The relevance of speciation in the remediation of soils and sediments contaminated by metallic elements—an overview and examples from Central Scotland, UK†
Andrew S.
Hursthouse
Department of Chemistry and Chemical
Engineering, University of Paisley, Paisley, UK PA1 2BE
First published on 5th December 2000
Abstract
The environmental impact of metallic contaminants in soils and sediments is dependent both on the chemical speciation of the metal and the response of the matrix to biological and physicochemical conditions. These factors are responsible for the mobilisation of the metal from the solid into the aquatic phase and hence transport within the immediate vicinity, impacting on the rate of dispersal, dilution, uptake and transfer into living systems. The impact of changing environmental conditions on the contaminant inventory can be to enhance or moderate these phenomena, with subsequent consequences for the broader risk assessment of the contaminants. Remediation of metallic contaminants can only be brought about by their removal from the site or by establishing conditions which favour their retention in the solid phase. A wide range of in situ and ex situ approaches are available and a summary overview is presented. The examples show assessment at both the field and laboratory scale and demonstrate an equally wide range of success in achieving remediation targets. This can be attributed to limitations in ensuring that the desired conditions for the initial removal or immobilisation process are met and maintained over a suitable period of time. Three areas are reviewed which include: the transport and release of metallic contaminants in estuarine sediments and the assessment of their potential to impact on biota; terrestrial contamination systems involving the release of chromium from waste ore contaminating urban environments; the response of metal-contaminated wastes to changing environmental conditions and the impact of natural bioremediation. The focus of the discussion is to highlight the generation of reliable speciation information and the problems associated with impact and risk assessment. Particular issues of concern are the laboratory to field scale evaluation of contaminant behaviour and the approach used to assess the reliability of remediation options. In conclusion, part of a recent initiative in risk assessment and the development of pilot scale experimental systems to study long-term behaviour are addressed as future goals to fill gaps in current research.
1. Introduction and background
Contamination of the surface of the Earth by the metallic elements from human activities has been significant for centuries.1–6 For example, many materials were processed in significant quantities to support the developing technologies underpinning the growth of the Roman Empire. The extraction and utilisation of metals as major components of infrastructure and in high value goods became the cornerstone of the development of human civilisation.With the advent of the industrial revolution in Western Europe, e.g., the availability of steam powered devices, the utilisation of fossil fuels increased the capacity of society to manipulate the Earth’s resources and process materials.1,6,7 The net impact was to stimulate population growth, increasing the demand for food and technological products, and, as a direct result of the incomplete efficiency of these processes, releases of residual materials to the atmospheric, aquatic and terrestrial environment occurred. Many of the technological developments were associated with improvements in iron and steel manufacture and associated heavy industry.8,9
Through human use of metallic elements, contamination of the Earth's surface developed from localised problems associated with mining and initial ore processing, through to larger scale manipulation and refining, construction and manufacture and finally to waste disposal.6,8,10–13 This, coupled with the focusing of population centres into industrialised cities, resulted in an increased burden on the local environment.14
In the latter part of the 20th century, a decline in manufacturing in traditional industrial areas, an increase in population worldwide and an increased awareness of human impacts on global environmental systems at the local and global scale were observed. This has resulted in efforts to improve the degraded quality of aquatic systems and an increase in the recycling of land in urban environments, with more sensitive end-uses.11,15,16
The increase in human stress on sensitive surface environments requires the development of reliable management options for soils and sediments, which are often contaminated with a mixed group of physical and chemical components. Contamination from processing residues, direct deposition and accidental releases of organic and metallic species create a wide spectrum of hazards under an equally wide spectrum of contexts.15,17 The recognition and remediation of these situations rely on an intimate mix of science, technology and socio-economic factors.
It is within this context that the speciation of metallic components provides both the potential hazard (mobility, toxicity, etc.) and the pathway to a technologically acceptable solution. The factors defining and influencing the speciation, and the changes with both time and environmental conditions, provide the greatest challenge and opportunity for environmental science and technology. Whilst the contamination of the surface of the Earth by human intervention is a relatively old phenomenon, the evaluation, assessment and remediation of negative impacts are relatively recent.15,18,19 There exists considerable scope for multi- and interdisciplinary research and development.
This paper provides an overview of the context associated with metal contamination and highlights the role of metal speciation. It is not a comprehensive treatment, but presents the influence of the main factors controlling metal speciation and reviews a number of circumstances where these are significant for the contamination scenario. Observations are made which identify gaps and opportunities for future research.
2. Context and influencing factors
The remediation of a particular metallic contamination context must involve the evaluation of a number of key factors.(a) Definition of speciation and reliable quantification
The basis for the discussion of this aspect relies on the definition of the term speciation and, more significantly, the translation of that term to define the field context. Numerous definitions exist,20–24 and whilst they do not form part of the discussion here, the use and acceptance of a particular definition are important for the study or discussion of results from often quite dispersed investigations. However, the generic, guiding principle is that the metallic elements undergo a series of transformations of their molecular association during their cycling in surface environments, as a consequence of their basic elemental characteristics and their interaction with geochemical cycles within the Earth.In the context of soil and sediment contamination and remediation, we need to consider all aspects of the transfer of metals between phases (absorption, solubility, coagulation, coprecipitation, volatilisation), transformation of species by electron transfer, ligand exchange, availability and bioaccumulation and physical transport processes affecting a particular metal or group of interest.22,25,26 This will impact on the influence of the contaminant source, reaction within the deposited environment, suitability and effectiveness of remediation processes and long-term behaviour of remediation result—essentially a truly four-dimensional context. To adequately address this, data are required that define as closely as possible the metal speciation to allow appropriate assessments to be made.27 However, most analytical techniques, particularly when applied to the field situation or on processed samples, do not provide anything other than data that are strongly influenced by the method of collection—the important “operational” influence.24,28–30 This covers information obtained from both extractive operations (e.g., metal content defined by total, pseudototal, reagent selective extraction and hyphenated analysis methods31–36) and the limited range of (applicable) direct speciation techniques [e.g., XRD, X-ray absorption near-edge structure spectroscopy (XANES), EXAFS, NMR, vibrational and magnetic methods37–40). Many concerns are raised about the validity and appropriate use of speciation techniques, and caution is always advocated. However, we should not disregard evidence from any method. Applied in an appropriate manner, information is always gained; it is the relative weight we apply to the information and its role in subsequent interpretation that should be the driving factors. In some cases, it seems that we need not be over-pessimistic about the most often criticised approaches.41,42 In particular, sequential extraction is a powerful technique and, when applied in a comparative sense, can provide information which highlights differences in elemental mobility or reactivity in a particular environmental context.43 As part of the analytical process, modifications to extraction schemes (so that they are more sensitive to variation in matrix composition) can advance the detailed assessment of metal mobility.34 Classic debates in the literature centre on, for example, metal associations with pyrite in anoxic sediments44 and how sensitive results are to modifications of extraction schemes. However, with a full appreciation of these limits, the data produced provide important supporting information for treatment proposals and risk assessments.
Considerable additional information on metal mobility can be gained from the simulation of extraction and release of metals from host phases—soils or sediments. Column and batch experiments are widely used45–47 to gain an insight into the response of contaminated material to groundwater flow or interaction with reactive mobile phases and also as a significant transport mechanism for relatively strongly held contaminants.48 Problems relating to hydraulic integrity and the relative contribution of dissolved and colloidal material to the transport of mobile contaminants are important considerations for column methods, but this approach is of choice for speed and experimental flexibility.45
(b) Environmental conditions are important in controlling metal speciation
The most important controlling factors defining metal speciation at a particular point in space and time are: pH; composition and amount of organic matter; clay minerals; the presence and nature of Fe/Mn/Al oxides and hydroxides; redox potential; concentrations of salts and complexing agents; anion and cation content of the soil/sediment solution.49–51 The perturbation of the system (sampling, dredging, groundwater flow) impacts on the measured speciation through one or more of these factors.The kinetic influence on changes to metal speciation during transport to potential receptors in biological systems has been shown to have a varying effect for different metals in non-equilibrium systems,25,26,52,53 thus defining the need for a detailed understanding of release potential from solid phases. Within anoxic sediments, metal distribution between operationally defined extracts of real and model sediment components has shown that the expected oxidative release of common contaminant metals from sulfides is buffered to changes in Eh51,54–57 by the presence of organic matter and precipitation from oxidation reactions. However, pH control appears to have a more rapid impact.58–63 These factors are not only relevant to the response of contaminants to natural mixing and bioturbation, but also to the situation in which sediment derived soils are developing. There are direct implications for time dependent increases in metal availability64 or, as in marginal marine environments, tidal and seasonal changes may be important.65 The particle size distribution of the solid phase is also relevant here, where physical control of metal distribution is influenced by chemical composition and is, in turn, affected to varying degrees by pH and redox perturbations.66–68
For soils, the interaction of the contaminants with major soil parameters suggests that pH and organic matter have a strong regulatory role on partition to the liquid phase and hence mobility.69–71 This also influences the partition between the free ion and soluble metal complexes.72 The roles of oxides and hydroxides of iron, manganese and aluminium are also very relevant here and have a much more sensitive response to major Eh and pH changes, in addition to having an intimate physical and chemical relationship with other solid phases (e.g., organic matter, clay minerals25,26,73–75).
Furthermore, there is a time dependence of metal mobility from fresh solid phases in soil systems (the “ageing effect”), which can both enhance and retard any step change in major environmental parameters (e.g., pH) depending on the metal involved.72,76,77 The impact of these factors on transfer to biological systems (bioavailability, plant uptake) is less clear from field observations.77–83 It appears that major soil nutrients have a greater role in influencing uptake, even for metal hyperaccumulating plants.84–86
Therefore, the remediation “context” must be assessed in terms of changes to or influence of these factors if the role of metal speciation is to be adequately assessed.
