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000.
000.
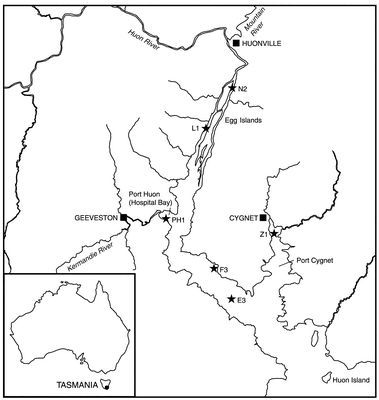 | ||
| Fig. 1 Map of the Huon Estuary in southeast Tasmania (the inset shows the location of the estuary). The sampling sites for the estuarine survey in August/September 1998 are shown. | ||
Past surveys of inorganic pollutants in the Huon Estuary have been sparse, with only a few trace metal measurements made as part of private or local government water quality assays. Further knowledge of the environmental status of the estuary will prove valuable. Trace elements such as Cu, As and Pb were considered important because they were used in pesticide formulations in the orchards of the catchment area for much of the 20th century.17
Sample sites were selected to cover the salinity gradient of the estuary, with particular reference to areas possibly affected by metal inputs. Fourteen stations for water sampling were chosen, with a subset of six selected (eight samples) for ICP-SMS, GF-AAS and HG-AFS analysis. These sites were designated L1, N2, PH1, E3, F3, Z1. Sediment samples were also collected from the same six sites in the estuary, so that the extent of sediment–water interaction might be discerned. Sites possibly contaminated from municipal or previous industrial use were PH1 and Z1. These sites are shown in Fig. 1.
Sample collection, treatment and storage
The water samples were obtained using ‘clean’ techniques. This involved the thorough cleaning of the sampling equipment prior to sample collection, a rigorous sampling protocol in the field, and finally the fastidious processing of the samples.18,19Samples were taken from a 7 m twin-hulled fibre glass vessel using 10 L Teflon-lined Niskin bottles (General Oceanics, Miami, USA), and then subsampled into 1 L low density polyethylene (LDPE) bottles (Nalgene, Rochester, USA). The samples were collected during the Huon ‘contaminants field survey’, from 31st August to 3rd September 1998.1 Samples were collected in the morning of each day and were then taken back to a certified Class 100 clean room laboratory where half of the sample was filtered through an acid-washed 0.45 µm HA filter (Millipore, Bedford, USA). The unfiltered and filtered samples were stored in this clean room laboratory. The samples were later acidified to pH 2 with doubly distilled hydrochloric acid (1 mL L−1; Seastar, Sidney, Canada) and stored for a minimum of 1 month before analysis. This aids in the decomposition of colloids, particles and metal–organic complexes in the sample and maximizes the complexation of the trace metals by the dithiocarbamate extracting agent.
Sediment samples were collected using a box corer. Cores were subsampled, with the top 2.5 cm sections being extruded into acid-washed polyethylene sealed containers. Sediment samples were stored in the dark in a cool room until digestion.
Water preconcentration/treatment prior to analysis
Samples were analysed for Cd, Cu and Co using a modified complexation and solvent extraction procedure described in detail elsewhere.20 Essentially, the method used a dithiocarbamate complexation and extraction into Freon at pH 5, and then back-extraction into nitric acid. The modification entailed an additional extraction of the sample to ensure that the metals of interest were completely recovered. The procedure was performed in a certified Class 100 clean room. Each batch of extractions was verified with the analysis of at least one CRM.The extraction batch destined for ICP-SMS analysis included eight samples, one blank and two CRMs {SLEW-2 and NASS-5 [National Research Council of Canada (NRCC), Ottawa, Canada]}. Replicates of two of the samples, one blank and another SLEW-2 CRM, were extracted for GF-AAS measurement. Previously, all eight samples had been extracted and analysed by GF-AAS. Extracted samples for ICP-SMS analysis were diluted to 5.0 mL with 3% HNO3 (Seastar, Sidney, Canada). Extraction efficiencies cannot be verified for Ni and Pb as these elements were not analysed by GF-AAS. However, it was expected that the extraction of Ni and Pb would be quantitative using dithiocarbamate complexation at pH 5.21
A 10 mL aliquot of each of the eight collected water samples was transferred into acid-cleaned 12 mL polypropylene tubes in the clean room laboratory before transfer to and dilution at the ICP facility. A field blank and four CRMs [SLEW-1 and -2, NASS-5 and SLRS-3 (NRCC)] were also supplied for ICP-SMS analysis. The water samples at pH 2 were diluted (1 + 19) with ultra-pure water, HNO3 (1 wt.%; Mallinckrodt, Paris, USA) and indium internal standard (High Purity Standards, Charleston, USA). All diluted samples were analysed using ICP-SMS for V, Mn, Fe, As, Mo, Ba and U.
For the determination of Mn, Fe and Zn by GF-AAS, a 10 mL aliquot of each sample was also transferred into acid-cleaned 12 mL polypropylene tubes. The samples were acidified with 250 µL of HNO3 (Seastar, Sidney, Canada) before GF-AAS analysis by direct injection.
It should be noted that, unlike the GF-AAS and HG-AFS techniques, ICP-SMS analyses of the water samples were made on a weight per weight basis. However, as the density of seawater is typically of the order of 1.025 g mL−1, any correction between the two unit systems would be less than the precision of the actual measurement. As such, for consistency and ease of understanding, all subsequent results are reported on a weight per volume basis. Environmental guidelines and element concentrations in CRMs for waters are typically expressed in this way.
