Development of a harmonised phosphorus extraction procedure and certification of a sediment reference material
V. Rubana, J. F. López-Sánchezb, P. Pardob, G. Rauretb, H. Muntauc and Ph. Quevauviller*d
aLaboratoire Central des
Ponts et Chaussées, Division Eau, BP 19, F-44340, Bouguenais, France
bUniversitat de Barcelona, Departament
de Química Analítica, Marti i Franquès, 1-11, E-08028, Barcelona, Spain
cEuropean Commission, Environment
Institute, I-21020, Ispra (VA), Italy
dEuropean Commission, Standards, Measurements and Testing Programme, 200 rue de la Loi, B-1049, Brussels, Belgium
First published on 30th November 2000
Abstract
A harmonised procedure for the determination of the forms of phosphorus in freshwater sediments, developed in the frame of the European Programme, Standards, Measurements and Testing (SMT) has been used for a certification campaign for a reference material. This operationally defined scheme is a good compromise between method performance and reproducibility. Furthermore, the method is rather simple to implement and could be used by water managers on a routine basis. A homogeneous and stable sediment reference material has been prepared and will be available before mid 2001. The so-called SMT protocol, together with the reference material, are useful tools in the field of water management, especially at a time when quality assurance is of paramount importance in laboratory analyses. Knowledge of the bioavailable forms of phosphorus is important not only for analysis of sediments but also for sludge and soils. Therefore, the SMT protocol could be extended to these materials.
Introduction
Single or sequential extraction procedures are often used in environmental studies in order to assess the mobility and the bioavailability of a given element.1–3 The determination of specific chemical species is difficult and often hardly possible. Therefore, determination of broader forms or phases defined by their function can be a reasonable compromise, e.g., “bioavailable” forms can give sufficient information to achieve a sound environmental policy.4 The proposed methods are operationally defined, related to specific reagents and procedures, i.e., results are interpreted as being related to a specific phase of the sediment (although sensus stricto they are related solely to a chemical procedure).Regarding phosphorus (P), sequential extraction schemes were first developed for soils and then extended to sediments.5 Many operationally defined schemes are available, allowing the fractionation of the following forms: exchangeable P;6–7 the fraction associated with Al, Fe and Mn oxides and hydroxides;8 and the fraction in Ca-bound compounds often referred to as apatite P.9–13 The lack of uniformity in the procedures used did not allow the results to be compared world-wide or the method to be validated (since the results are operationally defined). There is also considerable interest in the certification of reference materials for environmental analysis. However, the usefulness of a certified reference material (CRM) in the validation of an analytical methodology depends on how well the certified values are established.14
In order to improve this situation, the European Commission through the Standards, Measurements and Testing (SMT) programme has launched a collaborative project (SEPHOS, sequential extraction of phosphorus in freshwater sediment), which aimed to: (i) design a harmonised extraction scheme, (ii) test the selected scheme in interlaboratory studies involving expert European laboratories, and (iii) certify the extractable phosphorus content of a sediment CRM.15 The so-called SMT scheme allows the definition of the following forms: NaOH-extractable P (NaOH-P; P bound to Al, Fe, Mn oxides or hydroxides), HCl-extractable P (HCl-P; Ca-bound P), organic P (OP), inorganic P (IP), concentrated-HCl P (conc. HCl-P; total P). This paper presents the results of the certification campaign as well as the homogeneity and stability tests carried out on CRM 684, a freshwater sediment.
Origin and preparation of the CRM
The CRM was collected in the Po River (Italy), a large river influenced by agriculture and industries, large settlement, and extended rural run-off. The collection site is situated at the lower Po River, close to the city of Gorino. The sediment was collected at a depth of 2–3 m, by means of an INOX grab sampler.The sample was passed through a 2 mm INOX sieve and the fraction
less than 2 mm was collected. The sediment was then air-dried at
room temperature. The drying process was completed in a drying oven with circulating
air, not exceeding 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C in order to preserve the organic phosphorus
compounds. The dried material was passed through a Retsch jaw crusher set
at its smallest opening (1 mm) and ground in a Retsch hammer
mill equipped with tungsten carbide blades. The ground material was passed
through a 90 µm INOX sieve and the fraction less than 90 µm
was collected in a specially designed mixing drum. Table 1
gives the composition of the sample. The material was homogenised for 2 weeks.