(c) Structure of the contamination problem—the “source term” context
If the basic definitions of soil and sediment are considered, the true context for the remediation of metal contamination is emphasised. Soils and sediments differ primarily in their mode of deposition.87 Both contain mixtures of mineral, organic and aqueous phases. Soils include important gaseous components and sediments reflect more dynamic processes of transport and deposition in fluids (including air). The key features for this discussion are that both have a layered structure unless they have been disturbed (through natural mass movement processes or by human interactions). The theoretically layered nature of these media by default provides the contaminant assessor with the opportunity to make assumptions relating to the likely distribution, and enables the application of fundamental survey techniques to estimate contaminant distribution. In most cases, the distribution of the contamination does not follow the predicted distribution and causes fundamental problems for the reliability of site assessment processes.15,17,88,89The source of the contamination has a profound effect. Generally, discharges to the aquatic environment, whether from aerial deposition to a water body, diffuse seepage of contaminated groundwater or direct discharge from an industrial point source, will be subject to some degree of mixing and dilution prior to deposition in the sediment body. This will tend to disperse the contaminants and result in well-defined contamination layers within the sediment column. In a similar manner to ice-cores,4 with care, sediments can act as archives of contaminant inputs to the system.90–95 Remediation will therefore need to consider whether the sediment contamination follows the distribution predicted by well-understood environmental processes or is likely to have been disturbed by more catastrophic events—dredging, storm resuspension, etc.
In the case of soil contamination, the context is altered somewhat from that of sediments. This is due to the fact that soil is, on the most part, unsaturated with respect to water and potential contamination migration rates and soil processes tend to occur over much longer time scales. The terrestrial environment is subject to more intensive interaction with anthropogenic processes and to catastrophic inputs/disturbances with respect to contamination. This results in an increased probability that contaminant “hot spots” will be produced and increases the likely risk that the assessment of contamination will not make an adequate evaluation of the levels and distribution of pollutants. The structural integrity of layered materials can be compromised by buried structures, affecting both contaminant assessment and the engineering of remediation programmes.
The physical nature of the contamination source term has a role in both the short and long-term interaction of the contaminant with the environment at the point of introduction. This is likely to be more significant for soils than for sediments due to the integrating nature of mixing within the aquatic environment. The source term context may come from: (i) deposition as widely dispersed fallout/rainout from the atmosphere (e.g., regional contamination from Chernobyl, smelter fly ash); (ii) spillage from the storage of materials, ineffective processing or containment (leakage from drums, incomplete clearance of stored/stockpiled raw materials or waste); (iii) direct deposition of waste materials in large volumes to form new engineered surfaces (mine tailings, landfilled or abandoned primary metal processing sites).
With each system, the physicochemical nature of the source (as solid, liquid or gas) will then determine the immediate interaction with the prevailing environmental processes.
Contaminant interaction is also subject to a “degrees of scale” influence in both the short and long-term response to the conditions in the receiving environment. The more concentrated and the larger the scale of the deposition event, the more significant the impact of the contaminant source on the depositional environment. It may be that the waste material containing the contaminant determines the major environmental variables (e.g., pH, Eh, major soluble ions, hydraulic properties96–98 ), which, in turn, control contaminant behaviour [see Section 3(b) below]. Alternatively, the contaminant, whilst at levels of environmental significance, is influenced more strongly by the prevailing conditions within the host environment [see Section 3(a)]. These factors must be considered in terms of their influence on the likely mobility of the contaminants throughout the life cycle of the management process. The processes important for transport naturally include solubility, but must also consider colloid formation and physical mass movement of contaminants [e.g., Section 2(b)].99,100 The role of speciation here is to determine the likely partitioning between mobile and immobile phases and the potential for this factor to alter the long-term risk assessment and end-use for the site.
When defining contamination levels, an assessment must be made describing relevant baseline conditions. In the case of pollutants that are wholly anthropogenic, baseline levels are those at which measurement systems cannot detect the presence of the contaminant. In the case of metals, in nearly all circumstances, a significant background established by natural geochemical cycling will be present. This background will vary due to the geochemical cycling process and it is important to evaluate this in the context of the contamination.101,102 For severely contaminated sites, this is relatively straightforward. Numerous action and trigger levels have been supplied in the past to allow comparison of observed levels. In addition, relatively simplistic approaches to removing natural baselines from data sets have been incorporated in the assessment of the significance of contamination.103–108 However, it has been recognised that this approach does not adequately account for natural background variations, and the assumptions made cause under- and overestimation of the significance of the contamination levels.104,105 In addition, without a full understanding of the local interaction of the contamination and potential ecological impacts, the derivation of absolute concentration levels for contaminants is meaningless.109,110 In recognition of this issue, regulatory regimes worldwide are now moving towards advocating a risk-based assessment process for contaminated sites.17 This is a crucial point for the “speciation community” as it will drive the development of appropriate testing systems, and stimulate the collection of data linking speciation to environmental impact. By applying these criteria to the investigation process and incorporating them in the evaluation of remediation targets, the risk-based approach will allow a more effective remediation strategy to be defined and, within the context of limitations of the site investigation (sampling intensity versus cost), will provide a more realistic evaluation of the volumes of material required to be remediated.
(d) Remediate or not?
The identification of a contamination problem is only a small part of the contamination scenario. The decision to remediate implies that ownership of the contamination problem has been accepted. If the remediation exercise is undertaken, then the long-term performance of the solution must also be considered—does liability remain? What is required to demonstrate that the most appropriate methods used are really appropriate? As new remediation methods become available is there any need for retrospective application? These questions bring legal and economic considerations to the fore. Technically, most contaminants can be remediated (immobilised, degraded, extracted, etc.) and numerous reviews emphasise the degree of success/application achieved in field situations. The key features of the most commonly used remediation methods are summarised in Table 1, and the relevance of metal speciation to the remediation process is highlighted. The detail of many of the processes is difficult to review concisely, but from the range of demonstrated techniques it would appear that the majority of metal remediation methods rely on the mobilisation/leaching of contaminants under highly engineered conditions (ex situ or in situ) to demonstrate acceptable risk reduction.| Technique | Principle | Example | Metal speciation role/significance | Additional references |
|---|---|---|---|---|
| Biological | Biodegradation; biotransformation; accumulation; mobilisation | Ex situ—bioreactors; biopiles; windrow turning | No significant application to heavy metals unless release to leachate and there is an opportunity for aqueous phase removal | 118–122 |
| In situ—bioaccumulation; white rot fungi; rhizosphere biodegradation | Hyperaccumulator plants identified for metal contaminants. Enhancement in uptake due to amendments to solubilise or complex metals. Accumulation rates unlikely to provide short-term extraction | 123–129 | ||
| Chemical | Oxidation; reduction; immobilisation; extraction | Ex situ—solvent extraction; dehalogenation | Not generally applied to metal removal | |
| In situ—soil flushing; surface amendments | Wide application to metal removal. Various combinations of complexing agents, additives to control redox status to enhance mobility into soil solution or form immobile species. Subsurface FeO barriers allowing redox control | 130–143 | ||
| Physical | Volatility; density separations; soil washing; electroremediation | Ex situ | Soil washing based on a wide mix of chemical and physical properties—with extractants (see above); flotation of particles—based on density, size; electroremediation widely applied, requires ionic metal species, porewater flow | 138–151 |
| In situ | Applications of ex situ methods above | |||
| Solidification; stabilisation | Cement/pozzolanic; lime; vitrification | Ex situ | A wide variety of binding agents to solidify soil/sediment and prevent water migration and leaching. Thermal treatment to achieve the same properties | 152 |
| In situ | As per ex situ | |||
| Thermal | Thermal desorption; incineration; vitrification | Ex situ | Removal of volatile metals—limited to Hg with side effects being stabilisation of solid phase and reduced leachabilty. Vitrification impact as above. Secondary smelters applied where content high enough | 153 |
| In situ | Limited to thermal energy input |
In engineering terms, the application of technical observations to real contamination scenarios is often feasible—manipulation of soil or sediment, engineering barriers, directing groundwater flow, etc. However, economic or social factors (cost effectiveness, economic benefits, public acceptance, political will, etc.) will determine the final action for any particular contamination scenario.110 Within the cost benefit analysis of any remediation option, it is important to consider what is likely to happen if nothing is done. The natural recovery of the system is also an important question when remediation targets are to be set. Little data exist for the post-clean-up response if the source term is removed or reduced.110 In the case of sediment remediation, limited in situ techniques are available154 and, unless dredged materials (obtained from maintenance of shipping channels) are used for terrestrial soil amendment and landscaping,155 there is little motivation to dredge for remedial purposes alone. But, how quickly will the system recover once sources are reduced by, for example, point discharge control? Active intervention is only advocated when biological information suggests that natural recovery is not possible. But, at what stage and at what time are decisions made? In the case of soil remediation, progress has been made in the application of risk-based assessments of contaminant distribution to identify more realistic areas appropriate to remediate,156,157 and it offers considerable scope to advance the range of demonstrated remediation success.
