Total organic carbon analyzers as tools for measuring carbonaceous matter in natural waters†
Edward Todd
Urbansky
United States Environmental Protection
Agency, Office of Research and Development, National
Risk Management Research Laboratory, Water Supply and Water
Resources Division, 26 West Martin
Luther King Drive, Cincinnati, OH 45268, USA. E-mail: urbansky.edward@epa.gov; Fax: +1
513 469 7658; Tel: +1 513 569 7655
First published on 9th January 2001
Abstract
For some utilities, new US drinking water regulations may require the removal of disinfection byproduct (DBP) precursor material as a means of minimizing DBP formation. The Environmental Protection Agency's Stage 1 DBP Rule relies on total organic carbon (TOC) concentrations as a measure of the effectiveness of treatment techniques for removing organic material that could act as DBP precursors. Accordingly, precise and accurate methods are needed for the determination of TOC and dissolved organic carbon (DOC) concentrations in raw and finished potable water supplies. This review describes the current analytical technologies and summarizes the key factors affecting measurement quality. It provides a look into the fundamental principles and workings of TOC analyzers. Current peroxydisulfuric acid wet ashing methods and combustion methods are discussed. Issues affecting quality control, such as non-zero blanks and preservation, are covered. Some of the difficulties in analyzing water for TOC and DOC that were identified up to 20 years ago still remain problematic today. Limitations in technology, reagent purity, operator skill and knowledge of natural organic matter (NOM) can preclude the level of precision and accuracy desirable for compliance monitoring.
 Edward Todd Urbansky Edward Todd Urbansky | Ed Urbansky attended Allegheny College in Meadville, PA, and Purdue University in West Lafayette, IN. He joined EPA'sNational Risk Management Research Laboratory in July 1997. Although he is an inorganic chemist, much of his work at EPA has dealt with the analytical chemistry of disinfection byproducts or perchlorate. He has authored or coauthored 30 papers and edited a book, Perchlorate in the Environment. He also serves on the Editorial Board of Journal of Environmental Management. |
1 Introduction
Organic carbon compounds are subject to reaction with oxidizing disinfectants. When raw water is chlorinated, active chlorine compounds (Cl2, HOCl, ClO−) react to produce chlorinated disinfection byproducts (DBPs).1,2 Some of these have been associated with carcinogenesis in animal toxicology studies.3 As a consequence, the Environmental Protection Agency (EPA) has established limits for DBPs in US public water systems either individually or as classes in the Stage 1 DBP Rule, e.g., trihalomethanes, haloacetates, 2,2,2-trichloroethanediol (chloral hydrate).4,5 The identified DBPs comprise about 40% of the total organic halide that is adsorbable to activated carbon.6 It is not possible to identify and quantify all the possible DBPs, and there is probably limited benefit in continuing to look for as yet unidentified DBPs, as has been addressed elsewhere.7 Because nearly all regulated DBPs are chlorinated and/or brominated hydrocarbons that result from the direct reaction of halogen oxidants with carbonaceous matter, EPA's Office of Water has focused on the elimination of organic matter that acts as precursor material in its recent regulations.4,5 Although the importance of removing DBP precursors was pointed out by Australian authorities in the 1996 Drinking Water Guidelines, there was no indication of acceptable values for total organic carbon (TOC) or dissolved organic carbon (DOC) concentration. Instead, specific DBPs were mentioned, and precursor removal was listed as one of the means for controlling DBP formation.8 The European Union (EU) does not regulate TOC concentration specifically, saying only that there must be “no abnormal change.”9 Instead, the EU has established an analogous parameter, oxidizability by permanganic acid (acidified KMnO4) for 10 min at 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C.
This value is limited to 5.0 mg L−1
(as
O2),9 which corresponds to
a maximal TOC concentration of 1.9 mg L−1
(as
C) if complete reaction is assumed. Likewise, the UK Drinking Water Inspectorate (Anthony
Lloyd, 2000, personal communication) and the Guidelines for Canadian
Drinking Water Quality (published by the Ministry of Health for the Federal-Provincial
Subcommittee on Drinking Water) have no specific TOC regulation. Switzerland's
Agency for the Environment, Forests and Landscape does limit TOC concentration
to 2.0 mg L−1, but not strictly—groundwaters
with naturally higher TOC concentrations may be used as drinking water sources.
The Swiss Food Manual offers only an “experience value” of 1.0 mg L−1,
which is considered to reflect a naturally clean water; above 2.0 mg L−1,
the source of the TOC should be ascertained before the water is used for a
potable water supply (Konrad Stemmler, 2000, personal communication).
°C.
This value is limited to 5.0 mg L−1
(as
O2),9 which corresponds to
a maximal TOC concentration of 1.9 mg L−1
(as
C) if complete reaction is assumed. Likewise, the UK Drinking Water Inspectorate (Anthony
Lloyd, 2000, personal communication) and the Guidelines for Canadian
Drinking Water Quality (published by the Ministry of Health for the Federal-Provincial
Subcommittee on Drinking Water) have no specific TOC regulation. Switzerland's
Agency for the Environment, Forests and Landscape does limit TOC concentration
to 2.0 mg L−1, but not strictly—groundwaters
with naturally higher TOC concentrations may be used as drinking water sources.
The Swiss Food Manual offers only an “experience value” of 1.0 mg L−1,
which is considered to reflect a naturally clean water; above 2.0 mg L−1,
the source of the TOC should be ascertained before the water is used for a
potable water supply (Konrad Stemmler, 2000, personal communication).
It is worth pointing out that DBP minimization and microbe inactivation are competing public health goals, and actions taken to further one can upset the other.10 With the Interim Enhanced Surface Water Treatment Rule (IESWTR), the EPA has attempted to strike a balance, partly by advocating the use of the lowest effective disinfectant dose.11 DOC concentration continues to be a parameter used for DBP minimization studies.12 Given the emphasis that current US drinking water research and regulations place on removing natural organic matter (NOM) (DBP precursors), accurate and precise measurement of TOC concentrations is of obvious importance and provides the focus of this review.
Until recently, most advances in the determination of TOC concentration were made by oceanographers and marine chemists. For this reason, papers on freshwater analysis are still published in marine science journals. In 1993, Marine Chemistry devoted an entire issue to the measurement of TOC concentration. Most of the past research was dedicated to assessing carbon cycling in natural bodies of water, and the topic has been reviewed previously by Van Steenderen13 and Wangersky.14 The application of this technique to the regulation of potable water is a new driving force for research papers now appearing in analytical chemistry, environmental science and drinking water journals. Although the amount of work in TOC concentration determination is continually expanding, the field remains moderately immature due to the relatively few active researchers and modest funding that have been dedicated to it historically.
Because there are inorganic and organic carbon-containing substances in natural waters, the first step in any analysis is to remove the inorganic constituents. Next, all of the organic carbon must be converted to an identifiable form. Modern instruments convert it to carbon dioxide. Lastly, this must be measured. All instruments must go through a process like that shown in Scheme 1. The sections that follow discuss the individual steps in more detail.
 | ||
| Scheme 1 Flow chart of the process for the determination of total organic carbon (TOC) and total inorganic carbon (TIC) beginning with an aqueous liquid sample. Volatile organic carbon (VOC) is lost during the sparging step and is not usually detected (see text for more details). If a filtration step is inserted before the acidification, particulate carbon is removed so that dissolved inorganic carbon (DIC) and dissolved organic carbon (DOC) are measured instead. | ||
2 Inorganic and organic carbon
2.1 Inorganic carbon
Inorganic carbon refers to carbon(IV) oxides and hydroxides and their ions. With the exception of the carbon tetrahalides (tetrahalomethanes), carbon(IV) is found as carbon dioxide (CO2), its hydrate, carbonic acid (H2CO3) and the subsequent dissociation products, hydrogen carbonate (HCO3−) and carbonate (CO32−). Inorganic carbon is not known to lead to the formation of DBPs. Because carbon(IV) is not oxidizable, reaction with halogen oxidants is not known to occur.Inorganic carbon may be either dissolved (DIC) or particulate (PIC). Particulate CaCO3 is commonplace. Care must be taken to avoid exceeding the neutralizing capacity of acids added to drive off CO2, as might occur in samples containing large amounts of particulate carbonate minerals. Phosphoric acid is used in commercial TOC analyzers,15 but sulfurous acid16 has also been used. For typical water samples, Van Hall et al.17 showed that essentially all of the DIC could be eliminated with 3–5 min of sparging after acidification. Using 14C-radiolabeled salts, Sharp18 showed that 99.9% of the DIC could be removed by 10 min of sparging; however, few instruments use this long a time. Sievers produces an instrument that uses vacuum degassing instead of sparging, which could be beneficial.