In order to test the bulk homogeneity, ten sub-samples of about 10 g
each were taken from the mixing drum and bottled. From each bottle, a pellet
was prepared and analysed for a number of major, minor and trace elements.
The interbottle and intrabottle variability were measured, special attention
was drawn on total P. The homogeneity was good (see results below).
°C in order to preserve the organic phosphorus
compounds. The dried material was passed through a Retsch jaw crusher set
at its smallest opening (1 mm) and ground in a Retsch hammer
mill equipped with tungsten carbide blades. The ground material was passed
through a 90 µm INOX sieve and the fraction less than 90 µm
was collected in a specially designed mixing drum. Table 1
gives the composition of the sample. The material was homogenised for 2 weeks.
In order to test the bulk homogeneity, ten sub-samples of about 10 g
each were taken from the mixing drum and bottled. From each bottle, a pellet
was prepared and analysed for a number of major, minor and trace elements.
The interbottle and intrabottle variability were measured, special attention
was drawn on total P. The homogeneity was good (see results below).
Analytical techniques
The determination of major and trace elements was carried out by X-ray fluorescence spectrometry using a Siemens sequential spectrometer. Organic carbon was determined by applying both C, H, N elemental analysis and Wästhoff combustion analysis. Calibration was performed using CRMs.The SMT protocol is described in the Appendix, further details are given elsewhere.15 This operationally defined scheme comprises five steps i.e., NaOH-P (≈P bound to Al, Fe and Mn oxides and hydroxides), HCl-P (≈P associated with Ca), OP, IP, and conc. HCl-P (≈total P). In this procedure, NaOH and HCl are used as extractants and an intake of 200 mg of sediment is necessary for the extraction. For each form of P, all laboratories made five independent replicates on two different days and from two different bottles. All 15 laboratories used spectrophotometry, based on the Murphy and Riley spectrophotometric method16 as the final detection method for the phosphorus extracted by the SMT procedure.
Although the Williams scheme is an operationally defined extraction scheme, biotests were carried out (on Scenedesmus quadricauda) in order to test the bioavailability of NAIP (non-apatite inorganic phosphorus), the fraction bound to iron oxides and which is called NaOH-P in the SMT protocol. Therefore, the SMT protocol can be considered as a valuable tool in the estimation of the available P fraction in a sediment.
All the reagents used were of analytical reagent grade. Suprapure KH2PO4 was used to prepare standard solutions and suprapure NaOH and HCl were used for calibration. The glassware and plasticware were soaked in 0.3% HCl and rinsed with de-ionised water. A quality control procedure was applied throughout the different steps from sampling, to preparation and analysis.
The data were treated using TEDI software (Tractament Estadistic de Dades Interlaboratori, Statistical Treatment of Intercomparison Data) supplied by the University of Barcelona and designed by A. Padro. This treatment was used to calculate the mean values and the standard deviations of each of the laboratory sets, which were submitted to a series of statistical tests, e.g., the Nalimov test to detect outlying values in the population of results and the Cochran test to detect outlying values in the laboratory variances.
Homogeneity study
One of the prime conditions for acceptance of a candidate reference material is for it to be homogeneous. The between-bottle homogeneity of the extractable P content was verified by the application of the extraction procedure on sub-samples taken from 20 bottles selected at random from the total set. The within-bottle homogeneity was assessed by 10 replicate determinations on the well mixed content of one bottle.17 The relative standard deviation (RSD) and the total uncertainty (URSD) are presented in Table 2. Between-bottle RSDs are low, ranging from 1.5% for conc. HCl-P to 2.7% for HCl-P. For the within-bottle homogeneity, RSDs are also very low. The lowest values are for IP (1–1.6%), the highest for HCl-P (1.9–3.8%) and NaOH-P (1.7–4.2%).| Between-bottle RSD (%) | Within-bottle RSD (%) | ||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | ||
| NaOH-P | 2.5 | 2.2 | 4.2 | 1.7 | 1.8 |
| HCl-P | 2.7 | 2.7 | 3.8 | 3.3 | 1.9 |
| IP | 1.6 | 1.6 | 1.4 | 1.5 | 1.0 |
| OP | 1.9 | 2.9 | 2.6 | 3.1 | 2.0 |
| Conc. HCl-P | 1.5 | 2.1 | 1.6 | 1.7 | 2.0 |
RSDs obtained for the between-bottle homogeneity are sometimes lower than those for the within-bottle homogeneity but this is not systematic. Furthermore, the values obtained when applying the two-tailed F-test at a significance level of 0.05 (Table 3) showed no difference between the within- and between-bottle homogeneity variances.
