Ruthenium terpyridine complexes with mono- and bi-dentate dithiolene ligands
Received
(in Cambridge, UK)
18th September 2000
, Accepted 30th October 2000
First published on 6th December 2000
Introduction
Transition metal complexes with redox active ligands such as polypyridyl and dioxolene feature multi-step redox processes due to changes in oxidation state of both the metals and the ligands themselves.1 Especially, the redox behavior of rhenium, ruthenium, and osmium polypyridyl complexes has extensively been studied in connection with their characteristic CT bands depending on the electron distribution between the central metals and the ligands. Moreover, ruthenium(II) polypyridyl complexes with a good leaving group are widely used as homogeneous catalysts in electrochemical reduction of NO2−![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 2 and CO2,3 and oxidation of NH34 and H2O,5 where ligand localized and metal-centered redox reactions are utilized as electron reservoirs for these catalytic reactions. It is well known that dithiolenes behave as redox active ligands, and adopt dianion, anion radical and neutral oxidation states (Scheme 1). Metal–dithiolene complexes, therefore, are also feasible candidates for homogeneous catalysts for redox reactions of various substrates.
2 and CO2,3 and oxidation of NH34 and H2O,5 where ligand localized and metal-centered redox reactions are utilized as electron reservoirs for these catalytic reactions. It is well known that dithiolenes behave as redox active ligands, and adopt dianion, anion radical and neutral oxidation states (Scheme 1). Metal–dithiolene complexes, therefore, are also feasible candidates for homogeneous catalysts for redox reactions of various substrates.
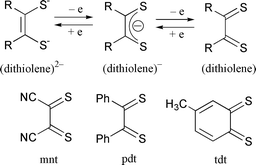 |
| | Scheme 1 | |
Metal–dithiolene complexes are usually synthesized as bis- and tris-chelate forms such as those of divalent Fe, Co, Rh, Ni, Pd and Pt and trivalent V, Cr, Re, Mo, W, Ru and Os, respectively.6–10 Introduction of dithiolene ligands to ruthenium–polypyridyl complexes would generate characteristic redox behavior because of strong interaction between unoccupied 3d orbitals of the sulfur of dithiolene with RuII. A few mixed chelate ruthenium complexes with polypyridyl and dithiolene ligands have been prepared so far.11 We have attempted introduction of dithiolenes to ruthenium polypyridyl complexes with a good leaving group. The present study reports the preparation and properties of new ruthenium terpyridine complexes with S2C2(CN)2 (mnt), S2C6H3Me (tdt) and S2C2Ph2 (pdt) ligands.
Experimental
Na2mnt,10 4,5-diphenyl-1,3-dithio-4-cyclopenten-2-one,12 and [Ru(dmso)Cl2(terpy)]13 and [Ru(CO)2Cl(terpy)]PF614 were prepared as described. All other commercially available reagents and solvents were used as purchased.
Preparation of complexes
[Ru(CO)2(mnt-κS![[hair space]](https://www.rsc.org/images/entities/b_char_200a.gif) )(terpy-κ3NN′N′′)] 1a and [Ru(CO)2(mnt-κ2SS′)(terpy-κ2NN′)] 1b.
[Ru(CO)2Cl(terpy)]PF6 (114 mg, 0.2 mmol) was added to a CH3OH solution (100 ml) of Na2mnt (37 mg, 0.2 mmol). The suspension gradually changed to a clear red solution in 2 h, during which time small amounts of yellow crystals of complex 1a precipitated. Then, red crystals of 1b precipitated when the solution was allowed to stand for another few hours. 1b was recrystallized from acetone. Yield 92 mg (87%). FAB-MS: m/z 531 ({M}+), 503 ({M − CO}+) and 475 ({M − (CO)2)}+). Calc. for C21H11N5O2RuS2: C, 47.54; H, 2.09; N, 13.20. Found: C, 47.31; H, 2.25; N, 12.95%. IR spectrum (KBr); ν(CN) 2197, 2170; ν(CO) 2093, 1962 cm−1. 1H NMR (CD3CN, R.T.): δ 9.06 (1H, d), 8.82 (1H, d), 8.4–8.6 (2H, m), 8.1–8.3 (2H, m), 8.01 (1H, t), 7.65–7.85 (3H, m) and 7.56 (1H, t). λmax/nm (ε/dm3 mol−1 cm−1) (CH3CN) 324 (12220) and 390 (5350).
)(terpy-κ3NN′N′′)] 1a and [Ru(CO)2(mnt-κ2SS′)(terpy-κ2NN′)] 1b.
[Ru(CO)2Cl(terpy)]PF6 (114 mg, 0.2 mmol) was added to a CH3OH solution (100 ml) of Na2mnt (37 mg, 0.2 mmol). The suspension gradually changed to a clear red solution in 2 h, during which time small amounts of yellow crystals of complex 1a precipitated. Then, red crystals of 1b precipitated when the solution was allowed to stand for another few hours. 1b was recrystallized from acetone. Yield 92 mg (87%). FAB-MS: m/z 531 ({M}+), 503 ({M − CO}+) and 475 ({M − (CO)2)}+). Calc. for C21H11N5O2RuS2: C, 47.54; H, 2.09; N, 13.20. Found: C, 47.31; H, 2.25; N, 12.95%. IR spectrum (KBr); ν(CN) 2197, 2170; ν(CO) 2093, 1962 cm−1. 1H NMR (CD3CN, R.T.): δ 9.06 (1H, d), 8.82 (1H, d), 8.4–8.6 (2H, m), 8.1–8.3 (2H, m), 8.01 (1H, t), 7.65–7.85 (3H, m) and 7.56 (1H, t). λmax/nm (ε/dm3 mol−1 cm−1) (CH3CN) 324 (12220) and 390 (5350).