2.2 Organic carbon (carbonaceous matter)
Carbonaceous material in natural water supplies varies dramatically between water sources. In part, this is due to the variability of NOM. A number of factors influence the make-up of NOM: upstream plant and animal life, temperature, climate/weather and soil type, to name a few. Although it is common to refer to all of the carbonaceous material as NOM, there are anthropogenic sources. For example, recharge or discharge from sewage treatment plants, agricultural farming, animal husbandry and local industry can all contribute to the organic matter. Consequently, herbicides, insecticides and a host of other compounds may be found either dissolved in the water or adsorbed to particulates. Particulate NOM can range in size from the supramolecular (e.g., colloids or viruses) to the microscopic (e.g., algae, protozoans or bacteria) to macroscopic (e.g., tree limbs/leaves, aquatic plants, insects, fish or other animals—alive or dead). In terms of analysis, filtration is the most common way of separating particulate organic matter (POM) from dissolved organic matter (DOM) in the liquid sample (e.g., glass fiber, alumina or membranes of nitrocellulose, polyethersulfone or cellulose acetate). It must be realized that such a definition of dissolved is necessarily operational and depends on the nominal pore size of the filter (usually 0.45 µm). Under the pressure of filtration, some colloidal materials, planktonic organisms and detritus may be physically degraded into pieces that can pass through the membrane. Even if filtration is omitted, the inner diameters of needles and tubing used in the instruments (often 1.59 mm = 0.0625 in) restrict the sizes of objects. Thus, zooplankton and visible colonies of phytoplankton, monera or fungi may be excluded. Lastly, some of these organisms may adhere to the walls of tubing and sampling needles.‡In terms of drinking water treatment, most macroscopic objects are removed by sieving or straining, while many smaller particles are removed by gravel/sand filtration or coagulation–sedimentation.19 Much of the interest in TOC/DOC measurement stems from controlling DBPs. Because large objects are normally removed before disinfection, they generally do not represent DBP precursors and are therefore excluded from TOC. However, unusual objects are occasionally observed in the chlorinators of utility plants that are not well maintained.
Descriptions of NOM fill whole volumes and are beyond the scope of this
review. However, it will be instructive to cover some key points. First, soluble
NOM can range in molecular mass from under 102 Da to over 106
Da.20 Second, NOM contains a mixture of aromatic
and aliphatic functionalities that react differently.21,22
Third, NOM has a mixture of hydrophilic and hydrophobic moieties that cause
it to exhibit surfactant-like properties. Molecules aggregate into pseudomicelles
that can perhaps become colloidal.23 This
can blur the line separating dissolved and particulate matter
from a strictly physicochemical standpoint. Fourth, most of the NOM in freshwater
is allochthonous
(produced outside the system by terrestrial
microbes, plants and animals), whereas most of the NOM in seawater is autochthonous
(produced
inside the system by aquatic microbes, plants and animals).23
Fifth, some compounds are of smaller mass (e.g., ethanedioate,
methanoate, ethanoate), but much of the carbonaceous matter comprises
compounds with molecular masses ranging from 100 u to 30000 u
or more. Some species (e.g., lignins, tannins, polypeptides,
polysaccharides and their derivatives) can have molecular masses reaching
into the millions. In addition to coming from detritus, larger molar mass
compounds are also derived from petrochemicals (e.g., polyacrylates
used as scale inhibitors, non-ionic surfactants used in laundry detergents
or pollution from refineries and oil drilling operations).
During the sparging process to remove inorganic carbonates (vide supra), volatile organic carbon (VOC) is normally lost. VOC comprises primarily low molecular weight hydrophobic species, such as short alkanes or small aromatic molecules [e.g., C6H6, CH3C6H5, CH3CH2C6H5, (CH3)2C6H4]. Estimates of VOC concentration usually fix it at less than 10% of the DOC concentration, and it is commonly neglected.14 However, VOC concentration can be determined on some instruments by modifying the program, and some investigators have in fact measured VOC concentrations.§24 Once the VOC has been sparged away, what remains is non-spargeable (non-purgeable) organic carbon (NSOC). NSOC can be further divided into the particulate (NSPOC or just POC) and the dissolved (NSDOC). Taken all together, the concentrations of VOC, NSDOC and POC sum to the TOC concentration.
3 Digestion methods for converting organic carbon to carbon dioxide
All TOC analyzers work by converting organic matter to carbon dioxide. Except for tetrahalomethanes, this is a redox process and involves further oxidation of the carbon atoms in the molecules. There are several different methods used to convert carbonaceous matter to carbon dioxide. As a result, there is considerable brand-to-brand design variability—unlike many modern analytical instruments in the chemistry laboratory. The relative effectiveness of the different digestion methods remains a source of debate to this day. Moreover, it is not clear which of these approaches gives the best estimate of DBP precursor material, which is the crux of the matter for potable water analysis, but not for oceanographic and marine chemical studies of carbon budget and balance. A number of manufacturers and their products are listed in Table 1.| Manufacturer | H2S2O8a | Combustion | Detector |
|---|---|---|---|
| a Method for promoting H2S2O8 reaction is signified by superscript: Δ = thermal; λ = UV light. b n.a. = not available from this manufacturer. c NDIR = non-dispersive infrared. | |||
| Anatel Corporation, Boulder, CO | A-2000λ | n.a.b | NDIRc |
| OI Analytical College Station, TX | 1010Δ | 1020 | NDIR |
| Shimadzu Scientific Instruments Inc., Columbia, MD | n.a. | 5000A, 5050A | NDIR |
| Sievers Instruments Inc., Boulder, CO | 400λ, 800λ, 2244APλ | n.a. | Conductivity |
| Tekmar-Dohrmann, Cincinnati, OH | Phoenix 8000λ | Apollo 9000 | NDIR |
| ThermoGLAS Thermo Environmental Instruments Inc., Franklin, MA | n.a. | 1200 | NDIR |
| UIC Inc., Joliet, IL | n.a. | CM5120 | Coulometric titrimetry |
3.1 Combustion
While a number have relied on sealed tube analysis, which is considered to be the reference technique for seawater,25–27 this approach has not generally been applied to freshwater. Sealed tube analyses are labor intensive and cannot be automated. For freshwater analysis, a variety of commercial TOC analyzers have been used. In these units, the aqueous sample is dispensed as a stream of liquid into a combustion tube filled with a catalyst that promotes the redox reaction with oxygen. A combustion tube for the Tekmar-Dohrmann Apollo 9000 is shown in Fig. 1. Oxidation–reduction takes place at temperatures ranging from 680 to 1000°C. Some units
also allow the sample to be loaded into a ceramic or glass boat which is then
moved into the hot part of the tube furnace.
 | ||
| Fig. 1 Diagram of the combustion tube used in the Apollo 9000 TOC analyzer shown during the filling process. The liquid aqueous sample is directed in a stream down onto the hot catalyst. Reproduced with permission of Tekmar-Dohrmann. | ||
One of the first applications of combustion analysis was to seawater samples,
which can be refractory to peroxydisulfuric acid digestion. Early combustion
methods, even with a cobalt oxide catalyst, used high temperatures, e.g.,
950°C.28,29 The use of transition
metal catalysts (e.g., Pt, Co, Cu, Ir, their oxides or alloys)
allows lower combustion temperatures to be used, specifically 680°C.
For example, a platinum catalyst was used by Sugimara and Suzuki30
to determine DOC concentration in seawater. The chemistry of these catalysts
has been studied, reviewed and summarized elsewhere.31–33
Some studies concluded that the more effective catalysts contain at least
5% platinum.14 In other studies, the
support appeared to influence the reaction efficiency, as platinized alumina
was shown to be less satisfactory than platinized quartz wool or quartz spheres.34 Other studies, however, found that equally good
results were obtained with non-catalytic packings.35,36
Besides effects due to the packing material, one of the problems with some
combustion tubes has been a dead volume/cold zone near the point of injection;
this can be a source of memory problems (residual carryover).35
Combustion TOC analyzers require a constant flow of high purity oxygen. This can be provided by purchasing the compressed gas in cylinders or through the use of an air compressor and a TOC air generator. For example, Balston makes a TOC air generator that contains a small furnace (formerly made by Whatman and now sold under the name Whatman–Balston, Tewksbury, MA, USA). The air flows through a particulate filter and over a heated catalyst (in a fashion very similar to a combustion TOC analyzer) so that hydrocarbons are completely burned. The carbon dioxide and water are removed via a regenerating desiccant system that functions similar to a molecular sieve. According to the technical support staff, the unit continually alternates between desiccant chambers to regenerate the desiccant, relying on absorption–desorption cycling.