Sediment leaching before analysis
Each collected sediment sample was dried overnight in an oven at 105°C. Samples were gently ground to break up large aggregates before a small subsample (∼0.5 g) was taken for leaching (partial digestion) with 5 mL concentrated HNO3
(Mallinckrodt, Paris, USA) for 4 h at 100°C. The final leach liquor was diluted gravimetrically (1 + 99) prior to analysis. Samples were digested in triplicate. Two CRMs were also taken through this mild digestion process, namely Buffalo River Sediment (NIST, Gaithersburg, USA) and BCSS-1 Marine Sediment (NRCC), a near-shore sediment from the Gulf of St. Lawrence. Comparison with the total metal concentration of the two CRMs enabled a measure of the nitric acid extraction efficiency to be calculated.
ICP-MS analysis
Measurements were carried out using an ELEMENT sector field ICP-MS (Finnigan, Bremen, Germany). This instrument has three spectral resolution settings available for analysis, namely low, medium and high (nominal m/Δm values = 300, 3000 and 7500, respectively), and is housed in a separate laboratory under a flowing stream of HEPA filtered air (but not clean room conditions). Technical information concerning the typical operation and performance of this instrument has been presented previously.7,22–24A number of elements (i.e., Mo, Cd, Ba, Pb and U) were analysed by ICP-SMS in low resolution mode, offering greater instrumental sensitivity. Other elements (V, Mn, Fe, Co, Ni, Cu and Zn for waters; also Al, Sc and Cr for sediments) were analysed using ICP-SMS in medium resolution mode (nominally m/Δm = 3000), while As was analysed using the highest spectral resolution available (adequate to separate As from polyatomic species such as ArCl and CaCl24). Isotopes considered in this study and associated interferences are shown in Table 1. Elements were chosen for analysis taking into consideration existing analytical expertise, and the recent history of human activity in the Huon Estuary.
| Isotope | Resolution | Common interferences for sample type shown | |
|---|---|---|---|
| Water | Sediment | ||
| a Not analysed in this particular sample type. b Interference not able to be separated from isotope of interest with any available instrumental resolution. c Negligible interference on this isotope for this sample type. | |||
| 27Al | 3000 | a | CN, Fe2+ |
| 45Sc | 3000 | a | CO2, CO2H, SiO, SiOH |
| 51V | 3000 | ClO, ClN, ArNH | ClO, ClN, ArNH |
| 52Cr | 3000 | a | ArC, ArO, ArN, ClOH |
| 55Mn | 3000 | ArN, ArNH, ArOH | ArN, ArNH, ArOH |
| 56Fe | 3000 | ArO, CaO | ArO, CaO |
| 59Co | 3000 | ArNa, CaO | ArNa, CaO |
| 60Ni | 3000 | CaO, NaCl, ArMg | CaO, NaCl, ArMg, Sn2+ |
| 63Cu | 3000 | ArNa, PO2, Na2OH, ClN2 | ArNa, PO2, Na2OH, ClN2, ArAl, ArMg |
| 66Zn | 3000 | ArMg, ArSi, CaO, CaOH | S2, SO2, ArMg, ArSi, CaO, CaOH |
| 75As | 7500 | ArCl, CaCl, ArK | ArCl, CaCl, ArK |
| 95Mo | 300 | ArMnb | a |
| 111Cd | 300 | MoOb | c |
| 137Ba | 300 | BaHb | a |
| 208Pb | 300 | c | c |
| 238U | 300 | c | a |
External calibration was used for the analysis of the waters, water extracts and sediment digests. Mixed standard solutions were prepared from a 100 mg L−1 multi-element solution (QCD Analysts-Environmental Science Solutions, Spring Lake, USA). Where necessary, standards and samples were diluted with ultra-pure Milli-Q deionized water (Millipore, Bedford, USA) that had been further purified in a quartz sub-boiling still. All standards and samples were acidified before analysis with equal amounts of HNO3 (Mallinckrodt, Paris, USA). Indium (10 or 100 µg L−1) was used as an internal standard (prepared from a 1000 mg L−1 single element solution; High Purity Standards, Charleston, USA).
Replicate analyses of CRM and Huon water samples by ICP-SMS showed analytical reproducibilities typically in the range 2–5% for elemental concentrations greater than 1 µg L−1 (depending on the instrumental resolution utilized), 5–20% for 0.1–1 µg L−1 and greater than 20% for concentrations <0.01 µg L−1.
Representative instrumental detection limits [based on three times the standard deviation (3σ) of 10 consecutive ultra-pure blank measurements] for the ICP-SMS instrument used in this work have been presented in detail elsewhere.22–24 Instrument and method detection limits were typically <0.2 µg L−1 (often <0.05 µg L−1) for all isotopes considered in this study. It should be noted that, as the instrumental sensitivity decreases with increased spectral resolution, poorer ICP-SMS detection limits result.7,23
GF-AAS analysis
The instrument used for all GF-AAS determinations was a Perkin-Elmer Zeeman 5000 (Germany). Optimum settings were used, based on the manufacturer's recommendations.25 Injections of 20 µL were employed for the determination of both Cu and Cd, with a 40 µL injection used for Co. Matrix modifiers were employed for the direct injection GF-AAS analyses (Mn, Fe and Zn), except for two river (low salinity) samples. For comparison with ICP-SMS results, all eight samples were analysed using a matrix modifier. For Mn and Fe, the modifier used was Mg(NO3)2 (Suprapur; Merck, Darmstadt, Germany)25,26 and, for Zn, 10% citric acid (Analar; Merck Pty. Ltd., Melbourne, Australia).27For each set of analyses performed using GF-AAS, at least one CRM was analysed. Blanks were taken in the field and during filtration. Extraction blanks, standard blanks and sample bottle blanks were also analysed by GF-AAS.18 Extraction recoveries were determined by comparing absorbances of extracted and non-extracted standards. Extracted standard values were used for the calculation of the GF-AAS results. During the analyses of the Huon samples by GF-AAS, repeat analysis of CRM waters and samples showed reproducibility to within approximately 5% (or the detection limit, whichever was the greater). Detection limits were estimated as 3σ of all blanks including reagent blanks, sample blanks and field blanks. The detection limits for Co, Cu and Cd (extracted metals) were 0.013, 0.009 and 0.0008 µg L−1, respectively. For the direct analyses of Mn and Fe, the detection limits were measured as 0.18 and 1.28 µg L−1, while, for Zn, values of 0.033 and 0.18 µg L−1 were measured for riverine and marine samples, respectively.