[Ru(CO)2(tdt)(terpy)] 2b.
To a solution of [Ru(CO)2Cl(terpy)]PF6 (114 mg, 0.2 mmol) in CH3CN (10 ml), tBuOK (45 mg, 0.4 mmol) and 3,4-toluenedithiol (31 mg, 0.2 mmol) in CH3OH (10 ml) were added. The solution was stirred until red microcrystals precipitated. The red solid was filtered off, washed with C2H5OH and dried. Recrystallization from DMF gave red single crystals. ESI-MS: m/z 545 ({M}+), 517 ({M − CO}+) and 489 ({M − (CO)2}+). Calc. for C24H17N3O2RuS2· H2O: C, 51.24; H, 3.40; N, 7.47. Found: C, 51.27; H, 3.11; N, 7.73%. IR spectrum (KBr); ν(CO) 2033 and 1979 cm−1. 1H NMR (DMF-d7, R.T.): δ 9.54 and 9.52 (2H, d), 8.92 (4H, t), 8.84 (2H, d), 8.47 (2H, t), 8.31 (2H, t), 8.17 (2H, t), 7.98 (4H, q), 7.77 (2H, q), 7.67 (2H, q), 7.04 (1H, d), 6.99 (1H, s), 6.74 (1H, d), 6.69 (1H, s), 6.42 (1H, d), 6.37 (1H, d), 2.07 (3H, s) and 1.99 (3H, s). λmax/nm (ε/dm3 mol−1 cm−1) (acetone) 323 (11170) and 395 (5420).
[Ru(CO)(C(O)OCH3)(SC(Ph)C(Ph)SC(O)OCH3)(terpy-κ3NN′N′′)]·0.5H2O 3·0.5H2O.
A methanolic solution (20 ml) of [Ru(CO)2Cl(terpy)]PF6 (228 mg, 0.4 mmol), 4,5-diphenyl-1,3-dithio-4-cyclopenten-2-one (108 mg, 0.4 mmol) and CsOH (150 mg, 0.8 mmol) was stirred for 2 h under aerobic conditions. The volume of the brown solution was reduced to 2 ml and allowed to stand for one night to give black-brown crystals of 3. Yield 60%. ESI-MS (DMF): m/z 692 ({M − OCH3}+), 664 ({M − COOCH3+}), 636 ({M − COOCH3 − CO+}), 605 ({M − COOCH3 − CO − OCH3+}) and 577 ({M − COOCH3 − (CO)2 − OCH3+}). Calc. for C34H28.5N3O5.5RuS2: C, 55.80; H, 3.86; N, 5.74. Found: C, 55.91; H, 3.77; N, 5.86%. IR spectrum (KBr): ν(CO) 1966, 1711 and 1684 cm−1.
[Ru(dmso)(mnt)(terpy)] 4.
An aqueous solution (5 ml) of [Ru(dmso)Cl2(terpy)] (48 mg, 0.1 mmol) was added to a CH3OH solution (10 ml) of Na2mnt (19 mg, 0.1 mmol). The yellow solution changed to a brown suspension and then gradually became a clear red solution. After 2 h, red microcrystals precipitated and were collected, washed with ethanol, and dried. Yield 40 mg (72%). ESI-MS: m/z 475 ({M − dmso}+). Anal. Calc. for C21H17N5ORuS3: C, 45.64; H, 3.10; N, 12.67. Found: C, 45.39; H, 3.35; N, 12.41%. IR spectrum (KBr): ν(CN) 2191 cm−1. 1H NMR (acetone-d6, R.T.): δ 8.80 (2H, d), 8.57 (2H, d), 8.52 (2H, d), 8.21 (1H, t), 8.12 (2H, t), 7.70 (2H, t) and 2.61 (6H, s). λmax/nm (ε/dm3 mol−1 cm−1) (CH3CN) 312 (20760) and 442 (6370).
[RuCl(tdt)(terpy)]BF45 BF4.
A methanolic solution (20 ml) containing [Ru(dmso)Cl2(terpy)] (200 mg, 0.4 mmol), H2tdt (63 mg, 0.4 mmol) and CsOH (150 mg, 0.8 mmol) was refluxed for 2 h. Concentration of the brown solution to ca. 2 ml under reduced pressure resulted in a dark brown precipitate of [Ru(dmso)(tdt)(terpy)], which was isolated by filtration and washed with water. Addition of 10 drops of concentrated HCl to [Ru(dmso)(tdt)(terpy)] (100 mg) suspended in 10 ml of CH3OH gave a clear red-purple solution. Further addition of NaBF4 (44 mg, 0.4 mmol) in 5 ml of water precipitated [RuCl(tdt)(terpy)]BF45, which was collected by filtration, washed with water and dried in vacuo. Yield 58 mg (47%). ESI-MS: m/z 524 ({M}+) and 244 ({M − Cl}2+). Calc. for C22H17BF4N3RuS2: C, 43.26; H, 2.81; N, 6.88. Found: C, 43.37; H, 3.01; N, 6.59%. 1H NMR (acetone-d6, R.T.): δ 9.08 (3H, t), 8.83 (2H, d), 8.73 (2H, d), 8.56 (1H, t), 8.08 (4H, dt), 7.72 (2H, d), 7.33 (1H, t), 6.83 (2H, m), 2.39, 2.35 (3H, s). The two singlets at δ 2.39 and 2.35 indicate two geometrical isomers of tdt. λmax/nm (ε/dm3 mol−1 cm−1) (CH2Cl2) 313 (23600), 328 (620700), 393 (20700) and 527 (8060).
[RuCl(pdt)(terpy)]ClO46ClO)4.