3.2 Peroxydisulfuric acid wet ashing
Many of the early works on measuring organic carbon in seawater used peroxydisulfuric acid.37 Peroxydisulfuric acid is a strong oxidant that reacts with organic matter. The gases evolved by digestion (redox reaction) are carried off by a stream of oxygen or an inert gas so that the carbon dioxide can be determined. Digestion cells for the OI and Tekmar-Dohrmann TOC analyzers are shown in Fig. 2. | ||
| Fig. 2 Diagram of the peroxydisulfuric acid wet ashing reaction chamber used in the (a) OI Analytical 1010 (thermal) and (b) Tekmar-Dohrmann Phoenix 8000 (ultraviolet) TOC analyzers. Reproduced with permission of Tekmar-Dohrmann . | ||
The peroxydisulfate anion (O3SOOSO32−)
contains a peroxy linkage which can be homolytically cleaved either thermally (∼100°C)
or photolytically [ultraviolet (UV) light]. Thus, peroxydisulfate
acts as a source of radical anions. These radical species either react directly
with organic matter or with water to produce hydroxyl radical. The hydroxyl
radical ferociously attacks organic matter. Regardless of the means used to
promote the reaction, all peroxydisulfuric acid-based techniques rely
on the formation of radical sulfate ions to begin the process. Peroxydisulfuric
acid decomposes as follows.38 Uncatalyzed
pathway:
 | (1) |
 | (2) |
 | (3) |
 | (4) |
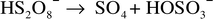 | (5) |
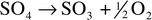 | (6) |
3.3 Other digestion methods
Any species that can form a radical can be used to initiate redox chemistry, and those radicals generated directly from water or oxygen can fulfill this need. Although not incorporated into any commercial instrument used in the analysis of untreated water supplies, an analytical instrument has been designed based on digestion with high intensity UV light. The UV light produces hydroxyl radicals which tear apart organic molecules, producing CO2, which is then detected by infrared absorbance.42,43 However, the Tekmar-Dohrmann UV light promoted peroxydisulfuric acid instrument can be operated in this mode for low level DOC concentration measurement.44 Presumably, the Anatel instrument could also be operated in this fashion. The effects of UV light on titanium dioxide are well known, and this phenomenon has been exploited for the digestion of organic matter.45 About 2–5% of the DOC was refractory to this approach; the authors attributed the difficulty to the formation of compounds that did not readily absorb UV light. This explanation is not especially satisfactory given the high reactivity of hydroxyl radicals and hydrated electrons which are probably responsible for a considerable portion of the reactivity as opposed to direct photolysis. Processes using ozone and hydroxyl radicals have also been applied to TOC concentration determination, but are used in only one commercially available instrument designed for online analysis.46Besides peroxydisulfuric acid, a host of other oxidants have been employed at various times for aqueous phase digestion. Some of these are worth mentioning, at least briefly. Chromic acid (H2Cr2O7/H2CrO4) was at one time used for wet ashing,47–54 but has fallen out of favor most likely due to a combination of its hazardous nature even when cool and inconvenient waste disposal. It is not incorporated into any commercial instrument, nor has it been used in recent sealed tube studies. Dry combustion has also been used to determine NSOC in seawater.55 Dry combustion loses even moderately volatile material because it requires evaporating off all the water first. Fry et al.55 relied on the natural marine sulfate salts to act as an oxidant in a fashion similar to a Kjeldahl determination. Due to the large sample volume required and the potential for contamination or loss in manipulations, this approach seems unlikely to catch on. Lastly, sealed tube combustion requires a mention for historical if not scientific purposes. This method has been used in one form or another as a reference.56 It requires sealing a glass tube with the sample and an oxidant. Because it cannot be automated, it will never catch on in a drinking water quality control laboratory, but will remain a standby in research settings.
3.4 Efficacy
One of the first comparisons between thermal peroxydisulfuric acid and combustion methods demonstrated that NOM from deep seawater was partly refractory to peroxydisulfuric acid oxidation.57 On the other hand, a comparison of combustion and UV–H2S2O8 digestion on Amazon River DOC isolates showed no statistically significant difference.58 Although some papers on combustion argued that additional carbonaceous matter was detected,30 the authors later retracted those results due to difficulties with the method.59 A number of difficulties were traced to the blank (vide infra) and peak integration.60 In the case of marine samples, the soluble metabolic byproducts of some plankton were demonstrated to be refractory to UV–H2S2O8 digestion and could build up over time.61 It is not clear whether this finding has significance for freshwater samples. A comparison of sealed tube combustion, automated combustion and thermally promoted peroxydisulfuric acid digestion showed differences of ≤10% for marine pore waters taken off the North Carolina coast62 and for vertical profiles from the Mediterranean Sea.36 Depending on the molecular weight of fractionated organic matter, comparative analyses of estuarine and coastal waters using combustion and thermal peroxydisulfuric acid yielded DOC concentrations that varied by a factor of up to two.63 UV–peroxydisulfuric acid digestion gave consistently lower (∼30% on average) results for TOC concentration than combustion when applied to the analysis of estuarine waters.64 The differences were most noticeable at lower salinities; thus, the chloride effect40,41 cannot be responsible for this phenomenon. The most recent tests of saline water showed differences among sealed tube, combustion (Shimadzu TOC-5000) and thermal peroxydisulfuric acid (OI 700) analyses in the neighborhood of 3%, but some of the peroxydisulfuric acid values were demonstrably lower.65Some of the most recent studies on freshwater samples also showed small (∼10%)66,67 differences between the different digestion methods, while another showed larger differences (∼25%).68 Laboratory tests on better characterized substances suggest that many materials are readily digested by either peroxydisulfuric acid or combustion.69 Nonetheless, there is continual concern over the efficacy of different digestion methods because: (1) the composition of organic matter is variable both geographically and temporally; (2) the frequency with which recalcitrant compounds may be found is unknown; and (3) the characteristics of such compounds are not well defined. The magnitude and verisimilitude of the differences between different digestion methods have been27 and will continue to be a matter of debate. The issue of completeness is further complicated by speculation that some material may be particularly susceptible to biodegradation prior to analysis. In my estimation, it is unlikely to be resolved anytime soon, partly due to other complicating factors (viz., quality control, vide infra).
4 Carbon dioxide measurement
4.1 Infrared absorbance photometry
The most popular detector in commercial instruments is the non-dispersive infrared (NDIR) detector. The term non-dispersive means that the light beam has not been subjected to a monochromator that would disperse it into individual wavelengths (e.g., grating or prism). Dispersive infrared spectrophotometers suffer from difficulties in obtaining an accurate and precise zero point.70,71 In addition to offering high throughput, NDIR detectors are rugged and inexpensive; this combination makes them attractive—especially since many marine chemists work aboard ships.62 Although filters can be placed before the photometer, this is not done in TOC analyzers. This type of detector has been widely used by investigators in this field.28,37,72,73 The linear carbon dioxide molecule has no net dipole moment at rest. Nonetheless, it does have stretching (symmetric: ν1 = 1388 cm−1 and antisymmetric: ν3 = 2349 cm−1) as well as non-linear vibrational modes (bending: ν2 = 667 cm−1) that produce a variable dipole moment and allow the absorption of infrared light.74,75 Fig. 3 shows diagrams of NDIR detectors used by OI Analytical and Tekmar-Dohrmann. | ||
| Fig. 3 Diagram of the non-dispersive infrared detectors used by (a) OI Analytical and (b) Tekmar-Dohrmann. Reproduced with permission of Tekmar-Dohrmann. | ||
Because there is no monochromator, the post-digestion gas stream must be devoid of other chromophores that would absorb infrared radiation. However, considerable water vapor is present in the post-digestion gases. A little water is produced by the redox reaction between hydrocarbons and oxygen, but most of it results from evaporation of the water in the sample. In peroxydisulfuric acid units, sparging and heating lead to evaporation. In the case of combustion units, the entire sample is essentially flash evaporated; accordingly, up to 200 µL of liquid can be converted to ∼250 mL of vapor. This must be removed prior to detection. High levels of sulfate or sulfite can also interfere, giving falsely high values, because both SO3 and SO2 absorb infrared radiation.16
The water can be removed from the gas stream in a number of ways. Permeation tubes are used in some instruments to accomplish this (e.g., Tekmar-Dohrmann and OI Analytical). The post-digestion gas stream flows through a tubular membrane while a countercurrent flow of dry gas is maintained outside. Water vapor penetrates through the semipermeable membrane into the waste gas stream, while carbon dioxide stays inside and flows into the detector. Unfortunately, these tubes have a finite lifetime. In addition, they can become saturated if used continuously for an extended period. This represents a technical difficulty in ensuring a water-free gas stream. At present, no instrument manufacturer seems to have surmounted this problem, and users must be alert to the possibility of baseline drift after hours of non-stop usage. Permeation tubes vary in performance due to innate quality in manufacture and operating conditions, especially the countercurrent gas flow rate.