HG-AFS analysis
Dissolved inorganic As(V + III) determinations were made on 0.45 µm filtered water samples and sediment digests using semi-automated HG-AFS. This method, described in detail elsewhere,28 couples batch hydride generation, using NaBH4, and hydride trapping with the detection of arsine by atomic fluorescence spectroscopy. Using this method, the detection limit (3σ of 10 replicates of the NaBH4 blank signal) for As(V + III) in a 5 mL sample was 0.0009 µg L−1.Method blanks, in which Milli-Q water was added to sample bottles in the field, then filtered and acidified, did not indicate any detectable arsenic contamination. The method accuracy was determined by analysing the seawater reference material NASS-4 (NRCC), with a reproducibility of approximately 1% found for triplicate analysis.
Results and discussion
Water samples
To provide a sound frame of reference for this study, two distinct ‘reference’ techniques (namely GF-AAS and HG-AFS) were used to corroborate the data obtained by ICP-SMS for the analysis of marine water samples. Both GF-AAS and HG-AFS have previously been fully tested and verified.2,3,28,29 CRM waters were also analysed to help establish the accuracy of the ICP-SMS technique for a broader range of elements.| Isotope/element analysed | Sample preparation method | SLEW-1 | SLEW-2 | NASS-5 | SLRS-3 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Certified | Measured | Certified | Measured | Certified | Measured | Certified | Measured | |||
| a Blank limited. b From ref. 30. c From ref. 14. d Information only value. e Detection limited. f Not analysed. g As measurements confirmed by the analysis of NASS-4, not NASS-5. | ||||||||||
| ICP-SMS | 59Co | Back-extraction | 0.055 ± 0.008 | 0.054 | 0.011 ± 0.003 | 0.013 | ||||
| 60Ni | Back-extraction | 0.709 ± 0.054 | 0.69 | 0.253 ± 0.028 | 0.32 | |||||
| 63Cu | Back-extraction | 1.62 ± 0.11 | 1.34 | 0.297 ± 0.046 | 0.32 | |||||
| 66Zn | Back-extraction | 1.10 ± 0.14 | a | 0.102 ± 0.039 | a | |||||
| 111Cd | Back-extraction | 0.019 ± 0.002 | 0.016 | 0.023 ± 0.003 | 0.029 | |||||
| 208Pb | Back-extraction | 0.027 ± 0.005 | 0.018 | 0.008 ± 0.005 | 0.014 | |||||
| 51V | Dilution (1 + 19) | 1.33b | 1.6 | 1.35c | 1.1 | [1.2]d | 1.25 | 0.30 ± 0.02 | 0.31 | |
| 55Mn | Dilution (1 + 19) | 13.1 ± 0.8 | 16.7 | 17.1 ± 1.1 | 14.4 | 0.919 ± 0.057 | a | 3.9 ± 0.3 | 4.24 | |
| 56Fe | Dilution (1 + 19) | 2.08 ± 0.34 | a | 2.37 ± 0.37 | a | 0.207 ± 0.035 | a | 100 ± 2 | 96.4 | |
| 75As | Dilution (1 + 19) | 0.765 ± 0.093 | e | 0.792 ± 0.082 | e | 1.27 ± 0.12 | e | 0.72 ± 0.05 | e | |
| 95Mo | Dilution (1 + 19) | — | 4.52 | [3.7]d | 3.53 | 9.6 ± 1.0 | 9.91 | 0.19 ± 0.01 | 0.21 | |
| 137Ba | Dilution (1 + 19) | — | 28.2 | 16.9c | 18.6 | — | 5.98 | 13.4 ± 0.6 | 16.9 | |
| 238U | Dilution (1 + 19) | 1.14b | 1.45 | [1.2]d | 1.21 | [2.6]d | 3.18 | [0.045]d | 0.052 | |
| GF-AAS | Co | Back-extraction | 0.055 ± 0.008 | 0.057 | 0.011 ± 0.003 | e | 0.027 ± 0.003 | f | ||
| Cu | Back-extraction | 1.62 ± 0.11 | 1.37 | 0.297 ± 0.046 | 0.31 | 1.35 ± 0.07 | 1.31 | |||
| Cd | Back-extraction | 0.019 ± 0.002 | 0.016 | 0.023 ± 0.003 | 0.021 | 0.013 ± 0.002 | 0.013 | |||
| Mn | Direct matrix modifier | 13.1 ± 0.8 | 13.1 | 17.1 ± 1.1 | 14.5 | 0.919 ± 0.057 | 0.87 | 3.9 ± 0.3 | 3.47 | |
| Fe | Direct matrix modifier | 2.08 ± 0.34 | 2.74 | 2.37 ± 0.37 | 2.74 | 0.207 ± 0.035 | f | 100 ± 2 | 104 | |
| Zn | Direct matrix modifier | 0.86 ± 0.15 | f | 1.10 ± 0.14 | 1.3 | 0.102 ± 0.039 | f | 1.04 ± 0.09 | 1.02 | |
| HG-AFS | As | Direct | 1.26 ± 0.09g | 1.18 | ||||||