3. Examples where speciation influences situation
A wealth of examples exists where the role of metal speciation is fundamental to the impact and remediation of contamination. Examples from three areas, with locally significant impacts, are reviewed here; experimental details and the validation of analytical data are described in the contributing references. The examples focus on the Clyde Estuary and industrial activities in the area around Glasgow, Central Scotland. However, whilst the context of the work has a strong geographical focus, the results illustrate the relevance of speciation in the remediation context. They do not represent a definitive answer in terms of the clean-up of a particular scenario, but assist in the translation of focused research on generic properties of contaminated soils and sediments to regional scale systems.(a) Cr behaviour in intertidal sediments in harbours and estuaries, River Clyde, UK
The Clyde Estuary on the west coast of Scotland represents one of the UK’s most contaminated estuarine environments.103,158–160 It has suffered from the continued input of multicomponent discharges which peaked in the mid-19th century. However, with changes in industrial activity and shifts in population and social structure, marked improvements in water quality have been seen, the general opinion being that the estuary is recovering ecologically in response to these improvements.161The quality of sediments within the estuary has been studied in detail over a number of years162 and, within a context of generally high levels of heavy metals and organic contaminants, concentration hot spots have been observed.158,163,164 These have been attributed to focused industrial, military and sewage discharge points. The studies have mainly monitored surface sediments and improvements in water quality are reflected in these.
The intertidal zone in particular has a role in the estuarine food web, acting as a breeding and feeding ground for a number of important components of the ecosystem.
Over the last 5–6 years, our studies have focused on the evaluation and assessment of changes in sediment quality, as defined by contaminant distribution and the response of the ecosystem to this factor.158,165,166 One element in particular—chromium—exhibits relatively high enrichment in the estuarine system,103,160 reflecting not only an important historical contamination context within the inner estuary and lower River Clyde [see Section 3(b)], but also long and as yet poorly quantified inputs from a variety of historical industrial activities, in particular, tanning and heavy industry (ship building, engineering and associated metals processing).159
Chromium is a well-documented example of an element that human society has used widely in technological progress and, in so doing, has increased population exposure. Its accessible hexa- and trivalent oxidation states are the key features of its beneficial and detrimental contribution and also its geochemical behaviour in surface environments. Simply, toxic Cr(VI) is generally found as a mobile component in surface environments, whilst Cr(III) is an essential nutrient and relatively immobile in these systems.167,168
Within the intertidal sediment zones, depth profiles of metal distribution appear to reflect relatively undisturbed historical influences on contaminant distribution. This has resulted in the observation of buried “peaks” in metal distribution. Fig. 1 summarises examples of this distribution169 and shows depth distribution profiles for Cr, with Pb and Cu for comparison. The data represent cores collected from intertidal sites on important tributaries and within the main channel of the estuary, and show the dilution of the magnitude of contamination moving seaward. The effect of normalising to expected metal-accumulating factors [identified in Section 2(b) as percentage organic matter and fine grain size] does not remove the variation, and the relatively high levels at depth and some preliminary radiometric assessments170 suggest that bioturbation and physical mixing processes do not account for the distribution. The concentration levels for Cr would cause concern in the context of terrestrial contamination.94,171 Whilst the contamination has been derived from sustained, historical effluent discharges to the estuary, active remediation of the sediments is unlikely. However, it is important to assess the relative mobility of the buried contaminant peaks, in particular, the influence of tidal cycles on pore water flushing and changes in the regional hydrology following climate change.172,173 Essentially, we must attempt to answer the question: will buried contamination buffer the recovery of the system?
 | ||
| Fig. 1 Examples of the vertical distribution of Cu, Pb, Cr in sediment cores collected from the Clyde Estuary.94,174 Cores (a) and (b) from the White Cart, a tributary of the Clyde; core (c) from the main river channel. Data for total aqua regia extractable metal content w/w (dry). | ||
As a part of this evaluation, a detailed assessment of the factors controlling Cr distribution is required. A review of the results of multiple correlation analysis of data from seven sites is summarised in Fig. 2. Regression analysis lines are omitted and the most significant relationships were observed for total (aqua regia extractable) Cr with total Fe and Mn. In the case of organic matter and fine sediments, a relatively poor correlation is observed. The results agree with other assessments160 [Section 2(b)] and indirectly point to the dominance of iron and manganese on Cr behaviour.
 | ||
| Fig. 2 Association of total sediment Cr with total sediment Mn and Fe, the percentage organic matter and percentage fine-grained sediment in cores collected from the Clyde and tributaries. All metal data aqua regia total, n = 140, from seven cores collected between 1994 and 1997.94,174 Correlation coefficients: (a) 0.550; (b) 0.816; (c) −0.265; (d) 0.158. | ||
Further evidence for controlling mechanisms is available from the analysis of pore waters. These were collected in situ by suction. Fig. 3 summarises typical data from one site.104,174 Whilst the samples are from relatively shallow depths and provided low vertical resolution, evidence of expected subsurface pore water release of Fe and Mn is obtained [Fig. 3(a)]. The pore water concentrations of Cr are relatively low for all the sites sampled (range, 2–70 µg l−1; mean, 30 µg l−1) and the total concentrations correlate well with both the total sediment Cr and Fe, but much less strongly with total sediment Mn. The ratio of Cr(III) to Cr(IV) also increases with depth [Fig. 3(b)], reflecting a response to the redox status of the sediments [Section 2(b)]. The ability of pore waters to support relatively high levels of Cr(III) is somewhat surprising and no evidence was obtained for levels of colloidal material which might support the insoluble reduced oxidation state. Analytically, the methodology seems to be free from operational artefacts.104 The mechanism must involve the release of sorbed Cr(III) from reactive phases in the sediments, and with strong associations with Fe (perhaps associated with sulfide or relict oxides) and to a lesser extent Mn. The control of Cr release is likely to be related to the redox mobility of Fe, which further emphasises the influence of sediment geochemistry on contaminant speciation and subsequent mobility [Section 2(b)].
 | ||
| Fig. 3 Typical pore water distribution of Fe, Mn and Cr in surface sediments. Samples collected by an in situ suction device over a 4 day period:104,174 (a) a typical vertical profile of total dissolved metals at Woodhall site, day 1; (b) downcore variation of Cr(III) versus Cr(VI) in the first extraction—three sites shown for clarity; (c) variation of correlation coefficient downcore for subsequent daily pore water extractions compared to day 1. All data pooled (five depths at four sites over 4 days). | ||
Further analysis of the pore water data provides simple kinetic information on the response of Cr to remobilisation. Data from consecutive extractions over a 4 day period at low tide are compared in terms of a correlation with levels for the first day. The results, shown in Fig. 3(c), show that for both dissolved Fe and Mn pore water concentrations tend to regain initial levels, but in the case of dissolved Cr show a much slower rate of re-equilibration with the system. It is unfortunate that the experiment was of a relatively short duration and seasonal factors could not be studied.65 However, it does highlight the potential response of sediment associated Cr to changes in the hydrology of the system and the likely dependence of Cr release on more than one parameter.
Data from the application of sequential extraction schemes partially support this remobilisation issue, emphasising the benefit of including data from many analytical techniques [Section 2(a)]. The scheme used in this study43,175 has been applied to intertidal systems elsewhere, but is not as sensitive to the more complex solid phases.44 These have been bulked to include sulfide and residual associations. Fig. 4(a) is typical of the associations found,169 with a significant portion of the total sediment Cr associated with exchangeable and reducible components. From multiple correlation analysis, a minor association is observed between the Cr held in the Fe and Mn oxide/hydroxide dominated components and the total pore water Cr. The behaviour of Fe and Mn, from similar analysis, shows very different trends; when moving from Mn to Fe, there is a progressive increase in the significance of the residual fraction. Some evidence is found for the surface layer to host a more available Fe fraction. This is not surprising, as material within the surface layer will be prone to resuspension and oxidation [Section 2(b)]. However, the main factor controlling the availability and potential release of Cr from the sediments appears to be total (aqua regia extractable) sediment Fe [Fig. 4(b)].
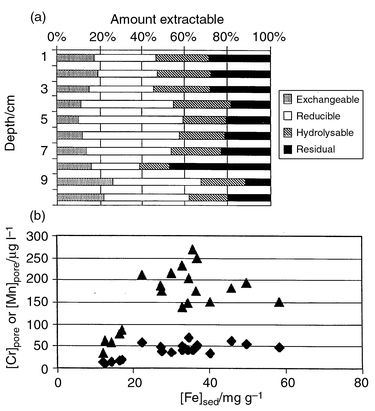 | ||
| Fig. 4 (a) An example of Cr sequential extraction data for a sediment core from Woodhall on the Clyde Estuary.94,169 (b) The association of total sediment Fe and dissolved Cr and Mn in pore waters, first extract (n = 20). Correlation coefficients: Fe versus Cr (◆) = 0.723 and Fe versus Mn (▲) = 0.589. | ||
The consequence of this potential mobility has been assessed for biological impact on simplified food webs.158,165,169 Whilst some evidence of bioaccumulation is found, the mechanism responsible for Cr uptake appears to be regulated by sediment dwelling biota.