A methanolic solution (20 ml) containing [Ru(dmso)Cl2(terpy)] (200 mg, 0.4 mmol), 4,5-diphenyl-1,3-dithio-4-cyclopenten-2-one (108 mg, 0.4 mmol) and CsOH (150 mg, 0.8 mmol) was refluxed for 2 h. Concentration of the brown solution to ca. 2 ml under reduced pressure precipitated dark brown [Ru(dmso)(pdt)(terpy)], which was isolated by filtration and washed with water (5 ml) to remove CsCl. To [Ru(dmso)(pdt)(terpy)] suspended in 10 ml of CH3OH were added 10 drops of concentrated HCl with stirring. Treatment of the resulting clear deep blue solution with aqueous NaClO4 (100 mg, 0.82 mmol) solution (5 ml) gave a blue microcrystalline powder, which was filtered off and dried in vacuo. Yield 104 mg (36%). ESI-MS: m/z 612 ({M}+) and 288.5 ({M − Cl}2+). Calc. for C29H21N3O4RuS2: C, 48.95; H, 2.97; N, 5.91. Found: C, 48.59; H, 3.12; N, 6.06%. 1H NMR (acetone-d6, R.T.): δ 9.02 (2H, d), 8.77 (3H, m), 8.22 (2H, t), 7.77 (2H, d), 7.64 (2H, q) and 7.2–7.6 (10H, m). λmax/nm (ε/dm3 mol−1 cm−1) (CH2Cl2) 315 (18130), 330 (20260) and 566 (9780).
[Ru(OSO2CF3)(pdt)(terpy)]CF3SO37CF3SO3.
Brown [Ru(dmso)(pdt)(terpy)] (100 mg) gradually dissolved in 10 ml of CH3OH after 10 drops of CF3SO3H were added to the suspension. The resultant clear blue solution was concentrated to 0.5 ml by evaporation. Addition of 5 ml of diethyl ether afforded blue microcrystals, which were collected by filtration and dried in vacuo. Yield 120 mg (68%). ESI-MS: m/z 726 ({M}+) and 288.5 ({M − CF3SO3}2+). Calc. for C31H21N3O6RuS2: C, 42.56; H, 2.42; N, 4.80. Found: C, 42.61; H, 2.72; N, 4.61%. 1H NMR (acetone-d6, R.T.): δ 9.02 (2H, d), 8.76 (3H, m), 8.21 (2H, t), 7.78 (2H, d), 7.58 (1H, t) and 7.2–7.55 (11H, m). λmax/nm (ε/dm3 mol−1 cm−1) (CH2Cl2) 329 (18080), 568 (7540).
X-Ray structural determinations
X-Ray data of complexes 1a, 1b and 2b were collected with graphite-monochromated Mo-Kα radiation on a Rigaku AFC-5S diffractometer. Crystallographic data are summarized in Table 1. All the calculations were performed with the TEXSAN crystallographic software package.15 The structures were solved by direct methods for 1a and heavy-atom methods for 1b and 2b and expanded using Fourier techniques. The structure of 3 was solved by direct methods, and the ruthenium, sulfur and nitrogen atoms, the atoms in the carbonyl and methoxycarbonyl ligands, and ethylene carbon atoms of Ph2C2S2COOCH3 were refined anisotropically.
| |
1a·CH3OH |
1b
|
2b
|
3·H2O |
| Formula |
C22H15N5O3RuS2 |
C21H11N5O2RuS2 |
C24H17N3O2RuS2 |
C33H27N3O6RuS2 |
|
M
|
562.6 |
530.5 |
544.6 |
726.8 |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2) (no. 2) |
P21/n (no. 14) |
P21/n (no. 14) |
P (no. 2) |
|
T/°C |
23 |
23 |
23 |
23 |
|
a/Å |
9.389(4) |
13.273(3) |
14.202(10) |
14.579(3) |
|
b/Å |
14.1223(4) |
9.301(3) |
9.408(7) |
19.743(4) |
|
c/Å |
8.817(3) |
17.059(2) |
18.47(1) |
12.948(3) |
|
α/° |
90.98(3) |
|
|
108.34(2) |
|
β/° |
103.42(3) |
92.40(1) |
108.52(5) |
94.30(2) |
|
γ/° |
96.32(3) |
|
|
98.16(2) |
|
V/Å3 |
1129.2(8) |
2104.2(8) |
2339(3) |
3473(1) |
|
Z
|
2 |
4 |
4 |
2 |
|
μ(Mo-Kα)/cm−1 |
9.14 |
9.73 |
8.74 |
6.16 |
| No. of reflections |
3174 |
5351 |
5910 |
13678 |
| No. of observed reflections |
2954 |
5136 |
5687 |
12946 |
|
R (%) |
6.0 |
5.4 |
5.8 |
8.3 |
|
Rw (%) |
5.4 |
5.2 |
6.5 |
7.6 |
CCDC reference number 186/2256.
See http://www.rsc.org/suppdata/dt/b0/b007541h/ for crystallographic files in .cif format.
Measurements
Cyclic voltammetry was performed with a BAS CV-100W voltammetry analyzer at a scan rate of 50 mV s−1. The sample solutions (ca. 1.0 mM) containing 0.1 M NBun4BF4 were deoxygenated with a stream of nitrogen gas. Redox potentials obtained were referenced to the ferrocenium–ferrocene couple. Electronic spectra were recorded on a Shimadzu UV-vis-NIR scanning spectrophotometer UV-3100PC. Spectroelectrochemistry was performed with a thin-layer electrode cell with a platinum mini grid working electrode sandwiched between two glass windows of an optical cell (path length 0.5 mm). 1H NMR spectra were measured on a JEOL-EX 270 (270 MHz) spectrometer, IR spectra on a Shimadzu FTIR-8100 spectrophotometer.