For those isotopes considered following simple dilution of the water samples, four CRMs for natural waters (SLEW-1 and -2, NASS-5 and SLRS-3) were used for verification purposes (Table 2). The analysis of Fe by ICP-SMS was limited by blank constraints (although the measured Fe concentration in the relatively ‘concentrated’ SLRS-3 fell within acceptable limits), while As was unable to be measured due to raised detection limits arising from the use of ICP-SMS in high resolution mode (∼1% signal transmission of that available in low resolution mode). A hydride generation apparatus (offering excellent sensitivity and matrix separation for As and other ‘hydride forming elements’) specifically dedicated to our ICP-SMS instrument was unfortunately not available for this work.15,32 The measured concentrations and associated uncertainties for Mo in NASS-5 and for V, Mn, Fe and Mo in SLRS-3 overlapped with certified limits. With the exception of SLRS-3, Mn showed the most variation between measured and reference values, with most ICP-SMS concentrations found to lie outside certified ranges. In many cases, Mn determinations using ICP-SMS were only slightly elevated above the detection limit. The absence of certified concentrations and uncertainties for V (except for SLRS-3), Mo and Ba prevents any definitive statements being presented about the relative accuracy of the ICP-SMS technique for the analysis of these elements. Recent literature values for V, Mo and Ba in SLEW-1 and -2 are also shown in Table 2.
Cobalt, Cu and Cd (following sample back-extraction) and Mn, Fe and Zn (direct analysis with a matrix modifier) in the reference waters were also determined by GF-AAS (Table 2). Measured concentrations overlapped with reference values (generally to within ±5–10%, taking the relative uncertainties of both sets of data into consideration), and with those obtained by ICP-SMS (where applicable). Both Cu and Mn were found to be ∼15–20% too low for SLEW-2, but similar values were also found using ICP-SMS. These measurements were repeated on three occasions with similar concentrations found. Certified Cu and Mn concentrations were measured for at least one other CRM water during these analytical sequences, suggesting that the composition of our SLEW-2 sample may differ from certified values, perhaps due to ageing. Manganese concentrations measured by GF-AAS generally agreed with certified values for SLEW-1, NASS-5 and SLRS-3. Zinc was measured by GF-AAS without the blank problems encountered using ICP-SMS (Table 2). This can be partly attributed to: (i) the Class 100 clean room in which the GF-AAS is housed; and (ii) the fact that the GF-AAS is a dedicated trace (or ultra-trace) instrument. Although slower (in that only one element can be measured at any one time), direct injection GF-AAS enables many elements to be measured in estuarine waters with minimal sample pretreatment. This is advantageous for those elements of low concentration or those with associated blank limitations.
Unlike the GF-AAS used in this study, the ICP-SMS was not housed in a certified clean room, and is used for the analysis of a variety of sample types, operating on a mixed commercial/research basis. As a result, blanks were found to be problematic for a number of elements, particularly Fe and Zn. These elevated blank levels were found to be related to: (i) background concentrations existing in a general instrument laboratory; (ii) the purity of the reagents used; and (iii) instrumental contamination. Work is currently in progress to remove or minimize some of this blank background. Working with smaller dilution factors would also help to elevate ICP-SMS signals above measured blank levels.
Arsenic was measured in the reference seawater NASS-4 by HG-AFS. A concentration of 1.18 µg L−1 was found, compared with a certified value of 1.26 ± 0.09 µg L−1 (Table 2), confirming that this technique is accurate and applicable for the determination of As in marine samples.