| F experimental (F-test for comparison of variances) | ||||
|---|---|---|---|---|
| B–W1 | B–W2 | B–W3 | B–W4 | |
| Critical values for a two-tailed test (P = 0.05): F(19.9) = 3.68; F(9.19) = 2.88. | ||||
| NaOH-P | 1.303 | 2.769 | 2.113 | 1.887 |
| HCl-P | 1.046 | 2.095 | 1.567 | 1.919 |
| IP | 1.025 | 1.361 | 1.107 | 2.813 |
| OP | 2.204 | 1.782 | 2.570 | 1.042 |
| Conc. HCl-P | 1.991 | 1.277 | 1.308 | 1.877 |
Stability study
The stability of the extractable phosphorus content was tested to determine the suitability of this kind of material as a candidate reference material. Sample bottles were stored at +4, +20 and +40°C
during a period of 12 months starting in December 1998 and the extractable
phosphorus contents were determined (in six replicates) after 1,
3, 6, and 12 months. Any change in the content of an analyte with time
indicates an instability provided that a good long-term analytical reproducibility
is obtained. Instability would be detected by comparing the contents of different
analytes in samples stored at different temperatures with those stored at
a low temperature, at the different stages of analysis.The samples stored at +4°C were used as the references
for the samples stored at +20 and +40°C. Table 4
gives the ratios (RT, where T denotes
the temperature of storage in °C) of the mean values (XT)
of six measurements made at both +20 and +40°C, and
the mean value (X4) from six determinations
made at the same time on samples stored at a temperature of +4°C:
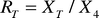 | (1) |
 | (2) |
| Fraction | Time/month | R20 ± U20 | R40 ± U40 |
|---|---|---|---|
| NaOH-P | 1 | 1.03 ± 0.02 | 1.01 ± 0.02 |
| 3 | 1.01 ± 0.01 | 1.00 ± 0.03 | |
| 6 | 0.98 ± 0.02 | 1.00 ± 0.01 | |
| 12 | 0.98 ± 0.01 | 0.94 ± 0.01 | |
| HCl-P | 1 | 0.98 ± 0.03 | 1.01 ± 0.03 |
| 3 | 1.00 ± 0.02 | 1.02 ± 0.02 | |
| 6 | 0.94 ± 0.02 | 0.97 ± 0.04 | |
| 12 | 0.99 ± 0.01 | 1.01 ± 0.01 | |
| IP | 1 | 0.98 ± 0.01 | 0.97 ± 0.02 |
| 3 | 1.01 ± 0.01 | 1.01 ± 0.01 | |
| 6 | 0.98 ± 0.01 | 0.99 ± 0.01 | |
| 12 | 1.02 ± 0.01 | 1.01 ± 0.01 | |
| OP | 1 | 0.97 ± 0.03 | 0.98 ± 0.03 |
| 3 | 1.00 ± 0.03 | 1.00 ± 0.02 | |
| 6 | 0.98 ± 0.01 | 0.96 ± 0.01 | |
| 12 | 0.98 ± 0.02 | 0.93 ± 0.02 | |
| Conc. HCl-P | 1 | 1.00 ± 0.02 | 1.00 ± 0.02 |
| 3 | 1.01 ± 0.01 | 1.00 ± 0.01 | |
| 6 | 1.01 ± 0.01 | 1.01 ± 0.01 | |
| 12 | 1.03 ± 0.02 | 1.00 ± 0.02 |
As can be seen from Table 4,
the ratio RT is close to 1 for all the forms
of P extracted ; at +20°C 0.94 < RT < 1.03
and at +40°C 0.96 < RT < 1.02.