Results and discussion
Preparation of complexes
Reactions of [RuCl3(terpy)] with S2C2(CN)22− (mnt2−), S2C7H62− (tdt2−) and S2C2Ph22− (pdt2−) in CH3OH gave unidentified insoluble solids probably due to irreversible oxidation of these “free” ligands by RuIII. To avoid such unfavorable reactions, [Ru(CO)2Cl(terpy)]+ and [Ru(dmso)Cl2(terpy)] were used as starting complexes for preparation of dithiolene complexes. When Na2mnt was allowed to react with [Ru(CO)2Cl(terpy)]PF6 suspended in CH3OH the suspension changed to a clear yellow solution at first and then became red. Yellow [Ru(CO)2(mnt-κS)(terpy-κ3NN′N′′)] 1a and red [Ru(CO)2(mnt-κ2SS′)(terpy-κ2NN′)] 1b (see below) were isolated as single crystals from the yellow and red solutions, respectively (eqn. 1). 1a was stable in the solid state, while it smoothly changed to red 1b in CD3CN. Low solubility of 1a in CH3OH led to isolation of the complex, but the smooth conversion from 1a into 1b in solutions made it difficult to detect the 1H NMR spectrum of 1a. The reaction of [Ru(CO)2Cl(terpy)]PF6 with the K+ salt of tdt2− in CH3OH gave red [Ru(CO)2(tdt-κ2SS′)(terpy-κ2NN′)] 2b, but [Ru(CO)2(tdt-κS)(terpy-κ3NN′N′′)] 2a was not confirmed in the reaction (eqn. 2). The reaction of [Ru(CO)2Cl(terpy)]PF6 with the Cs+ salt of pdt2− in CH3OH afforded only viscous products, and neither [Ru(CO)2(pdt-κS)(terpy-κ3NN′N′′)] 3a nor [Ru(CO)2(pdt-κ2SS′)(terpy-κ2NN′)] 3b was isolated under an N2 atmosphere. On the other hand, [Ru(CO)(C(O)OCH3)(SC(Ph)C(Ph)SC(O)OCH3)(terpy-κ3NN′N′′)] 3 (see below) selectively crystallized when the same reaction was conducted under aerobic conditions. Based on the yield of 3 (60%), it is not a degradation product of 3a and 3b. Complex 3 has two CH3OC(O) groups; one is attached to Ru and the other linked to the sulfur of pdt. The former apparently resulted from nucleophilic attack of CH3OH on a carbonyl carbon of the Ru(CO)2 moiety, while the latter is probably produced through esterification of the Ru–SC(Ph)C(Ph)SC(O)OH framework derived from attack of CO2 (from air) on the S2C2Ph2 group (eqn. 3). In fact, complexes having M–OH and M–S− units with high nucleophilicity have been shown to react with CO2 to give M–OCO2H and M–SCO2− moieties, respectively. For example, [Zn(L)][ClO4]2 (L = [14]aneN4 or [15]aneN4) take up CO2 in alcohol at room temperature to give the monoalkyl carbonato complexes, [Zn(L)(O2COR)],16 and [ML(OH)]n (M = Mn, Fe, Co, Ni, Cu or Zn, L = tris(3,5-diisopropyl-1-pyrazolyl)hydroborate, n = 1 or 2) have activity toward fixation of atmospheric CO2, changing to [LM(μ-CO3)ML].17 The reaction of [(Ir(η5-C5Me5))2Ir(η4-C5Me5CH2CN)(μ3-S)2] with CO2 produced C2O42−via [(Ir(η5-C5Me5))2Ir(η4-C5Me5CH2CN)(μ3-S)2]·2CO2.18 The formation of 3, therefore, is explained by attack of CO2 on the terminal sulfur of [Ru(CO)2(pdt-κS)(terpy-κ3-NN′N′′)] 3a followed by nucleophilic attack of CH3OH on a carbonyl carbon, and then esterification of the resultant [Ru(CO)(C(O)OCH3)(SC(Ph)C(Ph)SC(O)OH)(terpy-κ3NN′N′′)]. The selective crystallization of 3 rather than [Ru(CO)(C(O)OCH3)(SC(Ph)C(Ph)SC(O)OH)(terpy-κ3NN′N′′)] probably results from low solubility of the former in CH3OH.
| |
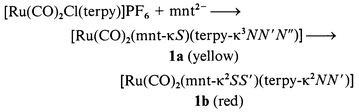
|
(1)
|
| | |
[Ru(CO)2Cl(terpy)]PF6 + tdt2− → ([Ru(CO)2(tdt-κS)(terpy-κ3NN′N′′)]) → [Ru(CO)2(tdt-κ2SS′)(terpy-κ2NN′)]
|
(2)
|
| |
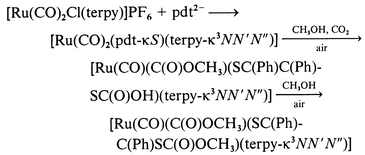
|
(3)
|
The reaction of [Ru(dmso)Cl2(terpy)] with mnt2− in CH3OH–water also generated neutral [Ru(dmso)(mnt)(terpy)] 4 (eqn. 4), but the similar reactions with tdt2− and pdt2− gave almost insoluble dark black and dark brown solids, respectively. In the case of tdt2−, treatment of the dark black solid with HCl in CH3OH resulted in a clear solution and [RuCl(tdt)(terpy)]+5+ was isolated as the BF4− salt. Similarly, in the case of pdt2−, addition of HCl or CF3SO3H to the dark brown solid suspended in MeOH produced cationic [RuX(pdt)(terpy)]+ (X = Cl 6+ or OSO2CF37+) under aerobic conditions. The mass spectrum of the insoluble dark brown solid obtained in the reaction of [Ru(dmso)Cl2(terpy)] with pdt2− showed the parent peak at m/z 655 ([Ru(dmso)(pdt)(terpy)]+) together with a strong fragment peak at m/z 577 ([Ru(pdt)(terpy)]+). Thus, the formation of cationic 5+, 6+, and 7+ is expressed by eqn. (5). ESR specctra of the solid states of 5+, 6+, and 7+ were silent and the 1H NMR spectra gave sharp signals in the range from δ 0 to 10. From these results it is clear that the oxidation state of the dithiolene is neutral and the electronic structures of 5+, 6+, and 7+ are denoted as [RuIIX(L0)]+ (L = tdt or pdt).