| Isotope/element nalysed | Sample preparation method | Z1 surface | E3 surface | E3 bottom | F3 surface | F3 bottom | PH1 bottom | L1 surface | N2 bottom | |
|---|---|---|---|---|---|---|---|---|---|---|
| a Blank limited. b Detection limited. | ||||||||||
| Salinity | 34.3 | 33.0 | 34.9 | 19.3 | 34.8 | 33.9 | 8.6 | 29.9 | ||
| ICP-SMS | 59Co | Back-extraction | 0.047 | 0.028 | 0.016 | 0.094 | 0.029 | 0.053 | 0.11 | 0.18 |
| 60Ni | Back-extraction | 0.24 | 0.24 | 0.22 | 0.29 | 0.25 | 0.28 | 0.33 | 0.32 | |
| 63Cu | Back-extraction | 0.21 | 0.13 | 0.11 | 0.22 | 0.13 | 0.16 | 0.28 | 0.17 | |
| 66Zn | Back-extraction | a | a | a | a | a | a | a | a | |
| 111Cd | Back-extraction | 0.006 | 0.003 | 0.002 | 0.005 | 0.004 | 0.005 | 0.003 | 0.008 | |
| 208Pb | Back-extraction | 0.05 | 0.023 | 0.019 | 0.051 | 0.027 | 0.073 | 0.082 | 0.041 | |
| 51V | Dilution (1 + 19) | 1.79 | 1.73 | 1.27 | 1.23 | 1.67 | 1.35 | 1.27 | 1.06 | |
| 55Mn | Dilution (1 + 19) | 2.53 | 1.13 | b | 4.76 | 1.84 | 1.88 | 5.66 | 16.6 | |
| 56Fe | Dilution (1 + 19) | 54.5 | 26.3 | 24.4 | 127 | 28.2 | 36.4 | 228 | 65.7 | |
| 75As | Dilution (1 + 19) | b | b | b | b | b | b | b | b | |
| 95Mo | Dilution (1 + 19) | 12.1 | 11.1 | 12.2 | 5.26 | 10.8 | 9.91 | 1.77 | 9.39 | |
| 137Ba | Dilution (1 + 19) | 6.83 | 6.34 | 5.74 | 8.12 | 6.11 | 6.01 | 12.8 | 7.62 | |
| 238U | Dilution (1 + 19) | 3.31 | 3.11 | 3.24 | 1.85 | 3.47 | 3.2 | 0.79 | 2.98 | |
| GF-AAS | Co | Back-extraction | 0.038 | 0.014 | 0.012 | 0.074 | 0.021 | 0.044 | 0.078 | 0.141 |
| Cu | Back-extraction | 0.18 | 0.13 | 0.11 | 0.23 | 0.11 | 0.14 | 0.24 | 0.15 | |
| Cd | Back-extraction | 0.0048 | 0.0035 | 0.0036 | 0.0038 | 0.0043 | 0.0045 | 0.0025 | 0.0057 | |
| Mn | Direct matrix modifier | 1.83 | 1.12 | 0.69 | 4.43 | 1.46 | 1.72 | 5.94 | 16.1 | |
| Fe | Direct matrix modifier | 47.8 | 25.6 | 19.3 | 142 | 31.3 | 39.4 | 235 | 73.4 | |
| Zn | Direct matrix modifier | 2.20 | 0.38 | 0.47 | 0.81 | 0.49 | 1.39 | 0.48 | 0.70 | |
| HG-AFS | As | Direct | 1.56 | 1.52 | 1.64 | 0.82 | 1.71 | 1.60 | 0.35 | 1.25 |
A high sensitivity method for As, HG-AFS, proved beneficial for the measurement of this element in the estuarine waters. Arsenic concentrations were measured directly in the Huon water samples, and were found to range from 0.023 to 1.8 µg L−1 (Table 3). As previously mentioned, these concentrations were near or below ICP-SMS detection limits using spectral resolution 7500 (after sample dilution), so no comparison between the two techniques was possible. To facilitate low level As measurements by ICP-SMS, the purchase of dedicated hydride generation attachments is suggested.
Vanadium, Ni, Mo, Ba and Pb in the certified and Huon Estuary waters were also measured by ICP-SMS, utilizing the multi-element capability of this instrument. Due to time and cost constraints, no comparison with other analytical techniques was possible for these elements. These elements are not normally analysed by workers in our laboratories using GF-AAS due to extraction or detection limitations. However, ICP-SMS data for these species are included in Tables 2 and 3 so that a broader picture of the trace element constituents of the estuarine system could be obtained.
The concentrations of all elements analysed in water samples collected from the Huon Estuary were found to lie well below recommended Australian environmental guidelines,33 and were comparable to concentrations measured in other local pristine waterways.1
Comparison with other estuarine/seawater studies using ICP-SMS
A number of studies have recently investigated the analysis of estuarine water or seawater samples using ICP-SMS.13,14,16 These studies have typically combined a careful choice of internal standard with dilution (1 + 4 or 1 + 9) to minimize matrix effects associated with the relatively high salt loading of the samples considered. They also report the need to minimize blank contamination by using ultra-pure reagents in association with ultra-clean instrumentation and laboratory environs.To minimize matrix effects in this work, we selectively removed some of the analytes of interest from the high salt matrix via a dithiocarbamate-chelation/back-extraction procedure. For other elements, the samples were simply diluted (1 + 19) to reduce matrix effects. Although this dilution factor is greater than that used in other studies, we chose to err on the side of caution. In many instances, ICP-SMS is still sensitive enough to accurately determine some elements while this increased dilution prevented the accurate analysis of other ultra-trace elements (e.g., As). For the number of samples analysed in this study, a single internal standard, indium, in combination with a sample matrix minimization procedure (extraction or dilution), was found to provide reliable results, based on measured CRM data. For lengthier analytical series looking at samples of lower dilution (i.e., more saline), multiple internal standards may prove valuable to compensate for any salt deposition near the ICP-SMS sampler and skimmer cones.14 Recent work in this laboratory has also investigated the analysis of marine and estuarine water samples after sample dilution (1 + 4),14 and preliminary results appear to be encouraging. This smaller dilution factor (than that used in this study) may increase the signal-to-background ratio for many elements, allowing for more accurate analysis. Further elements might also be successfully analysed (e.g., Cr, Sb). However, the increased sample matrix concentration would decrease the total number of samples to be analysed before the instrument must be cleaned or reconditioned.