There are other technologies for removing water vapor. Thermoglas has avoided the permeation tube problem by bubbling the post-digestion gas stream through concentrated sulfuric acid (98% w/w) to desiccate it. However, the sulfuric acid rapidly becomes saturated (and thus less effective) and the volume of the dehydrating chamber can be exceeded. This necessitates replacing the sulfuric acid, which is inconvenient, especially if it must be done periodically through a run sequence, thereby preventing unattended operation.
In my opinion, instrument makers would benefit by dropping permeation tubes
entirely in favor of another approach, perhaps a cold finger chilled to −20°C
to rapidly solidify water from the post-digestion gas stream. Periodically,
the cold finger could be warmed up and the water blown off. Because the overall
size of the cold finger would be small, it should be possible to cycle rapidly
through the necessary temperature range. A similar mechanism, using a U-tube
chilled to 0°C, has been employed by Peltzer and Brewer.76 The Shimadzu TOC-5000A uses a thermoelectric (Peltier)
dehumidifier for this purpose, which cools and dries the combustion gas; it
requires little maintenance.
All TOC analyzers using NDIR detectors rely on a constant flow of gas through the system. Flow-through systems raise detection limits relative to the sensitivity of the detector because only part of the analyte (further diluted in the carrier medium) is being detected per unit time. Consequently, a detector that could otherwise respond to a total amount of analyte has a lower apparent sensitivity in a flow-through system. This is a common phenomenon in chromatographic methods, where a mobile phase continuously flows through the system, spreading out the analyte peak. That notwithstanding, detection limits normally reach well below 500 µg L−1, which is generally about the limit of interest in raw water supplies. NDIR detectors offer high sensitivity, low cost, durability and low detection limits, which explains their omnipresence in the marketplace. OI Analytical, Shimadzu, Tekmar-Dohrmann and Thermoglas all use NDIR detectors. Some NDIR detectors are susceptible to noisy baselines from physical vibration, especially during shipboard use.76 On the other hand, the Licor (Lincoln, NE, USA) LI-6252 NDIR detector is insensitive to vibrations and thus has found favor among marine chemists.35,76,77
4.2 Coulometric and conductometric detectors
Unlike infrared absorbance detectors, coulometric detectors are unaffected by water vapor produced during the digestion. In the UIC analyzer, the carbon dioxide is titrated indirectly. The relevant chemical processes can be summarized as follows: (1) absorption of CO2; (2) electrochemical generation of hydroxide by reduction of water at the cathode; (3) acid–base reaction; and (4) oxidation of silver at the anode [eqns. (7)–(10)]:78 | (7) |
 | (8) |
 | (9) |
 | (10) |
Conductometric detection is used in Sievers instruments, which have a membrane-based sensor.79 CO2 permeates the membrane and redissolves into deionized water where it ionizes (hydrolyzes). The concentration is found from the conductivity, based on previous calibration with standards.
4.3 Other detection methods
Although not likely to play a major role in the determination of organic matter in natural waters (either saline or fresh), some alternative approaches merit a brief mention here. The earliest method of detecting carbon dioxide was gravimetry. The post-reaction gas stream was dried [e.g., with Mg(ClO4)2 or CaSO4] and the CO2 was absorbed to Ascarite® (NaOH on silica).80 This approach was also once used in combustion analysis to determine the elemental composition of organic carbon. Although other detectors are not commonly used today, alternative approaches to coulometry and absorption photometry exist. For example, reduction of the carbon dioxide to methane, followed by flame ionization detection was once used.81–86 In addition, carbon dioxide has been measured spectrophotometrically: the gas is driven off by heating, permeates a PTFE membrane and redissolves into a phenolphthalein solution.87 The shift in absorbance indicates the amount of carbon dioxide. Ion chromatography has also been applied to the determination of the TOC concentration; the carbonate is detected after combustion.885 Operation and use
5.1 Basics
Most of the units commercially available for benchtop use are controlled using a separate workstation (personal computer). Software for instrument control and data acquisition is Windows®-based (Microsoft Corp., Redmond, WA, USA). The use of a separate computer facilitates data archiving and retrieval and allows direct connection to a laboratory information management system (LIMS). Most instruments come with autosamplers or can be equipped that way. Because of concern over contamination from laboratory air and a desire to minimize loss of VOC or moderately volatile organics, most instruments are capable of using closed vials with PTFE-lined silicone septa. Septum-piercing needles are necessarily stronger and thicker and presumably present a safety hazard to hands. Consequently, some manufacturers have constructed autosamplers that prevent access to the vial rack once a sequence of analyses has begun. Unfortunately, this prevents the analyst from adding more sample vials to the rack during the sequence. It also prevents one from observing the operation to verify that there are no malfunctions, which can be very important. Because all of the vials must be loaded before the sequence is commenced, considerable time can be lost if a large number of samples are to be run (some racks can hold 80 vials). Moreover, additional blanks, check standards or replicates cannot be readily added to evaluate suspicious data. In my opinion, these factors outweigh the increased safety. Some of these features can be disabled with modest effort, actually resulting in a considerably more hazardous situation.As with any instrument, regular maintenance is required. In addition to the routine addition of fresh reagents, other tasks must be performed. Catalysts must be replaced in combustion TOC analyzers. Catalyst surfaces and support media are subject to scaling from dissolved minerals in natural waters, which are left behind as the water is evaporated. Even new catalysts and media must be conditioned to eliminate initially high background. When first used, a new catalyst will give high blanks until sorbed organic matter is destroyed. Some investigators have also attributed error to the amphoteric nature of alumina, which can sorb carbon dioxide and release it slowly.36,89 Any type of sorption particularly affects low level measurement, since there is a small sample signal added to a large and variable background signal. Usually this requires running the system with either a continuous flow of clean water for a few hours or a hundred separate injections. When room temperature sample water hits the heated catalyst and its support, there is an aging process (cracking, sintering, etc., from thermal effects) that must take place so that any occluded organic matter can be destroyed. Eventually, exposure to repeated injections of cool water, mineral scaling, ash and pyrolyzed matter takes its toll, and the catalyst must be replaced entirely. The rate of replacement depends both on the nature and number of samples. In addition, catalytic packings undergo continual chemical and physical deactivation. Part of the conditioning process speeds up the initial degradation so that a quasi-steady state is reached. These phenomena have been described in more detail by Bauer et al.32 and Cauwet.36
There are straightforward maintenance issues as well. Periodic checks for leaks (gas and liquid) must be performed since mechanical seals can fail. Detectors must be cleaned. Tubing and needles used for sample introduction must also be cleaned to prevent and remove biological growth. Remember that the continual flow of new samples provides fresh nutrients to hungry fungi, protistans and bacteria that are attempting to colonize surfaces. It also provides a supply of new organisms. After all, these are natural waters that should be expected to possess typical limnological characteristics. Otherwise, why would we disinfect them?
5.2 Quality control
°C furnace and then condensing it.90
Others have found that repeatedly adding small doses of hydrogen peroxide
and exposing the water to sunlight is sufficient to oxidize any organic carbon.65 Some water purification systems, for example Milli-Q (Waters,
Milford, MA, USA), appear to be capable of making very low DOC water,
especially if filtered through a 0.22 µm membrane to remove microorganisms
that pass through activated carbon or resins. A few instrument manufacturers
have incorporated programs that allow the production of a progressively cleaner
blank. For example, the Shimadzu TOC-5000A has a blank check
routine that allows the user to run water through the combustion furnace and
collect it as a condensate. In this fashion, organic matter is presumably
burned completely. The Tekmar-Dohrmann Phoenix 8000 UV–peroxysulfuric
acid analyzer has a routine that allows blank water to be cycled between the
sample syringe and the reaction chamber. Although contributions of organic
matter from the reagents are still possible, the oxidizable matter in the
bulk of the blank water is presumed to be converted to CO2 after
multiple digestion cycles as set by the user. Obviously, the required purity
of the blank depends on the TOC concentrations to be measured. Water purity
was still named as one of the key obstacles to accurate TOC and DOC measurement
in 1997.91 To measure accurately a concentration
of 1 ppm, a total blank containing about 0.10 ppm may be acceptable (as
provided by a reverse osmosis system). This contrasts with the pre-filtration
DOC blank, which must be lower to account for some leaching of organics from
the membrane.
The calibration standards are prepared in reagent water, but the sample is not. Consequently, a calibration curve with a non-zero y-intercept cannot be used as is. One might argue that a linear calibration curve could simply be adjusted by subtracting the value of the blank from each point. In principle, the resulting graph should indicate how the instrument responds to analyte. However, it ignores the possibility that a signal is measured in the absence of analyte (e.g., dark current). The process of deconvoluting contributions to the blank is further complicated by the possibility that individual reagents may be contaminated. Although combustion methods eliminate the need for an oxidant dissolved in reagent water, the phosphoric acid added to remove TIC may be a source of DOC. Because reagents are added to the sample and the blank, it is not possible to determine whether the blank or the H3PO4(aq) is responsible for contributing to the DOC on any given day. It is important to maintain perspective on the concentrations of interest, which are normally below 500 µg L−1 (as C) for all the compounds. It is not hard to imagine 100 compounds present at 5 µg L−1 (as C). Ambient levels of atmospheric methanal readily reach that concentration.