| | |
[Ru(dmso)Cl2(terpy)] + mnt2− → [Ru(dmso)(mnt)(terpy)]
|
(4)
|
| |
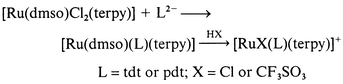
|
(5)
|
Molecular structures of complexes 1a, 1b, 2b and 3
The molecular structures of complexes 1a and 1b determined by X-ray diffraction analysis are shown in Fig. 1. Selected bond distances and angles are in Table 2. The ruthenium of 1a has an octahedral geometry with two carbonyl carbons, three nitrogens of terpyridine, and one sulfur of mnt. In most metal–mnt complexes the mnt coordinates as a bidentate ligand. The unusual monodentate coordination of mnt of 1a is ascribed to the stability of the Ru(terpy-κ3N,N′,N′′)(CO)2 framework. The Ru–C2 bond distance (1.85(2) Å) trans to S1 of the mnt ligand is slightly shorter than Ru–C1 (1.94(2) Å) trans to the center of the terpyridine. The relatively short Ru–C2 distance indicates a strong interaction between Ru and S1. The ruthenium of 1b is ligated by two nitrogens of terpyridine, two carbonyl carbons, and two sulfur atoms of mnt, and has a distorted octahedral environment. The two carbonyl ligands are situated in a cis position to each other. One (C1–O1) is trans to the sulfur atom (S1) of mnt and the other (C2–O2) is trans to a nitrogen atom (N1) of terpyridine. The ruthenium–carbonyl (1.924(10) and 1.88(1) Å) and the C–O distances (1.125(10) and 1.156(10) Å) are comparable with those found in other polypyridyl Ru(CO)2 complexes.14,19 Owing to the relatively strong interaction between Ru and the vacant d orbital of S, the Ru–N2 bond distance trans to S2 (2.160(5) Å) is clearly longer than the Ru–N1 distance trans to CO (2.129(7) Å). The Ru–C1 bond distance of 1.924(10) Å
trans to S1 is also slightly longer than the other Ru–C2 bond distance of 1.88(1) Å
trans to N1, suggesting that the interaction of Ru–S is stronger than that of Ru–CO ligand. The C19–C20 (1.372(9) Å) and C–S (1.726(7) and 1.726(8) Å) bond distances are similar to those found for [Ni(mnt)2]2−20 and [Me4N]2[V(mnt)3]21 that have dianionic mnt ligands. Based on these results, the electronic structure of 1b is expressed as [RuII(CO)2(mnt2−)(terpy)]. It is noteworthy that the central pyridine unit of terpy of 1b should occupy the trans position of one of the two carbonyl ligands if 1b is simply formed through the substitution of either terminal pyridine of terpy by the terminal sulfur of mnt in 1a. The 1H NMR spectra in CD3CN and DMF-d7 supported retention of the configuration of 1b in solution. The most reasonable rearrangement from 1a to 1b proceeds through five-coordinate [Ru(CO)2(mnt-κS)(terpy-κ2NN′)] formed by dissociation of either terminal pyridine of terpy prior to attack of the terminal sulfur of monodentate mnt on Ru (Scheme 2).
Table 2 Selected bond lengths (Å) and angles (°) for complexes 1a and 1b
|
1a
|
| Ru(1)–S(1) |
2.447(4) |
Ru(1)–N(1) |
2.08(1) |
| Ru(1)–N(2) |
2.04(1) |
Ru(1)–N(3) |
2.08(1) |
| Ru(1)–C(1) |
1.94(2) |
Ru(1)–C(2) |
1.85(2) |
| |
|
|
|
|
1b
|
| Ru(1)–S(1) |
2.408(2) |
Ru(1)–S(2) |
2.352(2) |
| Ru(1)–N(1) |
2.129(7) |
Ru(1)–N(2) |
2.160(5) |
| Ru(1)–C(1) |
1.924(10) |
Ru(1)–C(2) |
1.88(1) |