Sediment samples
The analysis of sediment samples from the Huon Estuary was also of interest. Samples were HNO3-leached (in triplicate) with analysis by ICP-SMS and HG-AFS (for As only). Analysis of the digested sediments was not undertaken using GF-AAS as this instrument is a dedicated ultra-trace analysis instrument, housed in a Class 100 clean room, and has not been used for sediment analysis in the past. The isotopes analysed using ICP-SMS are shown in Table 4. Cd and Pb were both analysed using spectral resolution 300, with MoO being the main concern for accurate Cd determination. The measured Mo concentration in the sediment leaches was generally <0.2 µg kg−1 (data not shown), so any contribution by MoO to the 111Cd signal would be minimal. Two or three isotopes of Cd (111 and 112), Ni (60, 61 and 62), Cu (63 and 65) and Zn (66, 67 and 68) were analysed and were found to agree to within ±5%, suggesting any unresolved spectral interference on these isotopes to be minor.| Isotope | NIST SRM 2704 Buffalo River Sediment | BCCS-1 Marine Sediment | L1 | N2 | PH1 | E3 | F3 | Z1 | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reference | Measured | % Extb | Reference | Measured | % Extb | |||||||
|
a Dried samples (∼0.5 g) were leached in concentrated HNO3
(5 mL) at 100°C for 4 h.
b Extracted metal concentration as a percentage of the total certified value.
c Values calculated from the Al2O3 and Fe2O3 values provided.
d Information only values.
e Not analysed.
|
||||||||||||
| 27Al | 61100 ± 1600 | 9840 ± 370 | 16 | 62600 ± 220c | 12400 ± 300 | 20 | 5400 ± 160 | 4660 ± 250 | 16100 ± 420 | 17500 ± 570 | 16600 ± 480 | 8620 ± 70 |
| 45Sc | [12]d | 2.51 ± 0.08 | 21 | — | 3.67 ± 0.05 | — | 1.41 ± 0.04 | 1.19 ± 0.04 | 7.50 ± 0.20 | 6.27 ± 0.16 | 6.25 ± 0.14 | 3.06 ± 0.03 |
| 51V | 95 ± 4 | 18.0 ± 0.6 | 19 | 93.4 ± 4.9 | 32.2 ± 0.8 | 34 | 15.1 ± 0.8 | 11.2 ± 0.7 | 61.9 ± 1.8 | 61.8 ± 1.6 | 66.0 ± 1.0 | 26.4 ± 0.5 |
| 52Cr | 135 ± 5 | 81.0 ± 1.7 | 60 | 123 ± 14 | 40.8 ± 0.2 | 33 | 8.10 ± 0.31 | 7.39 ± 0.50 | 31.6 ± 0.7 | 44.1 ± 1.4 | 39.0 ± 0.3 | 15.6 ± 0.7 |
| 55Mn | 555 ± 19 | 464 ± 17 | 84 | 229 ± 15 | 168 ± 3 | 73 | 41.7 ± 1.1 | 34.8 ± 1.2 | 86.8 ± 1.7 | 74.8 ± 1.4 | 77.5 ± 1.4 | 27.1 ± 0.6 |
| 56Fe | 41100 ± 1000 | 29500 ± 1040 | 72 | 32900 ± 98c | 22800 ± 300 | 69 | 6990 ± 80 | 5740 ± 190 | 28700 ± 600 | 32700 ± 600 | 32900 ± 600 | 13000 ± 20 |
| 59Co | 14 ± 0.6 | 11.2 ± 0.2 | 80 | 11.4 ± 2.1 | 9.8 ± 0.2 | 86 | 4.69 ± 0.17 | 3.74 ± 0.09 | 12.5 ± 0.4 | 7.77 ± 0.25 | 8.74 ± 0.11 | 3.92 ± 0.07 |
| 60Ni | 44.1 ± 3.0 | 35.2 ± 0.8 | 80 | 55.3 ± 3.6 | 47.6 ± 0.2 | 86 | 5.61 ± 0.35 | 6.25 ± 0.85 | 18.9 ± 1.0 | 21.6 ± 0.6 | 21.2 ± 0.7 | 7.34 ± 0.68 |
| 63Cu | 98.6 ± 5.0 | 91.6 ± 3.7 | 93 | 18.5 ± 2.7 | 13.3 ± 0.2 | 72 | 4.29 ± 0.26 | 3.59 ± 0.13 | 47.1 ± 1.1 | 19.9 ± 0.5 | 22.6 ± 0.4 | 13.0 ± 0.1 |
| 66Zn | 438 ± 12 | 382 ± 6 | 87 | 119 ± 12 | 92.3 ± 1.4 | 78 | 13.6 ± 2.6 | 28.3 ± 3.6 | 91.5 ± 7.2 | 61.5 ± 1.6 | 60.0 ± 2.0 | 46.2 ± 7.0 |
| 75As | 23.4 ± 0.8 | 21.0 ± 0.2 | 90 | 11.1 ± 1.4 | e | — | 3.09 ± 0.03 | 1.88 ± 0.16 | 17.4 ± 0.2 | 22.4 ± 0.6 | 27.8 ± 1.1 | 10.0 ± 0.3 |
| 111Cd | 3.45 ± 0.22 | 3.21 ± 0.03 | 93 | 0.25 ± 0.04 | 0.17 ± 0.01 | 68 | 0.08 ± 0.09 | 0.02 ± 0.01 | 0.25 ± 0.01 | 0.06 ± 0.03 | 0.05 ± 0.01 | 0.07 ± 0.04 |
| 208Pb | 161 ± 17 | 147 ± 1 | 91 | 22.7 ± 3.4 | 19.8 ± 0.2 | 87 | 4.68 ± 0.32 | 3.58 ± 0.21 | 27.7 ± 0.4 | 25.1 ± 0.3 | 29.2 ± 0.8 | 15.7 ± 0.1 |
A moderate nitric acid treatment of the sediments was chosen to enable the extractable metal contents of the samples to be determined and compared. Extraction efficiencies were assessed by comparison of measured and found concentrations for two reference sediment samples [one a fresh water river sediment (Buffalo River) and another from a marine environment (BCCS-1)]. Following the extraction procedure, a refractory residue was left that would be expected to contain a portion of most analytes. The data shown in Table 4 should be viewed in that light. It is well known that more aggressive acid attack (often employing HF) is necessary to provide a total metal concentration, but a less aggressive treatment is more likely to provide results representative of the ‘biologically available’ fraction.34,35 With the exception of Al, Sc, V and Cr (for BCCS-1), data are only shown in Table 4 for elements whose extraction efficiencies were >60% (ranging from Cr ≈ 60% to Cd ≈ 93%). Similar extraction efficiencies are reported for most elements for each reference sediment (exceptions are lower extractions of Cr, Cu and Cd for BCCS-1).