(b) Impact of chromite ore processing residues, Glasgow, UK
Within the urban area of Glasgow, Cr contamination can be particularly severe due to a number of deposits of chromite ore processing residue (COPR), with additional material from the extensive heavy metal working and processing industries. The COPR is essentially a complex mixture of reactants and wastes from a process that used a mixture of chromite ore, lime and soda ash to produce Cr(VI), which was then extracted by leaching with water. Disposal of residues has produced areas of contaminated land across Glasgow, estimated to be of the order of 1.3 × 106 m3.176Groundwater from these sites can be highly contaminated with Cr(VI). As groundwater drains into the River Clyde, transport and dilution rapidly reduce the pH and Cr concentration. The highest concentrations of Cr are >150 mg l−1 with pH ∼ 13, which reduces to 3.1–6.2 mg l−1 Cr(VI), pH 7–8 in surface streams and <1 mg l−1 in the open River Clyde [Fig. 5(a)]. However, the levels still represent significant contamination of surface waters and are reflected by elevated concentrations of Cr in local sediments.94,107,174 From the data presented in Fig. 5(b), the spatial extent of sediment contamination from the contaminated sites does not appear to be significant, reflecting the nature of the discharged Cr species. The form of Cr present in the system suggests that the mobile chromium is predominantly Cr(VI) and is associated with low molecular weight species, probably as small ionic molecules.107 Consequently, the contaminated discharges are fairly rapidly diluted when mixing with the main river. More detailed characterisation of the contamination source term, particularly in relation to Cr speciation, and an assessment of the potential impact of future weathering of the deposited wastes are being undertaken by others.177,178
 | ||
| Fig. 5 Impact of Cr-contaminated groundwater on the Clyde:104 (a) total dissolved Cr (bh = bore hole in central Glasgow; sw = surface stream draining one site; cc = confluence of stream with River Clyde; cds 1 and 4 = samples downstream in the main river); (b) residuals plotted from the normalisation of Cr to Li in surface sediments collected along the River Clyde from source (A) to lower estuary (Q); tidal limit lies between F and G. Confluence of contaminated surface stream with the River Clyde is at point H. | ||
However, there is still a considerable need to identify a means to remediate Cr contamination in Glasgow, and a variety of field scale trials have been undertaken for both in situ and ex situ methods. One of the most practical and technologically achievable methods is simply to pump and treat contaminated groundwater close to the point of contamination (Table 1). The most common approach to treat Cr-contaminated waters is the reduction of Cr(VI) to Cr(III) followed by precipitation of Cr(III).179–181
Much recent research has been directed towards the search for low cost, readily available substances which possess a significant affinity for the sorption of anionic Cr(VI) species, and which therefore may be used in the removal of chromium from polluted waters.182 Of particular interest is the use of microbial biomass to sequester dissolved toxic metals.183–185 In the environment, natural microbial activity provides some of the main pathways for the removal and/or immobilisation of heavy metals. Metal uptake arises through a variety of physicochemical mechanisms, including adsorption and ion exchange. With living biomass, metabolic mechanisms can also contribute to the uptake. Such materials have been used to remove toxic or valuable metals from water. For example, immobilised algal biomass has been used in the removal of a wide range of metals of different valence and solution chemistry, including Cr(III) and Cr(VI).184 The sorptive properties of the biomass can be regenerated and used repeatedly. Furthermore, given that the biomass is a waste product, it can be considered a low cost, economically attractive medium for the removal of metals from aqueous solution. Other low cost, materials are available which could potentially be used in the sorptive removal of chromium from aqueous streams. They include a variety of biomass and solid waste materials.186–189
We screened candidate sorbents using batch methods and assessed the direct reduction of Cr-contaminated groundwaters using ferrous sulfate, evaluating the applicability of a scaled up pilot plant system.120 The high pH conditions encountered in contaminated waters differ from those of common process applications, where the acidic conditions encountered tend to favour the stabilisation of ions in solution [Section 2(b)], and precipitation-based removal requires further chemical manipulation. The techniques assessed have been applied with reasonable success in this context, but fall into two groups: those suitable for treating moderately contaminated surface waters where the pH and total Cr content are relatively low; and those suitable for highly contaminated groundwaters (high pH and dissolved Cr). For the former, a variety of solid media are effective in solid–liquid contact systems—by physicochemical, sorptive reactions. Fig. 6(a) shows the results of screening experiments for batch sorption. The materials with the highest capacity for Cr(VI) offer potential as polishing steps for these less contaminated phases. In particular, Fe(0) seems an attractive option and has been used successfully as an active barrier in the field to treat Cr-contaminated groundwaters.142
 | ||
| Fig. 6 Treating Cr-contaminated groundwater:120 (a) screening of materials for sorptive removal of Cr(VI) from high pH contaminated groundwaters; (b) the influence of pH on the removal of Cr from contaminated groundwaters by reduction with Fe(II) in a bench scale experiment (Fe ■; Cr ●). | ||
Chemical treatment using ferrous sulfate is promising in the latter situation. The results of this study indicate that there is a fairly narrow range within which operating parameters should be set. The operating points identified show good agreement through the scale up process, which involved an initial 1 l volume mixing system and moved on to a > 100 l capacity system. The rapid reduction step for Cr(VI)–Cr(III) means that the residence time in any mixing system is short. The sensitivity of removal to pH is shown in Fig. 6(b) for the bench scale experiments. These conditions were found to transfer to larger pilot plant experiments, and the residence time and pH control on decontamination at the 100 l scale are summarised in Fig. 7.
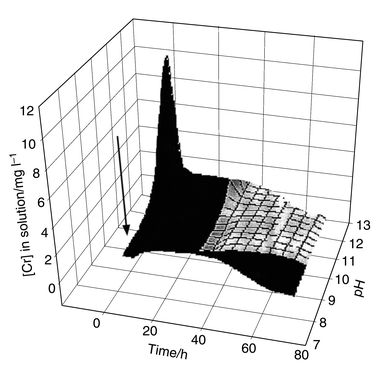 | ||
| Fig. 7 Most sensitive processing conditions for the removal of Cr(VI) from high pH contaminated groundwaters for a > 100 l mixing system.120 Target operating parameters (arrow) indicate short residence times and moderately alkaline pH. | ||
The production of ferric hydroxide sludge is the main operation parameter of concern for larger scale systems, and any field application would have to handle a fine floc, produced in reasonable quantities. Treatment methods for this component would require dewatering systems to be included. Clearly, the overriding factors are the reliability of the supply of contaminated ground and surface waters to the treatment point. A pump and treat system would only be suitable for reduction by ferrous sulfate where the chemical composition of the input waters could be constrained. Fluctuations in pH, particularly to lower, neutral or even slightly acidic conditions, will affect the settling and solubility of both the Fe and Cr components of the sludge.167 This would require an additional conditioning step in the scheme, which detracts from the potentially straightforward treatment system. Examples exist of fairly complex treatment schemes for Cr removal,185 which ultimately detract from field application. However, the hydrology and groundwater recharge, interaction of groundwater with the contaminated waste and rates of dissolution are needed before a realistic field-based facility could be designed.
Future options for the sites would include a mixture of ex situ, pump and treat methods for the highly contaminated waters. These could be established to run unattended using a pH-activated processing facility, which would be backed up by downstream “polishing” of surface waters using in situ sorptive materials.185 With numerous candidate materials, local supplies of biomass or scrap ferrous metal may prove to be economic options.
(c) Environmental behaviour of metal processing wastes—contribution to long-term risk assessment
The production of iron and steel has a long history of technological development. During the evolution of the manufacturing processes to current methods, the industry produced a wide range of slags and waste materials with little residual value. These are often environmentally of low significance and, in many cases, can be used as secondary raw materials in construction.190 The most significant releases from industrial processes are to the atmosphere and aquatic system. With advances in process control technology, a wide range of effective methods exist for the treatment of liquid effluents and the removal of fly ash from gaseous waste streams, which often incorporate wet scrubbing methods.191,192 This process produces a filter cake or sludge with little residual value, which has been disposed of to landfill or stockpiled.193As with many fly ash materials, the wastes are often high in heavy metal content and have a relatively high pH (pH > 8).96 Since 1998, we have been undertaking an evaluation of the long-term behaviour of typical filter cake residues that have been stockpiled for over a decade at a site in central Scotland.194,195 The site being studied has a complex depositional history, but provides an archived sample of waste materials that can be visually segregated. More significant is the observation that these materials support the development of vegetation cover. Can the development be evaluated as input into developing more effective long-term management of the material? How quickly can the material become a “soil” and what are the factors limiting plant growth and regeneration of the material?
Preliminary results show a measurable response of the material to environmental conditions.195 Samples of fresh filter cake and surface and subsurface deposits from the site (Table 2) show that, in terms of metal content, levels are not significant,171 although they would be of concern in a sensitive end-use. Laboratory scale batch leaching [Section 2(a)] was applied and the material has a relatively high buffer capacity (Fig. 8). Both leaching pH and leaching time have an impact on the waste. As the pH of the leaching solution decreases, however, the capacity is reduced and the more weathered (surface) material has a greatly reduced capacity. Part of the buffering of the system will be due to flow characteristics of the columns,196 but the decrease in pH (a major environmental parameter) indicates that changes to the bulk sample are occurring [Section 2(b)].
 | ||
| Fig. 8 Laboratory batch leaching of filter cake wastes from flue gas scrubbing:195 (a) comparison of leaching solution pH and leachate pH; (b) time dependence of leachate pH. Key: fresh cake ▲; surface sample ◆; 1 m depth ●; 3 m depth ■. | ||
| Sample | pH | Zn | Cu | Pb | Ca | Fe |
|---|---|---|---|---|---|---|
| Fresh cake | 9.4 | 2875 | 195 | 1651 | 13587 | 399243 |
| Surface | 8.6 | 1207 | 162 | 816 | 56191 | 313737 |
| 1 m depth | 8.9 | 1092 | 172 | 737 | 57662 | 320485 |
| 3 m depth | 8.6 | 1221 | 169 | 779 | 60635 | 320094 |
Further evidence of the effects of ageing of the waste is provided by sequential extraction. Results for lead and zinc are shown in Fig. 9 and, for both elements in all samples, substantial portions of the total metal content are found in the more mobile fractions. A trend is revealed moving down into the waste, with the surface material containing proportionally more exchangeable material, reflecting the breakdown (or weathering) of the residual phases.
 | ||
| Fig. 9 Zn (a) and Pb (b) distribution in weathered deposits of filter cake waste. Sequential extraction using the BCR method.34,195 | ||
This preliminary work is being supplemented by more detailed characterisation of the material and the site in an attempt to provide a comprehensive risk assessment model, including geochemical, hydrological and biological components. These results agree well with limited comparative studies.193,197 They highlight the influence of surface processes on the deposited waste matrices and the response to environmental conditions influencing the mobility of potential contaminants [Sections 2(b) and 2(c)]. In particular, the management of the natural regeneration of these wastes by plants relies on an understanding of soil profile development and waste weathering. It offers the most attractive and low cost route to the stabilisation of metallic contaminants in the wastes (Table 1).