| C(1)–O(1) |
1.125(10) |
C(2)–O(2) |
1.156(10) |
| S(1)–C(19) |
1.726(7) |
S(2)–C(20) |
1.726(8) |
| C(18)–C(19) |
1.44(1) |
C(19)–C(20) |
1.372(9) |
| C(20)–C(21) |
1.45(1) |
C(18)–N(4) |
1.125(10) |
| C(21)–N(5) |
1.122(8) |
|
|
| |
|
|
|
|
1b
|
| S(1)–Ru(1)–S(2) |
88.47(7) |
S(1)–Ru(1)–N(1) |
86.2(2) |
| S(1)–Ru(1)–N(2) |
86.2(2) |
S(1)–Ru(1)–C(1) |
177.6(2) |
| S(1)–Ru(1)–C(2) |
88.5(3) |
S(2)–Ru(1)–N(1) |
92.8(2) |
| S(2)–Ru(1)–N(2) |
168.7(2) |
S(2)–Ru(1)–C(1) |
89.2(2) |
| S(2)–Ru(1)–C(2) |
87.9(2) |
N(1)–Ru(1)–N(2) |
76.9(2) |
| N(1)–Ru(1)–C(1) |
94.6(3) |
N(1)–Ru(1)–C(2) |
174.6(3) |
| N(2)–Ru(1)–C(1) |
96.2(3) |
N(2)–Ru(1)–C(2) |
102.0(3) |
| C(1)–Ru(1)–C(2) |
90.8(4) |
Ru(1)–S(1)–C(19) |
100.7(3) |
| Ru(1)–S(2)–C(20) |
102.1(2) |
Ru(1)–C(1)–O(1) |
176.6(8) |
| Ru(1)–C(2)–O(2) |
177.5(8) |
N(4)–C(18)–C(19) |
178.5(9) |
| S(1)–C(19)–C(18) |
115.5(5) |
S(1)–C(19)–C(20) |
124.4(6) |
| C(18)–C(19)–C(20) |
120.0(6) |
S(2)–C(20)–C(19) |
124.3(5) |
| S(2)–C(20)–C(21) |
114.1(6) |
C(19)–C(20)–C(21) |
121.5(7) |
| N(5)–C(21)–C(20) |
174(1) |
|
|
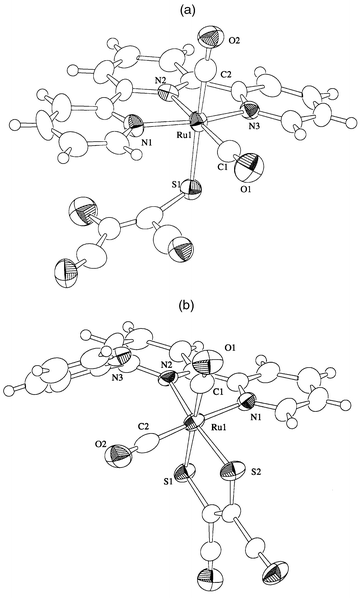 |
| | Fig. 1 Molecular structures of complexes 1a and 1b.
| |
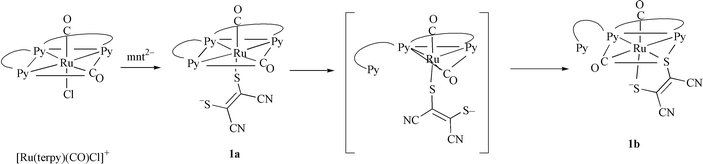 |
| | Scheme 2 | |
The molecular structure of complex 2b is shown in Fig. 2 and selected bond distances and angles are in Table 3. A significant difference in the bond distances and angles around the ruthenium atoms of 2b and 1b is not observed probably due to the same electronic structures of [RuII(CO)2(L2−)(terpy)] (L = mnt or tdt). The relatively long C–S bond distances of 2b compared with those of 1b is explained by the difference of the electron withdrawing ability between phenyl and two CN groups.
Table 3 Selected bond lengths (Å) and angles (°) for complex 2b
| Ru(1)–S(1) |
2.405(3) |
Ru(1)–S(2) |
2.362(3) |
| Ru(1)–N(1) |
2.148(7) |
Ru(1)–N(2) |
2.146(7) |
| Ru(1)–C(1) |
1.87(1) |
Ru(1)–C(2) |
1.847(10) |
| C(1)–O(1) |
1.16(1) |
C(2)–O(2) |
1.16(1) |
| S(1)–C(22) |
1.76(1) |
S(2)–C(21) |
1.77(1) |
| C(18)–C(19) |
1.50(2) |
C(19)–C(20) |
1.37(2) |
| C(20)–C(21) |
1.43(1) |
C(21)–C(22) |
1.39(1) |
| C(22)–C(23) |
1.39(1) |
C(23)–C(24) |
1.37(2) |
| C(24)–C(19) |
1.37(2) |
|
|
| |
|
|
|
| S(1)–Ru(1)–S(2) |
87.3(1) |
S(1)–Ru(1)–N(1) |
87.2(2) |
| S(1)–Ru(1)–N(2) |
86.5(2) |
S(1)–Ru(1)–C(1) |
175.7(3) |
| S(1)–Ru(1)–C(2) |
86.3(3) |
S(2)–Ru(1)–N(1) |
95.1(2) |
| S(2)–Ru(1)–N(2) |
169.9(2) |
S(2)–Ru(1)–C(1) |
88.7(3) |
| S(2)–Ru(1)–C(2) |
85.3(3) |
N(1)–Ru(1)–N(2) |
76.6(3) |
| N(1)–Ru(1)–C(1) |
94.8(4) |
N(1)–Ru(1)–C(2) |
173.5(4) |
| N(2)–Ru(1)–C(1) |
97.6(4) |
N(2)–Ru(1)–C(2) |
102.2(3) |
| C(1)–Ru(1)–C(2) |
91.7(5) |
Ru(1)–S(1)–C(22) |
102.8(4) |
| Ru(1)–S(2)–C(21) |
103.3(3) |
Ru(1)–C(1)–O(1) |
175(1) |
| Ru(1)–C(2)–O(2) |
174.9(9) |
S(1)–C(22)–C(21) |
122.5(7) |
| S(1)–C(22)–C(23) |
118.2(9) |
S(2)–C(21)–C(20) |
119.4(9) |
| S(2)–C(21)–C(22) |
122.7(7) |
C(19)–C(20)–C(21) |
117(1) |
| C(20)–C(19)–C(24) |
121(1) |
C(19)–C(24)–C(23) |
117(1) |
| C(22)–C(23)–C(24) |
122(1) |
|
|
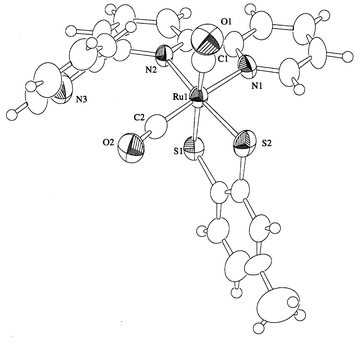 |
| | Fig. 2 Molecular structure of complex 2b.