Six sediment samples were collected and analysed from the Huon River (Table 4). Four sites (L1, N2, E3 and F3) were from the main arm of the estuary, with two sites (PH1 and Z1) from areas potentially affected by anthropogenic input (close to townships, human wastewater outfall, etc.). Depending on the analyte concentration, the reproducibility for each sample was typically 1–6% [but with some analytes of low concentration (e.g., Cd and Zn), it was as poor as 20–100%]. It is difficult to find trends in the data as presented. However, standardizing trace metal concentrations against Fe (or Al or Sc) is a recommended procedure for interpreting contaminant data in sediments.36 When trace metals were treated in this manner,1 it was found (results not shown) that Cu, Cd and Pb (and possibly Zn) were elevated by a factor of two to three in sediments from sites PH1 and Z1 over values found for other marine sediments from the estuary. This is not unexpected when it is recalled that these sites have been exposed to human input (from municipal sources, and also pulp-mill wastes for PH1) for many decades. Aside from Ni measured at sites PH1, E3 and F3, all elemental concentrations measured in the Huon sediments (Table 4) were below environmental guideline trigger levels.33 The higher Ni levels associated with some sites are unlikely to be an issue of contamination, but perhaps naturally linked with higher Ni concentrations associated with very fine-grained sediments.1
For comparison of analytical methods, As concentrations in the sediment digests were measured using ICP-SMS and HG-AFS. ICP-SMS data obtained using the maximum spectral resolution available (m/Δm = 7500) are shown in Table 4. Leachable As concentrations in the sediment samples ranged from 1 to 30 mg kg−1. An excellent correlation was found between the two techniques ([As]HG-AFS = 1.00 × [As]ICP-SMS + 0.08, r2 = 0.999), reinforcing earlier work from this laboratory indicating that accurate and interference-free As measurements can be made using ICP-SMS in high resolution mode, provided that the concentration of As is sufficiently above detection limits.24
Conclusions
This work has clearly shown that there is no one ‘best analytical technique’ for all elements in all sample types. A judicious choice of sample pretreatment method and analytical technique is required for each element when considering estuarine water and sediment samples, taking into consideration the environmental condition of the water body or sediment sample to be analysed (e.g., degraded vs. ‘substantially natural’ vs. pristine).The value of ICP-SMS as an instrument for the determination of ultra-trace metals in clean aquatic environments has been demonstrated. ICP-SMS data were validated for several elements (Mn, Co, Cu and Cd) by comparison with GF-AAS as a referee method, and by analyses of reference waters. Other trace metals (V, Ni, Mo, Ba, Pb and U) were evaluated on the basis of results from the analysis of CRM only. Iron and Zn were both found to be limited by the blank; direct analysis in a Class 100 clean room is considered to be advantageous for these two elements. Arsenic, provided it is present above ICP-SMS detection limits (as was found for sediments), was verified by HG-AFS measurements. In future work, ICP-SMS will be used to analyse a wider range of metals in estuarine samples. It is anticipated that, for waters, a smaller sample dilution than that used in this study might be beneficial. In agreement with other workers, preliminary results in this vein appear to be promising.
The elemental concentrations measured in water and sediment samples from the Huon River Estuary were (mostly) found to lie well within recommended limits when compared with the latest Australian environmental guidelines for estuaries.32 In terms of inorganic constituents and potential pollutants, the Huon Estuary was found to be a relatively ‘clean’ water system. In addition, there was no evidence in the estuary for contamination arising from the use of inorganic pesticides over ∼50 years in orchards in the Huon Valley (lead arsenate, copper salts, etc.). There is a possibility, however, that the Huon may contain elevated levels of an element not analysed in this study. Although this cannot be ruled out unequivocally, it is likely that any such ‘rogue species’ (arising from waste discharge or as a processing by-product) would also have had elevated partners that were considered in this work. The environmental implications of the results obtained from the Huon Estuary Study are discussed elsewhere,1 and will be the topic of other publications to come.
Acknowledgements
We are most grateful for the contributions of the Huon Estuary Study team members (from CSIRO Marine Research and the University of Tasmania) who made this paper possible. Access to the laboratory facilities of the Central Science Laboratory, CSIRO Marine Research and the School of Chemistry is also acknowledged. Gina Donnelly is thanked for her assistance with Fig. 1. Drs Barry O'Grady and Scott Stark, along with two anonymous referees, are thanked for their useful comments on the manuscript. The Huon Estuary Study was partly funded by the Fisheries Research and Development Corporation as project 96/284.References
- CSIRO Huon Estuary Study Team, Huon Estuary Study — Environmental Research for Integrated Catchment Management and Aquaculture. Final Report to Fisheries Research and Development Corporation. Project Number 96/284, June 2000, CSIRO Division of Marine Research, Marine Laboratories, Hobart, 2000, 285 pp.; also viahttp://www.marine.csiro.au/ResProj/CoastEnvMarPol/huonest/index.html. Search PubMed.