4. Role and future demands of metal speciation research in soil and sediment remediation
The influence of “speciation” on the behaviour of metals in contaminated soils and sediments is fundamental to the response of contaminants to remediation techniques. The examples reviewed here illustrate that fundamental biogeochemical operations rely on the dynamic interaction of metal species with a multidimensional and multicomponent system. Observations are based on the application of a wide range of measurement systems, which have variable and often significant limitations. However, they are the only means to provide us with the data required for statements to be made concerning the metal speciation in these phases. The range of papers in this volume demonstrates the evolution of our detailed knowledge. Yet the demands for robust analytical systems to provide reliable observations are as important as they have ever been.21,22,24 The greatest challenge remains to integrate this information gathering process into the evolving environmental management systems.198,199 The detailed information gained on metal speciation should increasingly direct assessment, remediation and aftercare of the sites identified as being contaminated or with a demand for change in use. Some progress is being made in this direction,17,200 but it is a relatively immature subject and limited experience exists on the consequences of the application of remediation technologies.Two main components must be considered in the future evolution of the role of speciation. These are: (i) site variability and the consequence of the scale up of assessment and remediation processes; and (ii) the impact of long-term changes in the major environmental factors influencing remediated sites. These issues are not new and are not just a problem in a speciation context. However, they should be considered and will influence the development of experimental techniques.
Scale up and site variability confound many remediation programmes, and there is much evidence for technical problems in the successful transfer of laboratory and small scale assessments of metal mobility/removal in soil/sediment systems to field applications.23,196,201 The research community is responding through the development of larger scale experimental systems and demonstration projects, with widely disseminated results.202
Changes in temperature and water balances are predicted for many geographical regions under a number of climate change scenarios.172,173 For example, in the context of the west of Scotland, minor but significant increases in rainfall have been predicted. There is some evidence from real monitoring data. Fig. 10 shows raw and moving average data for rainfall in central Paisley, Scotland since the late 19th century. Visually, the moving average data show a subtle but statistically significant increase in monthly average totals.203 The implications of a few per cent increase in precipitation to increases in leaching and runoff in soils and changes to river and estuarine water need to be evaluated. For some contaminants, increased flushing and dilution are likely, whilst for others the effects are less clear. At the centre of the evaluation is a full understanding of the influence of speciation, which will ensure that the speciation community has a sustained contribution to the management of metal contamination of the environment.
 | ||
| Fig. 10 Compilation of monthly average rainfall data for Paisley, 20 km southwest of Glasgow, UK.203 (a) 5 and (b) 20 year moving average plots. | ||
Thus, the speciation of metals in the remediation of soils and sediments has a pivotal role to play in hazard and risk assessment, remediation strategies and in the long-term management of remediated sites.
Acknowledgements
I am grateful to all past and present members of the Environmental Geochemistry Research Group at the University of Paisley for the experimental data described here, and for the funding and collaborative support from colleagues at the Universities of Strathclyde, Turin, Wroclaw, the City of Glasgow Council, CEFAS-Burnham-on-Crouch, FRS-Aberdeen and The CORUS Group UK Ltd. The organisers of Whistler 2000 provided the challenging topic of this paper.References
- A. Markham, A Brief History of Pollution, Earthscan Publications, London, 1994. Search PubMed.
- J. O. Nriagu, Science, 1996, 272, 223 CrossRef CAS.
- J. O. Nriagu, Science, 1998, 281, 1622 CrossRef CAS.
- S. Hong, J.-P. Candelone, C. C. Patterson and C. F. Boutron, Science, 1996, 272, 246 CrossRef CAS.
- W. Shotyk, Science, 1998, 281, 1635 CrossRef CAS.
- M. Macklin, in Managing the Human Impact on the Natural Environment: Patterns and Processes, ed. M. Newson, Belhaven Press, London, 1992, ch. 9. Search PubMed.
- K. H. Wedepohl, in Metals and Their Compounds in the Environment, ed. E. Merian, VCH, Weinheim, 1991, pp. 3–17. Search PubMed.
- R. C. Wilmoth, S. J. Hubbard, J. O. Burckle and J. F. Martin, in Metals and Their Compounds in the Environment, ed. E. Merian, VCH, Weinheim, 1991, pp. 19–65. Search PubMed.
- (a) W. Shotyk, S. A. Norton and J. G. Farmer, Water, Air Soil Pollut., 1997, 100, 213 CrossRef CAS; (b) D. D. Gilbertson, J. P. Gratan, M. Cressy and F. B. Pyatt, Water, Air Soil Pollut., 1997, 100, 327 CrossRef CAS.
- M. Newson, in Managing the Human Impact on the Natural Environment: Patterns and Processes, ed. M. Newson, Belhaven Press, London, 1992, ch. 2, pp. 14–36. Search PubMed.
- I. Thornton, Appl. Geochem., 1996, 11, 355 CrossRef CAS.
- K. Van de Velde, C. Barbante, G. Cozzi, I. Moret, T. Bellomi, C. Ferrari and C. Boutron, Atmos. Environ., 2000, 34, 3117 CrossRef CAS.
- E. M. Sunderland and G. L. Chmura, Sci. Total Environ., 2000, 256, 39 CrossRef CAS.
- M. S. Lowe and S. R. Bowlby, in Environmental Issues in the 1990s, ed. A. M. Manion and S. R. Bowlby, John Wiley, Chichester, 1992, ch. 7, pp. 117–130. Search PubMed.
- T. Cairney, The Re-use of Contaminated Land, John Wiley, Chichester, 1995. Search PubMed.
- A. Bora, in Green 2 Contaminated and Derelict Land, ed. R. W. Sarbsy, Thomas Telford, London, 1998, pp. 10–18. Search PubMed.
- The Risk Assessment for Contaminated Sites in Europe, Volume 1, Scientific Basis, ed. C. Ferguson, D. Darmendrail, K. Freier, B. K. Jensen, J. Jensen, H. Kasamas, A. Urzelai and J. Vegter, LQM Press, Nottingham, 1998. Search PubMed.
- S. Johnson, in Remedial Processes for Contaminated Land, ed. M. Pratt, Institution of Chemical Engineers, Rugby, Warwickshire, 1993, ch. 1, pp. 1–17. Search PubMed.
- D. M. Hamby, Sci. Total Environ., 1996, 191, 203 CrossRef CAS.
- M. Bernhard, F. E. Brinckman and P. J. Sadler, The Importance of Speciation in Environmental Processes, Springer Verlag, Berlin, 1996. Search PubMed.
- K. Irgolic, in Metal Speciation in the Environment, ed. J. A. C Broekaert, S. Gucer and F. Adams, Springer-Verlag, Berlin, Heidelberg, 1986, pp. 641–644. Search PubMed.
- J. J. Morgan and W. Stumm, in Metals and Their Compounds in the Environment, ed. E. Merian, VCH, Weinheim, 1991, pp. 67–103. Search PubMed.
- U. Forstner, Int. J. Environ. Anal. Chem., 1993, 51, 5 Search PubMed.
- A. M. Ure and C. M. Davidson, in Chemical Speciation in the Environment, ed. A. M. Ure and C. M. Davidson, Blackie, Glasgow, 1995, ch. 1, pp. 1–5. Search PubMed.
- D. L. Sparks, in Metal Speciation and Contamination of Soil, ed. H. E. Allen, C. P. Huang, G. W. Bailey and A. R. Bowers, CRC Press, Boca Raton, FL, 1995, ch. 2, pp. 35–58. Search PubMed.
- J. G. Hering, in Metal Speciation and Contamination of Soil, ed. H. E. Allen, C. P. Huang, G. W. Bailey and A. R. Bowers, CRC Press, Boca Raton, FL, 1995, ch. 3, pp. 59–86. Search PubMed.
- M. H. Mach, B. Nott, J. W. Scott, R. F. Maddalone and N. T. Whiddon, Water, Air Soil Pollut., 1996, 90, 269 CrossRef CAS.
- M. Stoeppler, in Hazardous Metals in the Environment, Techniques and Instruments in Analytical Chemistry, ed. M. Stoeppler, Elsevier, Amsterdam, 1992, vol. 12, ch. 6, pp. 97–132. Search PubMed.
- M. Stoeppler, in Hazardous Metals in the Environment, Techniques and Instruments in Analytical Chemistry, ed. M. Stoeppler, Elsevier, Amsterdam, 1992, vol. 12, ch. 2, pp. 9–18. Search PubMed.
- W.F. Pickering, in Chemical Speciation in the Environment, ed. A. M. Ure and C. M. Davidson, Blackie, Glasgow, 1992, ch. 2, pp. 9–32. Search PubMed.
- M. Sager, in Hazardous Metals in the Environment, Techniques and Instruments in Analytical Chemistry, ed. M. Stoeppler, Elsevier, Amsterdam, 1992, ch. 7, pp. 133–175. Search PubMed.