The uncertainty UT is small, generally less than
0.03. The variations of RT with time are slightly
higher for HCl-P. Note that the stability of another sample, rich in organic
matter, had previously been tested using the SMT protocol and proved satisfactory.18
Certification campaign
The sets of results were submitted to the following statistical tests: (i) a Kolmogorov–Smirnov–Lilliefors test, to assess the conformity of the distributions of individual results and of laboratory means to normal distributions; (ii) a Nalimov test, to detect “outlying” values in the population of individual results and in the population of laboratory means; (iii) a Barlett test to assess the overall consistency of the variance values obtained by the participating laboratories; (iv) a Cochran test to detect outlying values in the laboratory variances (s2i); and (v) a one-way analysis of variance (ANOVA; F-test) to compare and estimate the between- and within-laboratory components of the overall variance of all individual results.A summary of the statistical data, as obtained from computing (HOSTAN software, SMT), is given in Table 5. The sets of results found acceptable on statistical grounds were represented in the form of bar charts in which the length of a bar corresponded to the 95% confidence interval of the mean of laboratory means. The certified values were calculated as the arithmetic means of laboratory means (taking into account the number of sets accepted for certification after both statistical and technical scrutiny). This value is featured as a vertical dotted line on the bar graphs; its uncertainty is given by the half-width of the 95% confidence interval of the mean of laboratory means. Fig. 1 gives an example of a bar graph for IP.
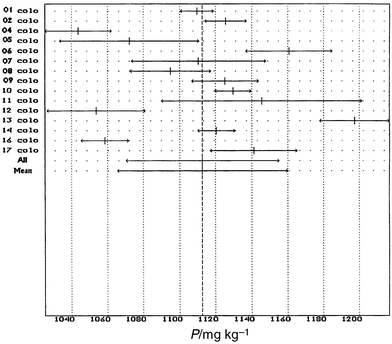 | ||
| Fig. 1 CRM 684. An example of a bar graph for IP. | ||
| NaOH-P | HCl-P | IP | OP | Conc. HCl-P | |
|---|---|---|---|---|---|
| a CI, confidence interval; sW within-laboratory and sB between-laboratory standard deviations, respectively. | |||||
| Number of data sets | 12 | 14 | 15 | 14 | 15 |
| Number of accepted replicates | 60 | 70 | 75 | 70 | 75 |
| All data sets compatible two by two? (Scheffe's multiple t-test) | Yes | Yes | Yes | Yes | Yes |
| Outlying data sets? (Dixon test, Nalimov t-test and Grubbs test) | No | No | No | No | No |
| Outlying variances? (Cochran test) | No | No | No | No | No |
| Means of means | 550 | 536 | 1113 | 209 | 1373 |
| sW | 17 | 17 | 26 | 6 | 27 |
| sB | 32 | 47 | 41 | 14 | 61 |
| Between-data s significant? (Snedecor test) | No | No | No | No | No |
| Variances homogeneous | No | Yes | No | Yes | No |
| s of means | 33 | 48 | 43 | 15 | 62 |
| Data sets means normally distributed? (Kolmogorov–Smirnov–Lilliefors test) | Yes | Yes | Yes | Yes | Yes |
| 95% CI of the mean of means | 21 | 28 | 24 | 8 | 34 |
Discussion
As can be seen from Table 3, no difference was detected between the within- and the between-homogeneity variances using an F-test. Therefore, the material was considered to be homogeneous. There was also no significant difference in the RT values, the ratio being close to 1, which showed the stability of the material both at 20 and 40°C. The CRM is homogeneous and stable and can
be used for the certification campaign.Before starting the statistical discussion a technical discussion was carried out to make sure that all laboratories strictly followed the SMT extraction protocol. A critical point that was stressed was the necessary calibration using extracting solutions (or external calibration with cross-check of calibrants in the extracting solutions).
All laboratories used colorimetry as the final method of determination. A wavelength of 880 nm was originally specified in the protocol. However, two laboratories used a wavelength of 700 nm and did not observe any difference with the other results based on a 800 nm wavelength. Although it is recognised that the choice of wavelength (700 or 880 nm) has an effect on the performance characteristics of the colorimetric method, in particular, its sensitivity, it had no detectable effect on the between-laboratory agreement (i.e., no detectable bias was observed for one particular wavelength). Therefore it was decided to accept both wavelengths in the analytical protocol.
The temperature of extraction was also discussed. A temperature of 21 ± 1°C
was required in the original protocol. It was pointed out that several laboratories
had lower or higher temperatures without their results being affected. Therefore,
the revised protocol was made more flexible and now stipulates a temperature
of 21 ± 3°C.