- P. D. Carpenter, E. C. V. Butler, H. W. Higgins, D. J. Mackey and P. D. Nichols, Aust. J. Mar. Freshwater Res., 1991, 42, 625 Search PubMed.
- D. J. Mackey, E. C. V. Butler, P. D. Carpenter, H. W. Higgins, J. E. O'Sullivan and R. B. Plaschke, Sci. Total Environ., 1996, 191, 137 CrossRef CAS.
- K. E. Jarvis, A. L. Gray and R. S. Houk, Handbook of Inductively Coupled Plasma Mass Spectrometry, Blackie Academic and Professional, Glasgow, 1992. Search PubMed.
- N. Bradshaw, E. F. H. Hall and N. E. Sanderson, J. Anal. At. Spectrom., 1989, 4, 801 RSC.
- M. Morita, H. Ito, T. Uehiro and K. Otsuka, Anal. Sci., 1989, 5, 609 Search PubMed.
- L. Moens, F. Vanhaecke, J. Riondato and R. Dams, J. Anal. At. Spectrom., 1995, 10, 569 RSC.
- L. Moens and N. Jakubowski, Anal. Chem., 1998, 7, 251A.
- J. S. Becker and H. J. Dietze, Spectrochim. Acta, Part B, 1998, 53, 1475 CrossRef.
- F. Vanhaecke and L. Moens, Fresenius' J. Anal. Chem., 1999, 364, 440 CrossRef CAS.
- M. Bensimon, M. Looser, A. Parriaux and N. Reed, Eclogae Geol. Helv., 1994, 87, 325 Search PubMed.
- M. Bensimon, M. Leuenberger and A. Parriaux, Analusis, 1996, 24, M8 Search PubMed.
- I. Rodushkin and T. Ruth, J. Anal. At. Spectrom., 1997, 12, 1181 RSC.
- I. Rodushkin, T. Ruth and D. Klockare, J. Anal. At. Spectrom., 1998, 13, 159 RSC.
- S. C. Peters, J. D. Blum, B. Klaue and M. R. Karagas, Environ. Sci. Technol., 1999, 33, 1328 CrossRef CAS.
- M. P. Field, J. T. Cullen and R. M. Sherrell, J. Anal. At. Spectrom., 1999, 14, 1425 RSC.
- R. H. Merry, K. G. Tiller and A. M. Alston, Aust. J. Soil Res., 1983, 21, 549 Search PubMed.
- J. E. O'Sullivan, R. J. Watson and D. J. Mackey, A Guide to Trace Metal Sampling in Estuarine Waters, 1996; http://www.environment.gov.au/marine/coastal_atlas/documentation/standards/oceanography.osullivan.html. Search PubMed.
- R. B. Plaschke, G. Dal Pont and E. C. V. Butler, Mar. Pollut. Bull., 1997, 34, 177 CrossRef CAS.
- D. J. Mackey, J. E. O'Sullivan, R. J. Watson and G. Dal Pont, Mar. Chem., 1997, 59, 113 CrossRef CAS.
- L.-G. Danielsson, B. Magnusson and S. Westerlund, Anal. Chim. Acta, 1978, 98, 47 CrossRef CAS.
- A. T. Townsend, K. A. Miller, S. McLean and S. Aldous, J. Anal. At. Spectrom., 1998, 13, 1213 RSC.
- A. T. Townsend, J. Anal. At. Spectrom., 2000, 15, 307 RSC.
- A. T. Townsend, Fresenius' J. Anal. Chem., 1999, 364, 521 CrossRef CAS.
- Perkin-Elmer, Techniques in Graphite Furnace Atomic Absorption Spectrophotometry,, Perkin-Elmer, 1985, Part No. 09938150. Search PubMed.
- W. Slavin, G. R. Carnrick and D. C. Manning, Anal. Chem., 1982, 54, 621 CrossRef CAS.
- R. Guevremont, Anal. Chem., 1981, 53, 911 CrossRef CAS.
- A. M. Featherstone, P. R. Boult, B. V. O'Grady and E. C. V. Butler, Anal. Chim. Acta, 2000, 409, 215 CrossRef CAS.
- A. M. Featherstone, E. C. V. Butler, B. V. O'Grady and P. Michel, J. Anal. At. Spectrom., 1998, 13, 1355 RSC.
- G. E. M. Hall, J. E. Vaive and J. C. Pelchat, J. Anal. At. Spectrom., 1996, 11, 779 RSC.
- J. M. Cook, J. J. Robinson, S. R. N. Chenery and D. L. Miles, Analyst, 1997, 122, 1207 RSC.
- B. Klaue and J. D. Blum, Anal. Chem., 1999, 71, 1408 CrossRef CAS.
- ANZECC/ARMCANZ, Australian and New Zealand Guidelines for Fresh and Marine Water Quality, National Water Quality Management Strategy, 1999; public comment draft available via WWW http://www.environment.gov.au/science/water/index.html.. Search PubMed.
- D. Florian, R. M. Barnes and G. Knapp, Fresenius' J. Anal. Chem., 1998, 362, 558 CrossRef CAS.
- G. Adami, P. Barbieri and E. Reisenhofer, Int. J. Environ. Anal. Chem., 1999, 75, 251 Search PubMed.
- D. H. Loring and R. R. T. Rantala, Earth Sci. Rev., 1992, 32, 325 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2001 |