| |
The molecular structure of complex 3 is shown in Fig. 3 and selected bond distances and angles are in Table 4. The distinct feature of 3 is that two methoxycarbonyl groups are linked to Ru and the terminal sulfur of monodentate pdt. Bond distances and angles around the Ru–C(O)OCH3 unit 3 (Ru(1)–C(2) 2.03(2), C(2)–O(2) 1.19(2), C(2)–O(3) 1.37(2) Å; C(2)–O(3)–C(3) 117(1), O(2)–C(2)–O(3) 116(1)°) are very similar to those previously reported for [Ru(CO)(C(O)OCH3)(bpy)2]+ (Ru–C 2.042(6), C–O 1.191(8), C–O 1.344(8) Å; C–O–C 116.4(6), O–C–O 119.2(6)°) which was prepared by reaction of CH3ONa with [Ru(CO)2(bpy)2]2+.19
Table 4 Selected bond lengths (Å) and angles (°) for complex 3
| Ru(1)–S(1) |
2.497(5) |
Ru(1)–N(1) |
2.05(2) |
| Ru(1)–N(2) |
2.03(1) |
Ru(1)–N(3) |
2.08(1) |
| Ru(1)–C(1) |
1.86(1) |
Ru(1)–C(2) |
2.03(2) |
| C(1)–O(1) |
1.15(2) |
C(2)–O(2) |
1.19(2) |
| C(2)–O(3) |
1.37(2) |
C(3)–O(3) |
1.38(2) |
| |
|
|
|
| Ru(1)–C(2)–O(2) |
128(1) |
Ru(1)–C(2)–O(3) |
115(1) |
| O(2)–C(2)–O(3) |
116(1) |
C(2)–O(3)–O(3) |
117(1) |
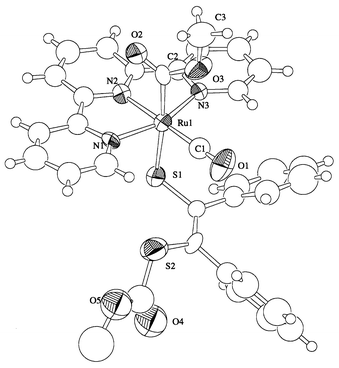 |
| | Fig. 3 Molecular structure of complex 3.
| |
Electrochemical properties
The redox behavior of the present complexes is summarized in Table 5. The cyclic voltammograms of 1b, 2b and 4 showed a couple of anodic and cathodic waves at E1/2 = +0.59, +0.02, and −0.17 V of the reversible 1b0/+ and 2b0/+ and 40/+ redox couples, respectively, in CH3CN. There is a large difference in the redox potentials of the two Ru–mnt complexes with CO (1b) and with dmso (4) (ΔE1/2 = 0.76 V). On the other hand, the redox potential of the 40/+ couple is close to that of a metal-centered RuII–RuIII redox reaction of the [Ru(mnt)(bpy)2]0/+ couple at E1/2 = −0.27 V.11 The one-electron redox reaction of 4, therefore, is reasonably assigned to the metal centered RuII–RuIII couple. The difference in the oxidation potentials of 1b, 2b and 4, therefore, is associated with the electron withdrawing ability of the dithiolene, carbonyl and dmso ligands of these complexes.
Table 5 Electrochemical data of [RuX(dithiolene)(terpy)]n+ (X = CO, dmso, Cl or OSO2CF3)a
| |
E
1/2/V vs. FeCp2+/FeCp2 |
| [complex]n+ |
(−/0) |
(+/0) |
|
E
1/2 = (Epa + Epc)/2, where Epa and Epc are anodic and cathodic peak potentials, respectively.
|
|
1b [Ru(CO)2(mnt)(terpy)] |
|
0.59 |
|
2b [Ru(CO)2(tdt)(terpy)] |
|
0.02 |
|
4 [Ru(dmso)(mnt)(terpy)] |
|
−0.17 |
|
5
+ [RuCl(tdt)(terpy)]BF4 |
−1.03 |
−0.13 |
|
6
+ [RuCl(pdt)(terpy)]ClO4 |
−1.30 |
−0.39 |
|
7
+ [Ru(OSO2CF3)(pdt)(terpy)]CF3SO3 |
−1.29 |
−0.39 |
In contrast to neutral complexes 1b, 2b and 4, the electronic structures of cationic 5+, 6+ and 7+ are denoted as [RuII(L0)X]+ (L = tdt or pdt; X = Cl or OSO2CF3). These complexes displayed two successive reversible cathodic processes in the range of −0.13 to −1.30 V in CH3CN as shown for 6+ (in Fig. 4). In addition, [RuCl(bpy)(terpy)]PF6 and [Ru(bpy)(terpy)(py)][PF6]2 do not undergo electrochemical reduction up to −1.5 V.22,23 The two successive reductions of 5+, 6+ and 7+ are associated with the ligand centered [RuII(L0)X]+–[RuII(L−)X]0 and [RuII(L−)X]0–[RuII(L2−)X]− redox couples.
![Cyclic voltammogram of [RuCl(pdt)(terpy)]+6+ in a CH2Cl2 solution of NBun4BF4 (0.1 M) at a glassy carbon electrode with a scan rate of 50 mV s−1.](/image/article/2001/DT/b007541h/b007541h-f4.gif) |
| | Fig. 4 Cyclic voltammogram of [RuCl(pdt)(terpy)]+6+ in a CH2Cl2 solution of NBun4BF4 (0.1 M) at a glassy carbon electrode with a scan rate of 50 mV s−1.
| |
The pdt localized redox reactions are hardly affected by the difference between Cl− and OSO2CF3− (6ClO4 and 7ClO4). On the other hand, the redox potentials of the 6+/0 and 60/− couples are observed at values more negative than those of 50/+ and 50/− (ΔE1/2 = 260–270 mV), indicating that the electron density (basicity) of pdt is higher than that of tdt. Strong basicity of pdt compared with tdt and mnt is associated with the unusual double addition of CO2 and CH3OH to the sulfur of pdt and carbon of CO of 3a, respectively, producing 3. It is noteworthy that a CO ligand of dicationic [Ru(bpy)2(CO)2]2+ undergoes nucleophilic attack by CH3ONa but not by CH3OH.