Regarding the certification campaign, the estimates of the within-laboratory standard deviation (sW) and the between-laboratory deviation (sB), as derived from the one-way ANOVA, demonstrated that the sB was not significant. For reasons of uniformity, it was decided to base the certification on the laboratory means rather than on all individual results. The half-width of the 95% CI of the mean of the data set means was adopted as the uncertainty.
For Cochran and Nalimov tests, a value is called an “outlier” when the hypothesis that it belongs to the population of results considered can be rejected with a risk of error of 0.01. The criterion was adopted that an outlier of variance would be eliminated only if the standard error of the mean (si/√ni) of the set exceeded the standard deviation of the distribution of all laboratory means.
The statistical evaluation of the results was carried out in order to ensure that the population of results accepted for certification had a normal distribution before the 95% CI was calculated. This was true in all cases (Kolmogorov–Smirnov–Lilliefors tests). In addition there were no outlying values (Nalimov). The set of variances was often not homogeneous (three cases of five), which was due to the different repeatability and reproducibility of the method as applied by the different laboratories.
A few laboratories were withdrawn: for NaOH-P, 3 labs; for HCl-P, 1 lab; and for OP, 1 lab. All the labs were accepted for IP and conc. HCl-P.
Certified values
The certified values (unweighted mean of P accepted sets of results) and their uncertainties (half-width of the 95% CIs) are given in Table 6 as mass fractions of the respective extracts obtained at the different steps (in mg kg−1 dry mass).| Certified value/mg kg−1 | Uncertainty/mg kg−1 | P (number of data sets) | |
|---|---|---|---|
| NaOH-P | 550 | 21 | 12 |
| HCl-P | 536 | 28 | 14 |
| IP | 1113 | 24 | 15 |
| OP | 209 | 9 | 14 |
| Conc. HCl-P | 1373 | 35 | 15 |
Follow-up
A homogeneous and stable sediment reference material for the certification of extractable phosphorus has been prepared and will be available before the middle of 2001 under the reference number CRM 684. This will allow laboratories to check and improve their results in this field. The harmonised SMT protocol, together with the reference material are useful tools in the field of water management, especially at a time when quality assurance is of paramount importance in laboratory analytical work.Finally, the results of the certification campaign carried out in the frame of the SEPHOS European project (EC Contract SMT4-CT96-2087) are promising. Since the knowledge of the bioavailable forms of P are important not only for the analysis of sediments but also for sludge and soils, the SMT protocol could be extended to these materials. New research will be launched in this direction.
Availability of CRM 684
CRM 684 will be available by the middle of 2001 from the Institute for Reference Materials and Measurements (IRMM), Retieseweg, B-2440 Geel, Belgium (Fax: +32 14 590406; E-mail: bcr.sales@irmm.jrc.be). Further information on other available CRMs can be obtained from the IRMM Website at http://www.irmm.jrc.be/mrm.html.Appendix: SMT protocol
A NaOH-extractable P and HCl-extractable P
(1) Weigh 200 mg of dry sediment in a centrifuge tube. It is important to keep the sediment ∶ volume ratio constant. 200 mg of sediment is the minimum required. (2) Add with a pipette, 20 ml of 1 M NaOH. (3) Cover the tube and stir overnight (16 h). Thorough mixing is necessary, the sediment must be kept in suspension (use, e.g., a magnetic stirrer, a shaker table). (4) Centrifuge at 2000g for 15 min.B Concentrated HCl-extractable P
(1) Weigh 200 mg of dry sediment in a porcelain crucible. (2) Calcine at 450°C for 3 h. (3) Pour the cool ash into a centrifuge tube.
(4) Add 20 ml of 3.5 M HCl with a pipette. HCl can be added
directly to the crucible to ease the transfer of the ash. (5) Cover the tube
and stir overnight (16 h). (6) Centrifuge at 2000g for 15 min.
(7) Collect the extract in a test-tube for the determination of concentrated
HCl-PC Inorganic and organic P
°C.
Put the tubes in an ultrasonic bath for 10 s and transfer to a porcelain
crucible. (5) Calcine at 450°C for 3 h. (6) Pour the cool ash
into the centrifuge tube. (7) Add 20 ml of 1 M HCl with a pipette.