Normally, calibration standards are prepared from potassium hydrogen ortho-phthalate (KHP). KHP is cheap and widely available as a primary acidimetric standard. However, KHP is readily oxidized and cannot therefore be used to gauge reaction efficiency.92 To evaluate the completeness of conversion of carbonaceous material to CO2, more refractory substances must be used. This was reiterated in a more recent study by Kaplan.67 Accuracy is clearly influenced by the efficacy of digestion methods (vide supra). Peak shape and rate of reaction are also issues; EDTA was found to give a much broader peak than phthalate, which was in turn broader than methanol or caffeine.35
Sharp91 has argued that carbon dioxide gas standards should be required to ensure an absolute and accurate calibration. Using gaseous standards completely dispenses with the issue of oxidation efficacy. This is a wonderful suggestion, but instrument design substantially hinders carrying it out in actual practice. Most commercial instruments do not have convenient ways to trigger measuring while injecting a gaseous standard. A separate gas phase calibration procedure and connection made to bypass the bulk of the plumbing would be desirable. With existing instruments, it would be possible to retrofit for injection of gaseous standards using a T-valve in the gas line, but some software modifications would be required to disable the liquid injection function (to prevent introduction of air or water from the tubing) and to ensure a sufficient purge time so that the system is entirely filled with the standard gas. Because most instruments give a real-time response, it should be possible to correlate the digitized signal with concentrations in the gas phase and work backwards to an amount of carbon dioxide by integrating a portion of the signal. For simplicity, however, the capability to inject a measured volume of gaseous standard (e.g., via a gas-tight syringe through a septum) would seem to be best.
In practice, potassium hydrogen phthalate, salicylic acid and benzoic acid are often used as standards. KHP is most common, being available as an inexpensive primary standard. Once an acceptable source of high purity water is available, it is used to make all calibration standards. Serial dilutions are routinely used to produce a calibration curve made up of 5–10 points, based on the desired level of precision and the concentration range of interest. Because of system changes (e.g., catalyst aging, permeation tube saturation, carryover, leaks, detector drift, biological growth), it is necessary to run periodic continual calibration check standards and blanks interspersed among sample replicates. As with any analytical technique, a suitable number of replicate measurements must be obtained and the data evaluated judiciously to eliminate outliers.
TOC analyzers measure only one parameter, infrared absorbance of CO2. As a result, it is not possible to add a surrogate or an internal standard as is frequently performed in chromatographic methods.93 One measure of method performance that is available to the analyst is the recovery of fortified analytes. Samples may be spiked with compounds designed to evaluate a particular phenomenon or step of the process. While KHP is an obvious choice, mono- and disaccharides, phenols, alcohols, fluorinated carboxylic acids, natural products (e.g., alkaloids, gelatin) and surfactants may be employed in this capacity. As long as the substance contains carbon and dissolves or disperses in the sample, it can be an acceptable choice. Materials may be chosen intentionally based on their resistance to digestion or the complexity of the matrix.
Clearly, running multiple replicates, analyzing check standards and performing careful calibrations are fundamental actions that can be taken to assess the quality of the data. However, some sample matrices are more amenable to analysis, while others are more problematic, as demonstrated by variable recoveries of spikes and low precision. The lack of stable, Standard (certified) Reference Materials33,94 makes laboratory-to-laboratory and instrument-to-instrument comparisons difficult, especially since dissolved organic matter is, by its very nature, poorly characterized and not readily replaced by some combination of pure reagents. There is no simple recipe; however, there are ongoing efforts to elucidate its make-up.95 Professional judgment and experience are necessary for evaluating all TOC or DOC experimental data.
Relatively little information is available on the precision of the measurements in different instruments. This appears to be partly due to the uncertainties introduced by irreproducibly pure gases and especially reagent water used for calibration standards. Sharp91 gives precision within 5%, but my limited experience is that multiple injections/replicates on combustion units can vary from 5% to 20% RSD—with 5% or so of the values as unexplainable outliers off by factors of 10–20! On the other hand, precision as good as 0.6% RSD has been obtained under optimal conditions.35 In our laboratory, UV–peroxydisulfuric acid digestion appears to be the most reproducible, but it is not clear why. Moreover, the response intensity for a given standard can drift over the course of hours. It should be noted that this does not reflect a baseline drift (e.g., detector dark current), but an actual change in net instrument response. Anecdotal evidence indicates that this drift is not introduced by the detector, but by a combination of memory effects (perhaps in the catalyst34) and incomplete water vapor removal by permeation tubes (vide supra). Much of it goes away if the permeation tube is replaced. It also appears that gas flow (dV/dt) is not adequate to attain turbulent mixing and to rapidly purge residual gases from the previous run. This can be particularly obvious when a blank is run after a modestly high sample, especially one that contains recalcitrant organic matter. With all of the tubing and valves, there are plenty of places for pockets of carbon dioxide to be trapped in some instrument designs.
As noted in Table 2, TOC is often calculated by subtracting TIC concentration from TC concentration. Some instruments permit acidification of the sample prior to injection so that the TIC concentration is not measured. Fukushima et al.96 compared sparging before injection with subtracting DIC concentration to obtain NSOC concentration, and concluded that subtracting was preferable. They believed that a portion of the humate, which became humic acid¶ at the lower pH used for sparging, was never actually injected into the combustion tube because it: (1) became colloidal; (2) precipitated and settled; or (3) flocculated and floated. Variability in the post-acidification behavior also led to imprecision, presumably due to the inability to inject the same amount of insoluble humic acid. If the DIC concentration is small, its quantification tends to be imprecise. Consequently, acidification and sparging prior to analysis may lead to better NSOC concentration results, depending on the NOM. The impact of DIC removal on measuring NSOC concentration is influenced by the nature of the organic matter; accordingly, careful investigation of the particular sample is required.
| a It must be understood that each term refers to a concentration rather than an amount of substance. The convention is to express C in mg L−1 (parts per million) rather than molarity. One reason for this is that not all of the material is soluble; some of it is particulate in nature. b The most common means for separating dissolved and particulate matter is filtration. Material that passes through a membrane filter with a nominal pore size of 0.45 µm is treated as dissolved. This is an operational definition of dissolved, and some investigators have chosen other cut-offs, such as 0.22 µm. With the development of dialysis membranes with assorted molecular weight cut-offs, it is possible to arbitrarily choose a large number of values. c Particulate NOM can range in size from microscopic to macroscopic; see text for more details. d Some investigators use the term non-purgeable instead of non-spargeable, thereby making the initials NPOC. e Some investigations have assessed VOC; see text for details. | |
|---|---|
| TC | Total carbon includes that carbon in any compound or particle. Both inorganic and organic sources are counted. TC = TOC + TIC |
| TIC | Total inorganic carbon includes all dissolved forms of carbonate and bicarbonate as well as suspended minerals containing these ions (particulate IC). TIC = DIC + PIC |
| DIC |
Dissolvedb
inorganic carbon includes only the soluble species: H2CO3/CO2,
CO32−, HCO3− and soluble
metal complexes of the anions. Because soluble (bi)carbonato complexes
are generally negligible in concentration relative to free anions, it is often
assumed that DIC ≈ [CO32−] + [HCO3−] + [CO2(aq)] |
| TOC | Total organic carbon includes all the organic sources, both NOM and manmade compounds. Particulates/suspended solidsc count as well as dissolved compounds. TOC = DOC + particulate OC. In practice, TOC is often calculated indirectly: TOC = TC − TIC. TOC = VOC + NSOC |
| DOC |
Dissolvedb
organic carbon includes all the soluble organic species. Because
of instrumental design and experimental convenience, most reported DOC values
are actually NSDOC (see which, below). DOC = NSDOC + VOC |
| NSOC |
Non-spargeabled
organic carbon includes the TOC not removed by sparging with a stream
of gas for 1–3 min. It is further divided into dissolved (NSDOC)
and particulate (NSPOC or just POC). NSDOC consists of ionizable
compounds with hydrophilic moieties, e.g., carboxylates, alcohols
and amines |
| VOC | Volatile organic carbon includes sources readily removed by sparging, including low molecular weight hydrocarbons or haloalkanes. Some common DBPs are somewhat volatile, e.g., trihalomethanes and haloethanenitriles. In practice, VOC is rarely measured and is usually assumed to constitute a small fraction of the DOCe |
Samples that contain large amounts of particulate carbonate minerals can be especially problematic as indicated by another multilaboratory study.97 The error was attributed primarily to a limiting reagent problem—insufficient acid was added to fully protonate the mineralized carbonate and drive off the carbon dioxide. When samples of high alkalinity are analyzed, complete removal of DIC must be verified. Operators must continually step back and look at the big picture rather than rely on canned methods and prepared reagents supplied by instrument manufacturers. Not all samples are amenable to the default settings. To deal with these situations, one must have a grasp of the processes involved in the analysis.