- A. Kot and J. Namiesnik, Trends Anal. Chem., 2000, 19, 69 CrossRef CAS.
- C. Sarzanini, J. Chromatogr., A, 1999, 850, 213 CrossRef CAS.
- S. E. Howe, C. M. Davidson and M. McCartney, J. Anal. At. Spectrom., 1999, 14, 163 RSC.
- J. Scancar, R. Milacic and M. Horvat, Water, Air Soil Pollut., 2000, 118, 87 CrossRef CAS.
- G. M. Greenway, in Chemical Speciation in the Environment, ed. A. M. Ure and C. M. Davidson, Blackie, Glasgow, 1995, ch. 4, pp. 65–85. Search PubMed.
- S. M. Glidewell and B. A. Goodman, in Chemical Speciation in the Environment, ed. A. M. Ure and C. M. Davidson, Blackie, Glasgow, 1995, ch. 3, pp. 33–64. Search PubMed.
- J. D. Ostergren, G. E. Brown, Jr., G. A. Parks and T. N. Tingle, Environ. Sci. Technol., 1999, 33, 1627 CrossRef CAS.
- A. Manceau, M.-C. Boisset, G. Sarret, J.-L. Hazemann, M. Mench, P. Cambier and R. Prost, Environ. Sci. Technol., 1996, 30, 1540 CrossRef CAS.
- C. T. Johnston, W. L. Earl and C. Erickson, in Metal Speciation and Contamination of Soil, ed. H. E. Allen, C. P. Huang, G. W. Bailey and A. R. Bowers, CRC Press, Boca Raton, FL, 1995, ch. 9, pp. 237–254. Search PubMed.
- M. D. Ho and G. J. Evans, Environ. Sci. Technol., 2000, 34, 1030 CrossRef CAS.
- D. K. Nordstrom, Water, Air Soil Pollut., 1996, 90, 257 CrossRef CAS.
- A. S. Hursthouse, M. S. Baxter, F. R. Livens and H. J. Duncan, J. Environ. Radioact., 1991, 14, 147 CrossRef CAS.
- M. Kersten and U. Forstner, in Chemical Speciation in the Environment, ed. A. M. Ure and C. M. Davidson, Blackie, Glasgow, 1995, ch. 9, pp. 234–275. Search PubMed.
- T. Theis and R. Iyer, in Metal Speciation and Contamination of Soil, ed. H. E. Allen, C. P. Huang, G. W. Bailey and A. R. Bowers, CRC Press, Boca Raton, FL, 1995, ch. 7, pp. 207–226. Search PubMed.
- W. Wenzel and W. Blum, in Metal Speciation and Contamination of Soil, ed. H. E. Allen, C. P. Huang, G. W. Bailey and A. R. Bowers, CRC Press, Boca Raton, FL, 1995, ch. 8, pp. 227–236. Search PubMed.
- S. Roy and D. Dzombak, Environ. Sci. Technol., 1997, 31, 656 CrossRef CAS.
- D. Grolimund, M. Elimelech, M. Borovec, K. Barmettler, R. Kretzschmar and H. Sticher, Environ. Sci. Technol., 1998, 32, 3562 CrossRef CAS.
- H. W. Schmitt and H. Sticher, in Metals and Their Compounds in the Environment, ed. E. Merian, VCH, Weinheim, 1991, pp. 311–331. Search PubMed.
- M. Manz, L. Weissflog, R. Kuhne and G. Schuurmann, Ecotoxicol. Environ. Saf., 1999, 42, 191 CrossRef CAS.
- S. L. Simpson, S. C. Apte and G. E. Batley, Environ. Sci. Technol., 1998, 32, 620 CrossRef CAS.
- D. Donnert, S. H. Eberle and J. Horst, in Metal Speciation in the Environment, ed. J. A. C. Broekaert, S. Gucer and F. Adams, Springer-Verlag, Berlin, Heidelberg, 1990, pp. 121–136. Search PubMed.
- R. J. M. Hudson, Sci. Total Environ., 1998, 219, 95 CrossRef CAS.
- T. Guo, R. D. DeLaune and W. H. Partick, Jr., Environ. Int., 1997, 23, 305 CrossRef CAS.
- W. Chen, S. K. Tan and J. H. Tay, Water, Air Soil Pollut., 1996, 92, 273 CAS.
- D. C. Cooper and J. W. More, Environ. Sci. Technol., 1998, 32, 327 CrossRef CAS.
- M. Astrom and K. Nylund, Appl. Geochem., 2000, 15, 807 CrossRef CAS.
- S. E. J. Buykx, M. Bleijenberg, M. A. G. T. van den Hoop and J. P. G. Loch, J. Environ. Monit., 2000, 2, 23 RSC.
- W. Calmano, W. Ahlf and U. Forstner, in Metal Speciation in the Environment, ed. J. A. C. Broekaert, S. Gucer and F. Adams, Springer-Verlag, Berlin, Heidelberg, 1990, pp. 503–522. Search PubMed.
- W. Calmano, J. Hong and U. Forstner, Water Sci. Technol., 1993, 28, 223 CAS.
- U. Forstner, in Metals and Their Compounds in the Environment, ed. E. Merian, VCH, Weinheim, 1991, pp. 379–398. Search PubMed.
- C. Savvides, A. Papadopoulos, K. J. Haralambous and M. Loizidou, Water Sci. Technol., 1995, 32, 65 CrossRef CAS.
- S.-Z. Lee, L. Chang, C.-M. Chen, M.-C. Liu and L.-J. Tsai, Water Sci. Technol., 1998, 38, 131 CrossRef CAS.
- F. M. G. Tack, S. P. Singh and M. G. Verloo, Environ. Pollut., 1998, 103, 109 CrossRef CAS.
- P. Ugo, A. Bertolin and L. M. Moretto, Int. J. Environ. Anal. Chem., 1999, 73, 129 Search PubMed.
- I. Ujevic, N. Odzak and A. Baric, Fresenius' Environ. Bull., 1998, 7, 183 Search PubMed.
- K. E. Murray, D. Cauvet, M. Kybeer and J. C. Thomas, Environ. Sci. Technol., 1999, 33, 987 CrossRef CAS.
- W. H. Patrick, Jr. and M. Verloo, Water Sci. Technol., 1998, 37, 165 CrossRef.
- M. L. Berrow and J. C. Buridge, in Metals and Their Compounds in the Environment, ed. E. Merian, VCH, Weinheim, 1991, pp. 399–410. Search PubMed.
- G. S. P. Ritchie and G. Sposito, in Chemical Speciation in the Environment, ed. A. M. Ure and C. M. Davidson, Blackie, Glasgow, 1995, ch. 8, pp. 201–233. Search PubMed.
- S. Sauve, W. Hendershot and H. E. Allen, Environ. Sci. Technol., 2000, 34, 1125 CrossRef CAS.
- S. Sauve, W. A. Norvell, M. McBride and W. Hendershot, Environ. Sci. Technol., 2000, 34, 291 CrossRef CAS.
- C. E. Martinez and M. B. McBride, Environ. Sci. Technol., 1999, 33, 745 CrossRef CAS.
- R. Roberts, A. M. Scheidegger and D. L. Sparks, Environ. Sci. Technol., 1999, 33, 3749 CrossRef.
- J. W. Stucki, G. W. Bailey and H. Han, in Metal Speciation and Contamination of Soil, ed. H. E. Allen, C. P. Huang, G. W. Bailey and A. R. Bowers, CRC Press, Boca Raton, FL, 1995, ch. 5, pp. 113–181. Search PubMed.
- B. Lothenbach, G. Furrer, H. Scharli and R. Schulin, Environ. Sci. Technol., 1999, 33, 2945 CrossRef CAS.
- P. Murray, Y. Ge and W. H. Hendershot, Environ. Pollut., 2000, 107, 127 CrossRef CAS.
- J. Hertz, in Metals and Their Compounds in the Environment, ed. E. Merian, VCH, Weinheim, 1991, pp. 221–231. Search PubMed.
- C. L. Schultz and T. C. Hutchinson, in Metals and Their Compounds in the Environment, ed. E. Merian, VCH, Weinheim, 1991, pp. 411–418. Search PubMed.
- Y. Ge, P. Murray and W. H. Hendershort, Environ. Pollut., 2000, 107, 137 CrossRef CAS.
- G. Wozniak, in Green 2 Contaminated and Derelict Land, ed. R. W. Sarbsy, Thomas Telford, London, 1998, pp. 60–63. Search PubMed.
- A. Barona and F. Romero, Water, Air Soil Pollut., 1997, 95, 59 CrossRef CAS.
- I. Convery and A. Mellor, Land Contam. Reclam., 1996, 4, 255 Search PubMed.
- M. Clusner Godt, in Element Concentration Cadasters in Ecosystems, ed. H. Lieth and B. Markert, VCH, Weinheim, 1990, pp. 345–356. Search PubMed.
- U. Forstner, in Metal Speciation and Contamination of Soil, ed. H. E. Allen, C. P. Huang, G. W. Bailey and A. R. Bowers, CRC Press, Boca Raton, FL, 1995, ch. 1, pp. 1–33. Search PubMed.
- W. H. O. Ernst, Appl. Geochem., 1996, 11, 163 CrossRef CAS.
- E. Lawrence, A. R. W. Jackson and J. M. Jackson, Dictionary of Environmental Science, Longman, Harlow, 1998. Search PubMed.