HCl can be added directly to the crucible to ease the transfer of the ash.
(8) Cover the tube and stir overnight (16 h). (9) Centrifuge at 2000g
for 15 min. (10) Collect the extract in a test-tube for OP determination.D Calculation
The concentration, C, in mg g−1 (dry weight) is: | (A1) |
For NaOH-P:
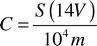 | (A2) |
Acknowledgements
This project was carried out under EC contract no. SMT4-CT96-2087 and was co-ordinated by the Laboratoire Central des Ponts et Chaussées. The following laboratories participated in the interlaboratory studies: Bundesamt und Forschungszentrum für Landwirtschaft, Vienna (Austria); CEMAGREF (France); Geological Survey of Finland, Espoo (Finland); Institut National de Recherche Agronomique, Villenave d'Ornon (France); Joint Research Centre, Environment Institute, Ispra (Italy); Laboratoire Central des Ponts et Chaussées, Bouguenais (France); Macaulay Institute for Land Use Research, Aberdeen (UK); Universitat de Barcelona, Departament de Química Analítica (Spain); Universidad de Cordoba (Spain); University of Gent (Belgium); Universität Hamburg (Germany); Universidad de Huelva (Spain); University of Lisboa (Portugal); Université Montpellier I (France); University of Uppsala, Erken laboratory (Sweden); University of Wageningen (The Netherlands).References
- A. Ure, Ph. Quevauviller, H. Muntau and B. Griepink, Int. J. Environ. Anal. Chem., 1993, 51, 135 Search PubMed.
- A. Ure, Ph. Quevauviller, H. Muntau and B. Griepink, EUR-Report 14763 EN, European Commission, Brussels, 1993, 86 pp. Search PubMed.
- G. Rauret, J. F. Lopez-Sanchez, A. Sahuquillo, R. Rubio, C. Davidson, A. Ure and Ph. Quevauviller, J. Environ. Monit., 1999, 1, 57 RSC.
- Ph. Quevauviller, A. Ure, H. Muntau and B. Griepink, Int. J. Environ. Anal. Chem., 1993, 51, 129 Search PubMed.
- S. C. Chang and M. L. Jackson, Soil Sci., 1957, 84, 133 CAS.
- A. H. M. Hieltjes and L. Lijklema, J. Environ. Qual., 1980, 3, 405 Search PubMed.
- K. C. Ruttenberg, Limnol. Oceanogr., 1992, 7, 1460 Search PubMed.
- R. Psenner and R. Puckso, Archiv. Hydrobiol., 1988, 30, 43 Search PubMed.
- J. D. H. Williams, J. K. Syers, S. S. Shukla, R. F. Harris and D. E. Armstrong, Environ. Sci. Technol., 1971, 11, 1113.
- J. D. H. Williams, J.-M. Jaquet and R. L. Thomas, J. Fish. Res. Bd. Can., 1976, 33, 413 Search PubMed.
- J. D. H. Williams, T. Mayer and J. O. Nriagu, Soil Sci. Soc. Am. J., 1980, 44, 462 Search PubMed.
- H. L. Golterman, Hydrobiologia, 1982, 92, 683 Search PubMed.
- H. L. Golterman, Hydrobiologia, 1996, 5, 1 Search PubMed.
- A. Sahuquillo, J. F. Lopez-Sanchez, R. Rubio, G. Rauret, R. P. Thomas, C. M. Davidson and A. Ure, Anal. Chim. Acta, 1999, 382, 317 CrossRef CAS.
- V. Ruban, J. F. Lopez-Sanchez, P. Pardo, G. Rauret, H. Muntau and Ph. Quevauviller, J. Environ. Monit., 1999, 1, 51 RSC.
- J. Murphy and J. P. Riley, Anal. Chim. Acta, 1962, 27, 31 CrossRef CAS.
- J. F. Lopez-Sanchez, A. Sahuquillo, H. D. Fiedler, R. Rubio, G. Rauret, H. Muntau and Ph. Quevauviller, Analyst, 1998, 123, 1675 RSC.
- P. Pardo, J. F. Lopez Sanchez, G. Rauret, V. Ruban, H. Muntau and Ph. Quevauviller, Analyst, 1999, 124, 407 RSC.
| This journal is © The Royal Society of Chemistry 2001 |