Because of the difficulties associated with accurate measurement of TOC, NSOC and DOC concentrations, the technique is sometimes more useful for the identification of trends as opposed to absolute determination. If the values for a particular sample can be corroborated by all the methods, there is greater confidence that the number is accurate. For most laboratories, running multiple analyses on multiple instruments is not an option due to the sample load, staffing resources and instrumentation costs.
Virtually every precaution that can be taken adds a new source of error.
Even the old standby of freezing has come under fire, seemingly lowering the
DOC concentrations of some marine samples.99
It has been suggested that salting out occurs upon freezing,100
so perhaps this is less of an issue with freshwater. That notwithstanding,
colloids, precipitates or flocs formed during freezing may undergo irreversible
changes or may be slow to redissolve upon melting. Alternatively, adsorption
to the container wall is also a risk. Acidification is not considered to be
satisfactory unless coupled with filtration, and even then only for a few
hours.30 Mercuric chloride (0.1%
HgCl2) after filtration (0.45 µm) and
with subsequent chilling to 2°C has been recommended as a preservative,90 but it can only be used for combustion analysis
because of the halide. Sodium azide (NaN3) has also been
used,67 but not for peroxydisulfuric acid
TOC analyzers for reasons similar to those for halides. Filtering samples
through suitable media (e.g., a membrane with a nominal pore
size of ≤0.45 µm) may be one of the most useful approaches
by virtue of removing monerans and protistans that can metabolize DOC. Obviously,
filters must be chosen with care to avoid those materials that might leach
DOC into the sample or adsorb it from the sample.
The choice of a container is not easy. Porous plastics may permit absorption or adsorption of hydrophobic compounds, especially small volatile ones. Being less porous, polycarbonate is preferable to polypropylene or polyethylene. In glass vials or bottles, charged species may adsorb to the surface. Nonetheless, glass has generally been adjudged preferable to plastic. Stainless steel has not been reported, but might work. Here is another example of the problem associated with analyzing an ill-defined mixture of compounds with an assortment of functionalities. Precautions taken to protect one may inadvertently affect another in an adverse fashion.
Contamination during sampling is generally not a problem for freshwater systems. Usually, influent treatment plant water can be taken from a port in the plant, the reservoir or even the source. The matter is unquestionably not so simple for marine chemists and oceanographers on seafaring vessels who must contend with the engine exhaust and traces of fuel, but further consideration of their sampling problems and possible curative actions is beyond the scope of this review.
Assuring quality results in the environmental analytical chemistry laboratory is a complicated process. The time, cost and effort in ensuring a specific level of accuracy and precision must be weighed against the intended use of the data and the needs associated therewith. A full discussion of these considerations could run to many pages. It is therefore fortunate that quality control issues have been nicely addressed elsewhere.111
5.3 Instrument design
Instruments in laboratories that analyze potable water must be workhorses, continuously running with little maintenance. They must be durable and not delicate. Ideally, they will require a minimum of technical knowledge to operate while still producing accurate and precise measurements. In order to be truly useful in the drinking water industry, a technique must be rugged, reliable and reproducible. In my opinion, current instrumentation has a number of design deficiencies. The most problematic of these are too many moving parts, large footprints, clumsy autosamplers and excessive plumbing that requires at least 10 mL of liquid to analyze a sample as small as 200 µL.I would favor autosamplers similar to those on gas chromatographs, but using a wide bore needle on the syringe. This would eliminate large amounts of tubing. In addition, it would obviate the need for multiport valves, which are prone to failure (i.e., clogging or abrasion) due to particulates present in natural waters (e.g., suspended sediment, algae and biological detritus).
Although TOC analyzers have been manufactured for about 40 years, the market appears to have been driven substantially by those users who do not need accurate and precise measurements at low concentrations (e.g., waste water, solid waste). Moderate technological progress in low level determination has taken place over the past 20 years. Perhaps the emphasis on DOC and NSOC concentration in new and upcoming drinking water regulations will lead to further advances in TOC analyzers.
6 Other techniques
Although not in widespread use for the analysis of potable water supplies, other techniques have also been applied to the determination of organic carbon. Complex aqueous radioactivity-containing heavy metal carbonates have been analyzed by near-infrared spectrometry.112 UV absorption spectrometry (λ = 250, 254 nm) and fluorescence measurement (λex = 340, 365 nm) have been similarly used.113,114 However, optical techniques are of limited utility in turbid waters. The specific UV absorbance has been included in the Stage 1 DBP Rule because some data suggest that it is a better indicator of the proclivity to form DBPs.115 A summary of approaches can be found elsewhere.1167 Conclusions
Analysts must maintain a holistic perspective in dealing with samples. The concentration and nature of the organic and inorganic constituents affect the precision and accuracy of the results. Questions about the efficacy of different digestion methods for oxidizing organic carbon to carbon dioxide remain unanswered, as do questions about the existence of biologically labile and refractory organic matter. Furthermore, it is not clear whether the concentration of DBP precursor material is better correlated with TOC or DOC concentration determined by a specific method. If a particular compound is refractory to peroxydisulfuric acid wet ashing, it may also be resistant to chlorination and therefore not a DBP precursor. Complete answers will require more improvements in not only instrumentation, but also operator skill. Perhaps even more important in assuring good data is the availability of certifiably pure oxygen and reagent water. Attention to the blank is clearly critical in making low level measurements. It is disheartening that modest to no progress has been made in solving the problems identified by Wangersky 6–7 years ago.14,117 Variable interlaboratory agreement is also an issue for organic carbon measurements.118 What does it mean when skilled investigators using identical instruments and methods obtain statistically different values for the same quantity? This has been addressed in some detail,94 but will probably remain problematic. The publication of each new research paper suggests that the science is more complex and multivariate than had previously been thought. Each raises new and interesting questions to be answered. Obtaining good values for DOC and TOC concentrations will continue to challenge the skills of everyone who uses a TOC analyzer. The difficulties that lie therein will remain a source of consternation for those engaged in disciplines and operations that rely on these parameters—whether ensuring safe drinking water or determining the balance of carbon in natural bodies of water.Acknowledgements
The assistance of Dacia Drury, Jennifer Heffron, Jennie Thomas, Michael R. Schock and Raymond A. Hauck in obtaining references for this review is duly noted. I acknowledge helpful discussions with Billy B. Potter of the EPA's National Exposure Research Laboratory (Microbial and Chemical Exposure Assessment Research Division, Chemical Exposure Research Branch). I appreciate comments received from Professor Peter J. Wangersky, a pioneer in measuring TOC and DOC concentrations in ocean water. Two reviewers are also acknowledged for their constructive comments. The US Environmental Protection Agency, through its Office of Research and Development, completed the work described here. It has been subjected to agency review and approved for publication.References
- S. D. Richardson, in Encyclopedia of Environmental Analysis and Remediation, ed. R. A. Meyers, Wiley, New York, 1998, pp. 1398–1421. Search PubMed.
- S. W. Krasner, in Formation and Control of Disinfection Byproducts in Drinking Water, ed. P. C. Singer, AWWA, Denver, CO, 1999, ch. 2, and references cited therein. Search PubMed.
- J. O. Zavaleta, F. S. Hauchman and M. W. Cox, in Formation and Control of Disinfection Byproducts in Drinking Water, ed. P. C. Singer, AWWA, Denver, CO, 1999, ch. 5, and references cited therein. Search PubMed.
- EPA, Fed. Regist., 1998, 63, 69390 Search PubMed.
- F. Pontius and W. R. Diamond, J. Am. Water Works Assoc., 1999, 91, 16 Search PubMed.
- H. Weinberg, Anal. Chem., 1999, 71, 801A CrossRef CAS.
- E. T. Urbansky, Anal. Chem., 2000, 72, 439A CAS.
- Australian Drinking Water Guidelines, National Health and Medical Research Council and Agriculture and Resource Management Council of Australia and New Zealand, Canberra, ACT, 1996, ch. 3. Search PubMed.
- Council Directive 98/83/EC of 3 November 1998 on the Quality of Water Intended for Human Consumption, Off. J. Eur. Communities, 1998, 330, 32 Search PubMed.
- P. A. Murphy and G. F. Craun, in Formation and Control of Disinfection Byproducts in Drinking Water, ed. P. C. Singer, AWWA, Denver, CO, 1999, ch. 6, and references cited therein. Search PubMed.
- EPA, Fed. Regist., 1998, 63, 69478 Search PubMed.
- R. P. Galapate, A. U. Baes, K. Ito, K. Iwase and M. Okada, Water Res., 1999, 33, 2555 CrossRef CAS.