- J. M. Oosterbaan, P. V. Jamet and E. Souto, in Green 2 Contaminated and Derelict Land, ed. R. W. Sarbsy, Thomas Telford, London, 1998, pp. 89–96. Search PubMed.
- D. Smith and P. Tucker, Environ. Geochem. Health, 1999, 21, 311 Search PubMed.
- W. M. Fox, M. S. Johnson, S. R. Jones, R. T. Leah and D. Copplestone, Mar. Environ. Res., 1999, 47, 311 CrossRef CAS.
- M. H. Bothner, M. Buchholtz ten Brink and F. T. Manheim, Mar. Environ. Res., 1998, 45, 127 CrossRef CAS.
- R. Carignan, S. Lorrain and K. Kum, Can. J. Fish. Aquat. Sci., 1994, 51, 1088 CAS.
- P. N. Owens, D. E. Walling and G. J. L. Leeks, Catena, 1999, 36, 21 CrossRef.
- A. S. Hursthouse, M. Adamczyk, L. Adamczyk, P. Iqbal and F. J. Smith, Mar. Pollut. Bull., 1994, 28, 765 CrossRef CAS.
- J. E. Rae and A. Parker, Appl. Geochem., 1996, 11, 211 CrossRef CAS.
- D. Venditti, S. Durecu and J. Berthelin, Arch. Environ. Contam. Toxicol., 2000, 38, 411 CrossRef CAS.
- E. C. Buck, N. R. Brown and N. L. Dietz, Environ. Sci. Technol., 1996, 30, 81 CrossRef CAS.
- L. Fanfani, P. Zuddas and A. Chessa, J. Geochem. Explor., 1997, 58, 124 CrossRef CAS.
- L. Liang and J. F. McCarthy, in Metal Speciation and Contamination of Soil, ed. H. E. Allen, C. P. Huang, G. W. Bailey and A. R. Bowers, CRC Press, Boca Raton, FL, 1995, ch. 4, pp. 87–112. Search PubMed.
- W. Stumm and J. J. Morgan, Aquatic Chemistry: Chemical Equilibria and Rates in Natural Waters, John Wiley, New York, 3rd edn., 1996. Search PubMed.
- F. M. Kindler and H. E. Sevim, in Metal Speciation in the Environment, ed. J. A. C. Broekaert, S. Gucer and F. Adams, Springer-Verlag, Berlin, Heidelberg, 1990, pp. 601–611. Search PubMed.
- A. Kabata-Pendias and S. Dudka, in Element Concentration Cadasters in Ecosystems, ed. H. Lieth and B. Markert, VCH, Weinheim, 1990, pp. 265–280. Search PubMed.
- P. W. Balls, S. Hull, B. S. Miller, J. M. Pirie and W. Proctors, Mar. Pollut. Bull., 1997, 34, 42 CrossRef CAS.
- C. M. Whalley, S. Rowlatt, M. Bennet and D. R. Lovell, Mar. Pollut. Bull., 1999, 38, 394 CrossRef CAS.
- S. M. Rowlatt and D. R. Lovell, Mar. Pollut. Bull., 1994, 28, 324 CrossRef CAS.
- B. G. Lottermoser, Aust. J. Soil Res., 1997, 35, 1165 Search PubMed.
- C. M. Whalley, S. Rowlatt, A. S. Hursthouse, P. Iqbal-Zahid, R. Durant and D. H. Vaughan, Water, Air Soil Pollut., 1999, 112, 389 CrossRef CAS.
- K. E. Murray, Environ. Geol., 1996, 27, 54 Search PubMed.
- P. M. Chapman, F. Wang, W. J. Adams and A. Green, Environ. Sci. Technol., 1999, 33, 3937 CrossRef CAS.
- G. Krantzberg, J. H. Hartig and M. A. Zarull, Environ. Sci. Technol., 2000, 34, 22A CrossRef CAS.
- T. F. Rees, Contaminated Land Treatment Technologies, Elsevier Applied Science, London, 1992. Search PubMed.
- M. Pratt, Remedial Processes for Contaminated Land, Institution of Chemical Engineers, Rugby, Warwickshire, 1993. Search PubMed.
- I. Martin and P. Bardos, A Review of Full Scale Treatment Technologies for the Remediation of Contaminated Soil, EPP Publications, Richmond, Surrey, 1995. Search PubMed.
- R. W. Peters and L. Shem, in Metal Speciation and Contamination of Soil, ed. H. E. Allen, C. P. Huang, G. W. Bailey and A. R. Bowers, CRC Press, Boca Raton, FL, 1995, ch. 2, pp. 255–274. Search PubMed.
- P. A. Wood, in Contaminated Land and its Remediation. Issues in Environmental Science and Technology, 7, ed. R. E. Hester and R. M. Harrison, Royal Society of Chemistry, Cambridge, 1997, pp. 47–71. Search PubMed.
- T. Nedwed and D. A. Clifford, Wastes Manage., 1997, 17, 257 Search PubMed.
- W. H. Rulkens, R. Tichy and J. T. C. Grotenhuis, Water Sci. Technol., 1998, 37, 27 CrossRef CAS.
- L. E. Macaskie and G. Basnakova, Environ. Sci. Technol., 1998, 32, 184 CrossRef CAS.
- I. Kuziemska and B. Quant, in Green 2 Contaminated and Derelict Land, ed. R. W. Sarbsy, Thomas Telford, London, 1998, pp. 308–312. Search PubMed.
- (a) A. S. Hursthouse, K. Forster, G. Murney, P. Tucker, C. Whalley, C. Wiltshire, S. Gallagher, J. Osinski and K. Wargocki, Contam. Land Reclam., 2000, 8, 271 Search PubMed; (b) A. S. Hursthouse, C. Whalley, S. Rowlatt, P. Iqbal-Zahid and K. Forster, Contaminated Soil 2000, Thomas Telford, London, 2000, pp. 783–784. Search PubMed.
- S. Wallace and J. P. Cork, in Green 2 Contaminated and Derelict Land, ed. R. W. Sarbsy, Thomas Telford, London, 1998, pp. 317–323. Search PubMed.
- A. B. Shandyba, S. V. Vakal and V. D. Chivanov, in Green 2 Contaminated and Derelict Land, ed. R. W. Sarbsy, Thomas Telford, London, 1998, pp. 313–316. Search PubMed.
- R. L. Chaney, M. Malik, Y. M. Li, S. L. Brown, E. P. Brewer, J. S. Angle and A. J. M. Baker, Curr. Opin. Biotechnol., 1997, 8, 279 CrossRef CAS.
- H. Z. Felix, Pflanzenernahr. Bodenk., 1997, 160, 525 Search PubMed.
- D. E. Salt, M. Blaylock, N. P. B. A. Kumar, V. Dushenkov, B. D. Ensley, I. Chet and I. Raskin, Bio/Technology, 1995, 13, 468 Search PubMed.
- D. E. Salt, R. D. Smith and I. Raskin, Annu. Rev. Plant Physiol. Plant Mol. Biol., 1998, 49, 643 Search PubMed.
- A. Kayser, K. Wenger, A. Keller, W. Attinger, H. R. Felix, S. K. Gupta and R. Schulin, Environ. Sci. Technol., 2000, 34, 1778 CrossRef CAS.
- W. H. O. Ernst, in Element Concentration Cadasters in Ecosystems, ed. H. Lieth and B. Markert, VCH, Weinheim, 1990, pp. 17–40. Search PubMed.
- M. C. Negri and R. R. Hinchman, Lab. Med., 1996, 27, 36 Search PubMed.
- K. Fischer, H.-P. Bipp, R. Riemschneider, P. Leidmann, D. Bieniek and A. Kettrup, Environ. Sci. Technol., 1998, 32, 2154 CrossRef CAS.
- C. G. Rampley and K. L. Ogden, Environ. Sci. Technol., 1998, 32, 987 CrossRef CAS.
- G. Ho and L. Qiao, Water Sci. Technol., 1998, 38, 17 CrossRef CAS.
- L. Qiao and G. Ho, Water Res., 1997, 31, 951 CrossRef CAS.
- M. A. M. Kedziorek, A. Dupuy, A. C. M. Bourg and F. Compere, Environ. Sci. Technol., 1998, 32, 1609 CrossRef CAS.
- R. W. Peters, J. Hazardous Mater., 1999, 66, 151 Search PubMed.
- N. Lu, S. Kungm, C. F. V. Mason, I. R. Triary, C. R. Cotter, A. J. Pappas and M. E. G. Pappas, Environ. Sci. Technol., 1998, 32, 370 CrossRef CAS.
- R.-A. Doong, Y.-W. Wu and W.-G. Lei, Water Sci. Technol., 1998, 37, 65 CrossRef CAS.
- J. Thoming, H. Stichnothe, S. Mangold and W. Calmano, Land Contam. Reclam., 2000, 8, 19 Search PubMed.
- W. Admassu and T. Breese, J. Hazard. Mater., 1999, B69, 187 CrossRef.
- W. R. Berti and S. D. Cunningham, Environ. Sci. Technol., 1997, 31, 1359 CrossRef CAS.
- R. M. Powell and R. W. Puls, Environ. Sci. Technol., 1997, 31, 2244 CrossRef CAS.
- R. W. Puls, C. J. Paul and R. M. Powell, Appl. Geochem., 1999, 14, 989 CrossRef CAS.
- E. C. Thornton and J. E. Amonette, Environ. Sci. Technol., 1999, 33, 4096 CrossRef CAS.
- P. Cauwenberg, F. Verdonckt and A. Maes, Sci. Total Environ., 1998, 209, 121 CrossRef CAS.
- I. M.-C. Lo and X.-Y. Yang, Wastes Manage., 1998, 18, 1 Search PubMed.
- D. J. Kelsh and M. W. Parsons, J. Hazard. Mater., 1997, 55, 109 CrossRef CAS.
- J. M. Dzenitis, Environ. Sci. Technol., 1997, 31, 1191 CrossRef CAS.
- L. M. Ottosen, H. K. Hansen, S. Laursen and A. Villumsen, Environ. Sci. Technol., 1997, 31, 1711 CrossRef CAS.
- Z. Li, J.-W. Yu and I. Neretnieks, Environ. Sci. Technol., 1998, 32, 394 CrossRef CAS.
- K. G. Senneset and J. M. Strout, in Green 2 Contaminated and Derelict Land, ed. R. W. Sarbsy, Thomas Telford, London, 1998, pp. 281–285. Search PubMed.
- C. Boyle, in Remedial Processes for Contaminated Land, ed. M. Pratt, Institution of Chemical Engineers, Rugby, Warwickshire, 1993, ch. 3, pp. 33–52. Search PubMed.
- W. P. Pisi, in Green 2 Contaminated and Derelict Land, ed. R. W. Sarbsy, Thomas Telford, London, 1998, pp. 334–335. Search PubMed.
- S. W. Paff and B. E. Bosilovich, J. Hazard. Mater., 1995, 40, 139 CrossRef CAS.
- M. M. A. F. Ferdinady-van Vlerken, Water Sci. Technol., 1998, 37, 345 CrossRef.
- A. Hauge, R. M. Konieczyn, P. O. Halvorsen and A. Eikum, Water Sci. Technol., 1998, 37, 299 CrossRef CAS.
- C. Arquiett, M. Gerke and I. Datskou, Water, Air Soil Pollut., 1996, 90, 83 CrossRef CAS.
- T. S. Bowers, N. S. Shifrin and B. L. Murphy, Environ. Sci. Technol., 1996, 30, 1437 CrossRef CAS.
- J. Figures, PhD Thesis, University of Paisley, Paisley, UK, 1997..
- P. W. Balls, R. E. Owens and F. L. L. Muller, Coast. Zone Top., 1997, 3, 46 Search PubMed.
- A. Turner, Estuarine, Coast. Shelf Sci., 2000, 50, 355 Search PubMed.
- D. J. Curtis and R. O. McLean, Coast. Zone Top., 1997, 3, 115 Search PubMed.
- B. S. Miller, in Heavy Metals in the Environment, ed. J. G. Farmer, CEP Consultants, Edinburgh, 1991, pp. 199–203. Search PubMed.
- B. S. Miller and D. J. Pirie, Coast. Zone Top., 1997, 3, 98 Search PubMed.
- P. E. Edgar, I. M. Davies, A. S. Hursthouse and J. E. Matthews, Mar. Pollut. Bull., 1999, 38, 486 CrossRef CAS.
- J. Figures, J. E. Matthews and A. S. Hursthouse, Coast. Zone Top., 1997, 3, 66 Search PubMed.
- D. J. Curtis, J. Figures, J. Matthews and P. Tatner, Coast. Zone Top., 1997, 3, 182 Search PubMed.
- F. C. Richard and A. C. Bourg, Water Res., 1991, 25, 807 CrossRef CAS.
- S. E. Fendorf, Geoderma, 1995, 67, 55 CrossRef CAS.
- A. S. Hursthouse, J. Matthews, J. Figures, P. Iqbal-Zahid, I. M. Davies and D. H. Vaughan, Environ. Geochem. Health, in press. Search PubMed.
- J. Thorburn, 1993, unpublished results..
- Royal Commission on Environmental Pollution, 19th Report, Sustainable Use of Soil, HMSO, London, 1996. Search PubMed.
- N. W. Arnell and N. S. Reynard, J. Hydrol., 1996, 183, 397 CrossRef.
- T. J. Marsh and F. J. Sanderson, Sci. Total Environ., 1997, 194–197, 59 CrossRef.
- A. S. Hursthouse, P. Iqbal and R. Denman, Analyst, 1993, 118, 1461 RSC.
- A. S. Hursthouse and F. R. Livens, Environ. Geochem. Health, 1993, 15, 163 Search PubMed.
- R. Jeffries and G. S. Hamilton, Contamination Investigation of 15 Sites in South and South East Glasgow, Known or Suspected to be Contaminated by Cr Waste, Glasgow City Council, Glasgow, 1994. Search PubMed.
- J. G. Farmer, M. C. Graham, R. P. Thomas, C. Licona-Manzur, E. Patterson, C. D. Campbell, J. S. Geelhoed, D. G. Lumsdon, J. C. L. Meeussen, M. J. Roe, A. Conner, A. E. Fallick and R. J. F. Bewley, Environ. Geochem. Health, 1999, 21, 331 Search PubMed.
- J. S. Geelhoed, J. C. L. Meeussen, D. G. Lumsdon, M. J. Roe, R. P. Thomas, J. G. Farmer and E. Patterson, Contam. Land Reclam., 1999, 7, 271 Search PubMed.
- K. Zotter and I. Licsko, Water Sci. Technol., 1992, 26, 207 CAS.
- J. W. Patterson, E. Gasca and Y. Wang, Water Sci. Technol., 1994, 29, 275 CAS.
- A. Ozer, H. S. Altundogan, M. Erdem and F. Tumen, Environ. Pollut., 1997, 97, 107 CrossRef CAS.
- S. E. Bailey, T. Olin, M. Brika and D. D. Adrian, Water Res., 1999, 33, 2469 CrossRef CAS.
- H. Eccles and S. Hunt, Immobilisation of Ions by Biosorption, Ellis Horwood, Chichester, 1986. Search PubMed.
- G. M. Gadd and C. White, Trends Biotechnol., 1993, 11, 353 CrossRef CAS.
- M. Nourbakhsh, Y. Sag, D. Ozer, Z. Aksu, T. Kutsal and A. Calgar, Process Biochem., 1994, 29, 1 CrossRef CAS.
- A. Netzer and M. R. Donahue, Metal Removal from Wastewater by Lignite Flyash, University of Dallas at Texas, Richardson, TX, 1993. Search PubMed.
- D. C. Sharma and C. F. Forster, Water Res., 1993, 27, 1201 CrossRef CAS.
- I. Gaballah, D. Goy, G. Kilbertus and J. Thauront, Resour. Conserv. Recycl., 1994, 10, 97 CrossRef.
- M. M. Alves, C. G. G. Beca, R. Guedes de Carvalho, J. M. Castanheira, M. C. Sol Pereia and L. A. T. Vasconcelos, Water Res., 1993, 27, 1333 CrossRef CAS.
- N. L. Nemerow, Zero Pollution for Industry—Waste Minimisation Through Industrial Complexes, John Wiley, New York, 1995. Search PubMed.
- A. S. Hursthouse, in Introduction to Industrial Chemistry, ed. C. A. Heaton, Blackie, Glasgow, 3rd edn., 1996, ch. 9, pp. 251–288. Search PubMed.
- J. J. Pierce, R. F. Weiner and P. A. Vesilind, Environmental Pollution and Control, Butterworth-Heinemann, Boston, 4th edn., 1998. Search PubMed.
- A. G. Khan, T. M. Chaudhry, W. J. Hayes, C. S. Khoo, L. Hill, R. Fernandez and P. Gallardo, Water, Air Soil Pollut., 1998, 104, 389 CrossRef.
- A. S. Hursthouse, P. Tucker, G. Witshire, D. Stark and C. Whalley, Fernigair Research Site, S. Lanarkshire: A Pilot Study of Contaminant Distribution, Soil Development and the Scope for Studies of Phytoremediation, Report to British Steel plc, University of Paisley, Paisley, UK, 1998. Search PubMed.
- D. M. Hepple, A. S. Hursthouse, P. Tucker and T. Henman, Contaminated Soil 2000, Thomas Telford, London, 2000, pp. 689–696. Search PubMed.
- F. Plassard, T. Winiarski and M. Petit-Ramel, J. Contam. Hydrol., 2000, 42, 99 CrossRef CAS.
- S. Nagano, S. Iwai, Y. Miura and Y. Morita, in Green 2 Contaminated and Derelict Land, ed. R. W. Sarbsy, Thomas Telford, London, 1998, pp. 483–487. Search PubMed.
- S. Zayane, A. Hursthouse, D. Cooke and R. Willey, Wastes Manage., 1994, 14, 33 Search PubMed.
- C. Whalley, Wastes Manage., 1997, 17, 41 Search PubMed.
- P. J. Young, S. Pollard and P. Crowcroft, in Contaminated Land and its Remediation. Issues in Environmental Science and Technology, 7, ed. R. E. Hester and R. M. Harrison, Royal Society of Chemistry, Cambridge, 1997, pp. 1–24. Search PubMed.
- U. Forstner, W. Calmano, J. Hong and M. Kersten, in Reviews on Analytical Chemistry—Euroanalysis VIII, ed. D. Littlejohn and D. Thorburn-Burns, Royal Society of Chemistry, London, 1994, pp. 83–102. Search PubMed.
- Web sites: http://www.nato.int/cms; http://www.epareachit.org; http://www.iws.uni-stuttgart.de/vegas; http://environment.paisley.ac.uk.
- M. Mansell, Nordic Hydrol., 1997, 28, 37 Search PubMed.
Footnote |
| † Presented at the Whistler 2000 Speciation Symposium, Whistler Resort, BC, Canada, June 25–July 1, 2000. |
| This journal is © The Royal Society of Chemistry 2001 |