- R. A. Van Steenderen, Water SA, 1981, 7, 28 Search PubMed.
- P. J. Wangersky, Mar. Chem., 1993, 41, 61 CrossRef CAS , and references cited therein.
- I. S. Al-Aasm, B. E. Taylor and B. Smith, Chem. Geol. (Isotope Geosci.), 1990, 80, 119 Search PubMed.
- G. Heron, M. J. Barcelona, M. L. Andersen and T. H. Christiansen, Ground Water, 1997, 35, 6 Search PubMed.
- C. E. Van Hall, D. Barth and V. A. Stenger, Anal. Chem., 1965, 37, 769 CrossRef CAS.
- J. H. Sharp, Limnol. Oceanogr., 1977, 22, 381 Search PubMed.
- K. Ruehl, Water Eng. Manage., 1999, 146, 22 Search PubMed.
- S. A. Andrews and P. M. Huck, in Disinfection Byproducts in Water Treatment: The Chemistry of Their Formation and Control, ed. R. A. Minear and G. L. Amy, CRC/Lewis, Boca Raton, FL, 1996, ch. 20, and references cited therein. Search PubMed.
- J.-P. Croué, J.-F. Debroux, G. L. Amy, G. R. Aiken and J. A. Leenheer, in Formation and Control of Disinfection Byproducts in Drinking Water, ed. P. C. Singer, AWWA, Denver, CO, 1999, ch. 4, and references cited therein. Search PubMed.
- C. Lee and S. M. Henrichs, Mar. Chem., 1993, 41, 105 CrossRef CAS.
- R. von Wandruszka, Geochem. Trans., 2000, 2 Search PubMed (http://www.rsc.org/ej/gt/2000/b001869o/index.htm), and references cited therein.
- M. J. Barcelona, Ground Water, 1984, 22, 18 Search PubMed.
- B. Fry, C. S. Hopkinson, Jr., A. Nolin and S. C. Wainwright, Chem. Geol. (Isotope Geosci.), 1998, 152, 113 Search PubMed.
- J. I. Hedges and J. Farrington, Mar. Chem., 1993, 41, 5 CrossRef.
- P. J. le B. Williams, J. Bauer, R. Benner, J. Hegeman, V. Ittekkot, A. Miller, B. Norrman, Y. Suzuki, P. Wangersky and M. McCarthy, Mar. Chem., 1993, 41, 11 CrossRef CAS.
- C. E. Van Hall, J. Safranko and V. A. Stenger, Anal. Chem., 1963, 35, 315 CrossRef CAS.
- P. D. Goulden, Water Res., 1976, 10, 487 CrossRef CAS.
- Y. Sugimara and Y. Suzuki, Mar. Chem., 1988, 24, 105 CrossRef.
- A. E. J. Miller, R. F. C. Mantoura and M. R. Preston, Mar. Chem., 1993, 41, 215 CrossRef CAS.
- J. E. Bauer, M. L. Occelli, P. M. Williams and P. C. McCaslin, Mar. Chem., 1993, 41, 75 CrossRef CAS.
- G. Spyres, M. Nimmo, P. J. Worsfold, E. P. Achterberg and A. E. J. Miller, Trends Anal. Chem., 2000, 19, 498 CrossRef CAS.
- C. Thomas, G. Cauwet and J.-F. Minster, Mar. Chem., 1995, 49, 155 CrossRef CAS.
- J. Qian and K. Mopper, Anal. Chem., 1996, 68, 3090 CrossRef CAS.
- G. Cauwet, Mar. Chem., 1994, 47, 55 CrossRef CAS.
- D. W. Menzel and R. F. Vaccaro, Limnol. Oceanogr., 1964, 9, 138 Search PubMed.
- I. M. Kolthoff and I. K. Miller, J. Am. Chem. Soc., 1951, 73, 3055 CrossRef CAS.
- G. Peyton, Mar. Chem., 1993, 41, 91 CrossRef CAS.
- G. R. Aiken, Environ. Sci. Technol., 1992, 26, 2435 CAS.
- T. Sakamoto and T. Miyasaka, Ultrapure Water, 1987, 4, 24 Search PubMed.
- S. A. Huber and F. H. Frimmel, Anal. Chem., 1991, 63, 2122 CrossRef CAS.
- J. E. Bauer, P. M. Williams and E. R. M. Druffel, Nature, 1992, 357, 667 CrossRef CAS.
- M. Miller, J. Furlong and D. Neal, Proc. Water Qual. Technol. Conf., AWWA, Denver, CO, 1997, P12C/1–P12C/5 (on CD-ROM). Search PubMed.
- M. I. Abdullah and E. Eek, Water Res., 1996, 30, 1813 CrossRef CAS.
- D. L. Wyatt and T. A. Petteroe, Proc. Annu. ISA Anal. Div. Symp., 2000, 33, 27 Search PubMed.
- E. K. Duursma, Neth. J. Sea Res., 1961, 1, 1 Search PubMed.
- R. Kieselbach, Anal. Chem., 1954, 26, 1312 CrossRef CAS.
- W. A. Moore, F. J. Ludzack and C. C. Ruchboft, Anal. Chem., 1951, 23, 1297 CrossRef CAS.
- N. F. Schulz, Anal. Chem., 1953, 25, 1762 CrossRef CAS.
- E. E. Archer, Analyst, 1954, 79, 30 RSC.
- D. D. Van Slyke, Anal. Chem., 1954, 26, 1706 CrossRef CAS.
- A. Krogh and A. Keys, Biol. Bull., 1934, 67, 132 Search PubMed.
- H. Kay, Kiel. Meeresforsch., 1954, 10, 26 Search PubMed.
- B. Fry, E. T. Peltzer, C. S. Hopkinson, Jr., A. Nolin and L. Redmond, Mar. Chem., 1996, 54, 191 CrossRef CAS.
- B. Fry, S. Saupe, M. Hullar and B. J. Peterson, Mar. Chem., 1993, 41, 187 CrossRef CAS , and references cited therein.
- J. H. Sharp, Mar. Chem., 1973, 1, 211 CrossRef CAS.
- R. Benner and J. I. Hedges, Mar. Chem., 1993, 41, 161 CrossRef CAS.
- Y. Suzuki, Mar. Chem., 1993, 41, 287 CrossRef.
- J. H. Sharp, R. Benner, L. Bennett, C. A. Carlson, R. Dow and S. E. Fitzwater, Limnol. Oceanogr., 1993, 38, 1774 Search PubMed.
- J. J. Ridal and R. M. Moore, Mar. Chem., 1993, 42, 167 CrossRef CAS.
- M. J. Alperin and C. S. Martens, Mar. Chem., 1993, 41, 135 CrossRef CAS.
- H. Ogawa and N. Ogura, Nature, 1992, 356, 696 CrossRef CAS.
- A. E. J. Miller, R. F. C. Mantoura, Y. Suzuki and M. R. Preston, Mar. Chem., 1993, 41, 223 CrossRef CAS.
- E. T. Peltzer, B. Fry, P. H. Doering, J. H. McKenna, B. Norrman and U. L. Zweifel, Mar. Chem., 1996, 54, 85 CrossRef CAS.
- L. A. Kaplan, Limnol. Oceanogr., 1992, 37, 1119 Search PubMed.
- L. A. Kaplan, J. Am. Water Works Assoc., 2000, 92, 149 Search PubMed.
- J.-F. Koprivnjak, J. G. Blanchette, R. A. Bourbonniere, T. A. Clair, A. Heyes, K. R. Lum, R. McCrea and T. R. Moore, Water Res., 1995, 29, 91 CrossRef CAS.
- M. Miller, J. Furlong and D. Neal, Proc. Water Qual. Technol. Conf., AWWA, Denver, CO, 1997, P12D/1–P12D/4 (on CD-ROM). Search PubMed.
- D. A. Skoog and J. J. Leary, Principles of Instrumental Analysis, Saunders, Ft. Worth, TX, 4th edn., 1992, pp. 270–271, and references cited therein. Search PubMed.
- H. A. Strobel and W. R. Heineman, Chemical Instrumentation: A Systematic Approach, Wiley, New York, 3rd edn., 1989, pp. 628–629. Search PubMed.
- K. J. Collins and P. J. L. B. Williams, Mar. Chem., 1977, 5, 123 CrossRef CAS.
- C. E. Van Hall and V. A. Stenger, Anal. Chem., 1967, 39, 503 CrossRef CAS.
- R. S. Drago, Physical Methods for Chemists, Saunders, Ft. Worth, TX, 2nd edn., 1992, pp. 154–156. Search PubMed.
- P. W. Atkins, Physical Chemistry, Freeman, New York, 4th edn., 1990, pp. 491–492. Search PubMed.
- E. T. Peltzer and P. G. Brewer, Mar. Chem., 1993, 41, 243 CrossRef CAS.
- X. A. Álvarez-Salgado and A. E. J. Miller, Mar. Chem., 1998, 62, 325 CrossRef CAS.
- Operation Manual for the CM5012 CO2 Coulometer, Rev. B, UIC, Joliet, IL, 1996. Search PubMed.
- TOC Model 800/810/820 Operation and Maintenance Manual, Sievers Instruments, Boulder, CO, 1999. Search PubMed.
- W. P. Pickhardt, A. N. Oemler and J. Mitchell, Jr., Anal. Chem., 1955, 27, 1784 CrossRef CAS.
- R. A. Dobbs, R. H. Wise and R. B. Dean, Anal. Chem., 1967, 39, 1255 CrossRef CAS.
- Y. Takahashi, R. T. Moore and R. J. Joyce, Am. Lab., 1972, 4, 31 Search PubMed.
- G. Cauwet, Mar. Chem., 1984, 14, 297 CrossRef CAS.
- F. T. Eggertsen and F. H. Stross, Anal. Chem., 1972, 44, 709 CrossRef CAS.
- I. Lysyj and K. H. Nelson, J. Gas Chromatogr., 1968, 6, 106 Search PubMed.
- K. Nakajima, Water Res., 1984, 18, 555 CrossRef CAS.
- R. T. Edwards, I. D. MacKelvie, P. C. Ferrett, B. T. Hart, J. B. Bapat and K. Koshy, Anal. Chim. Acta, 1992, 261, 287 CrossRef CAS.
- Y. S. Fung, Z. Wu and K. L. Dao, Anal. Chem., 1996, 68, 2186 CrossRef CAS.
- R. Benner and M. Strom, Mar. Chem., 1993, 41, 153 CrossRef CAS.
- W. Chen and P. J. Wangersky, Mar. Chem., 1993, 41, 167 CrossRef CAS.
- J. H. Sharp, Mar. Chem., 1997, 56, 265 CrossRef CAS.
- R. A. van Steenderen and J.-S. Lin, Anal. Chem., 1981, 53, 2157 CrossRef CAS.
- E. T. Urbansky, J. Environ. Monit., 2000, 2, 285 RSC.
- P. J. Wangersky, in The Handbook of Environmental Chemistry, ed. P. J. Wangersky, Springer-Verlag, Berlin, 2000, vol. 5, part D, ch. 7. Search PubMed.
- V. Thoss, M. S. Baird and M. A. Lock, J. Environ. Monit., 2000, 2, 398 RSC.
- T. Fukushima, A. Imai, K. Matsushige, M. Aizaki and A. Otsuki, Water Res., 1996, 30, 2717 CrossRef CAS.
- M. E. Caughey, M. J. Barcelona, R. M. Powell, R. A. Cahill, C. Gron, D. Lawrenz and P. L. Meschi, Environ. Geol., 1995, 26, 211 Search PubMed.
- W. H. Chen and P. J. Wangersky, J. Plankton Res., 1996, 18, 1521 Search PubMed.
- S. E. Fitzwater and J. H. Martin, Mar. Chem., 1993, 41, 179 CrossRef CAS.
- E. T. Peltzer and P. G. Brewer, Mar. Chem., 1993, 41, 91 CrossRef CAS.
- Standard Test Method for Total Organic Carbon in Water, ASTM, West Conshohocken, PA, 1996, Designation D2579-93e1. Search PubMed.
- Standard Test Method for Total, Organic, and Inorganic Carbon in High Purity Water by Ultraviolet (UV) or Persulfate Oxidation, or Both, and Infrared Detection, ASTM, West Conshohocken, PA, 2000, Designation D4779-93. Search PubMed.
- Standard Test Method for Total and Organic Carbon in Water Oxidation by Coulometric Detection, ASTM, West Conshohocken, PA, 2000, Designation D4129. Search PubMed.
- Standard Test Method for Total and Organic Carbon in Water by High Temperature Oxidation and by Coulometric Detection, ASTM, West Conshohocken, PA, 2000, Designation D4129-98. Search PubMed.
- Standard Test Method for Total Carbon and Organic Carbon in Water by Ultraviolet, or Persulfate Oxidation, or Both, and Infrared Detection, ASTM, West Conshohocken, PA, 2000, Designation D4839-94. Search PubMed.
- Standard Test Method for Total and Dissolved Carbon Dioxide in Water, ASTM, West Conshohocken, PA, 1996, Designation D513-92. Search PubMed.
- Standard Methods for the Examination of Water and Waste Water, ed. A. D. Eaton, L. S. Clesceri and A. E. Greenberg, APHA/AWWA/WEF, Washington, DC, 20th edn., 1998, Method 5310. Search PubMed.
- Methods for Chemical Analysis of Water and Wastes, US EPA, Environmental Monitoring and Support Laboratory, Cincinnati, OH, 1983, EPA-600/4-79-020, Method 415.1. Search PubMed.
- Methods for Chemical Analysis of Water and Wastes, US EPA, Environmental Monitoring and Support Laboratory, Cincinnati, OH, 1983, EPA-600/4-79-020, Method 415.2. Search PubMed.
- Methods for solid waste analysis, US EPA, Washington, DC, 2000, Method 9060, SW-846, http://www.epa.gov/epaoswer/hazwaste/test/9060.pdf Search PubMed.
- Ph. Quevauviller and O. F. X. Donard, J. Environ. Monit., 1999, 1, 503 RSC , and references cited therein.
- L. H. Espinoza, D. Lucas, D. Littlejohn and S. Kyauk, Appl. Spectrosc., 1999, 53, 103 Search PubMed.
- B. MacCraith, K. T. V. Grattan, D. Connolly, R. Briggs, W. J. O. Boyle and M. Avis, Sens. Actuators B, 1994, 22, 149 CrossRef.
- H. De Haan and T. De Boer, Water Res., 1987, 21, 731 CrossRef CAS.
- I. N. Najm, N. L. Patania, J. G. Jacangelo and S. W. Krasner, J. Am. Water Works Assoc., 1994, 86, 98 Search PubMed.
- K. Robards, I. D. McKelvie, R. L. Benson, P. J. Worsfold, N. J. Blundell and H. Casey, Anal. Chim. Acta, 1994, 287, 147 CrossRef CAS.
- P. J. Wangersky, in The Biology of Particles in Aquatic Systems, ed. R. S. Wotton, Lewis, Boca Raton, FL, 2nd edn., 1994, ch. 2. Search PubMed.
- P. King, H. Kennedy, P. P. Newton, T. D. Jickells, T. Brand, S. Calvert, G. Cauwet, H. Etcheber, B. Head, A. Khripounoff, B. Manighetti and J. C. Miquel, Mar. Chem., 1998, 60, 203 CrossRef CAS.
Footnotes |
| † This is the work of a United States government employee engaged in his official duties. As such it is in the public domain and exempt from copyright. ©US government. |
| ‡ At times, algae or mildew can be seen inside the plastic tubing of TOC analyzers. For this reason, periodic cleaning and replacement are indicated. Ideally, cleaning should take place before biological growth is visibly evident. Solutions of NaOCl (e.g., Clorox® bleach, Oakland, CA, USA) are usually satisfactory for this purpose. |
| § In practice, VOC concentration can be measured on combustion instruments. There are two ways to go about this. (1) The sample is maintained at neutral pH and sparged with oxygen. The gas stream is then directed through the furnace, where combustion occurs. VOC concentration is then measured directly. This approach assumes that weak bases are sufficiently protonated to be carried into the vapor phase. Some experimenting may be required to ensure that semivolatile moderately strong acids are accounted for. (2) Cd2+ or Pb2+ (or any metal cation for which the Ksp of the carbonate salt is small) is added to the sample, and the insoluble carbonate precipitate is filtered off. Next, the filtrate is injected. Caution must be exercised to ensure that recovery is satisfactory, because some carboxylates or phenoxides may be precipitated by heavy metal cations or occluded in the metal carbonate. In this approach, VOC is not lost before analysis. |
| ¶ NOM is sometimes divided into categories of humic acid, fulvic acid and humin. Humic acid is insoluble when pH ≤ 2. As the pH is raised, humic acid is deprotonated to form soluble humate. Fulvic acid and fulvate are soluble regardless of pH. It is assumed that the pH is raised with LiOH, NaOH or KOH. Both humate and fulvate can coordinate to transition metal cations and form insoluble species. Humin is insoluble over the whole pH range. For more details, see D. Langmuir, Aqueous Environmental Geochemistry, Prentice-Hall, Upper Saddle River, NJ, USA, 1992, pp. 161–162, and S. E. Manahan, Environmental Chemistry, Lewis/CRC, Boca Raton, FL, USA, 6th edn., 1994, pp. 80–82. |
| This journal is © The Royal Society of Chemistry 2001 |