Electronic spectra
The neutral dicarbonyl complexes 1b and 2b, and dmso complex 4, have ruthenium to terpy π* transitions at 390, 395, and 442 nm, respectively. These complexes possess the most reduced form of the dithiolene ligands and do not have characteristic strong absorption bands in the visible region. Controlled potential electrolysis of 2b at +0.2 V in acetone resulted in a new absorption band at 569 nm with decreasing intensity of the band at 395 nm in the visible region. Similarly, 4+ prepared by controlled potential electrolysis of 4 at +0.1 V in CH3CN displays a strong new band at 693 nm in the visible region, and the band at 442 nm of 4 decreased with increasing intensity of the band at 399 nm (Fig. 5). The new bands at 569 and 693 nm of 2b+ and 4+, respectively, are assigned to charge transfer (CT) within the ruthenium–dithiolene unit.
![Absorption spectra of [Ru(dmso)(mnt)(terpy)] 4 (dashed line) and [Ru(dmso)(mnt)(terpy)]+4+ (solid line) in CH3CN.](/image/article/2001/DT/b007541h/b007541h-f5.gif) |
| | Fig. 5 Absorption spectra of [Ru(dmso)(mnt)(terpy)] 4 (dashed line) and [Ru(dmso)(mnt)(terpy)]+4+ (solid line) in CH3CN.
| |
The electronic spectra of complex 6+ and the one-electron reduced 6 obtained by controlled potential electrolysis of the former at −0.7 V in CH2Cl2 are given in Fig. 6. Attempts to measure the electronic spectra of 5−, 6−, and 7− under controlled potential electrolysis of 5+, 6+ and 7+ at −1.40 V in CH2Cl2 were not successful due to the lability of these highly reduced complexes. The oxidized form 6+ has a ruthenium to terpy π* transition near 400 nm and a strong absorption band at 566 nm. The latter is the most characteristic feature and assigned to ruthenium to dithiolene, pdt, MLCT transition because the π* level of pdt is lower than that of terpy, since dithiolene ligands are reduced more easily than terpy. One electron reduction of 6+ to generate 6 brought about strong new bands at 500 and 720 nm with decreasing inensity of the 566 nm band. This red shift is an indication of an increase in the electron density of the complex. Similarly, the triflate complex 7+ showed a strong absorption band at 568 nm, which shifted to 720 nm upon one-electron reduction at −0.7 V in CH2Cl2. The similarity of electrochemical and spectroscopic properties between 6n+ and 7n+ is an indication of the similar basicity of Cl− and OSO2CF3− as monoanionic ligands. In the case of 5+ the ruthenium(II) to tdt π* MLCT band was observed at 527 nm. The blue shift of the band by 39 nm compared with that of 6+ (λmax 566 nm) is explained by the high electron density of pdt in 6+ compared with tdt in 5+. In fact the tdt moiety in 5 is reduced more easily than the pdt one in 6. When 5 was formed by controlled potential electrolysis of 5+ at −0.2 V the strong band at 527 nm decreased, and a new band appeared at 720 nm as found in the spectrum of 6.
![Absorption spectra of [RuCl(pdt)(terpy)]+6+ (solid line) and [RuCl(pdt)(terpy)] 6 (dashed line) in CH2C2.](/image/article/2001/DT/b007541h/b007541h-f6.gif) |
| | Fig. 6 Absorption spectra of [RuCl(pdt)(terpy)]+6+ (solid line) and [RuCl(pdt)(terpy)] 6 (dashed line) in CH2C2.
| |
References
- C. G. Pierpont and C. W. Lange, Prog. Inorg. Chem., 1994, 41, 331 Search PubMed; C. G. Pierpont and R. M. Buchanan, Coord. Chem. Rev., 1981, 38, 35 CrossRef; A. B. P. Lever, P. R. Auburn, E. S. Dodsworth, M. Haga, W. Liu, M. Melnik and A. Nevin, J. Am. Chem. Soc., 1988, 110, 8706 CrossRef; M. Haga, E. S. Dodsworth and A. B. P. Lever, Inorg. Chem., 1986, 25, 447 CrossRef CAS; H. Masui, A. B. P. Lever and P. R. Auburn, Inorg. Chem., 1991, 30, 2402 CrossRef CAS; C. J. da Cunha, S. S. Fielder, D. V. Stynes, H. Masui, P. R. Auburn and A. B. P. Lever, Inorg. Chim. Acta, 1996, 242, 293 CrossRef; M. Kurihara, S. Daniele, K. Tsuge, H. Sugimoto and K. Tanaka, Bull. Chem. Soc. Jpn., 1998, 71, 867 CAS.
- W. M. Rorer, Jr., K. J. Takeuchi and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 5817 CrossRef.
- H. Nagao, T. Mizukawa and K. Tanaka, Inorg. Chem., 1994, 33, 3415 CrossRef CAS; T. Mizukawa, K. Tsuge and K. Tanaka, Angew. Chem., Int. Ed., 1999, 38, 362 CrossRef CAS.
- O. Ishitani, P. S. White and T. J. Meyer, Inorg. Chem., 1996, 35, 2167 CrossRef CAS.
- T. J. Meyer, Acc. Chem. Res., 1989, 22, 163 CrossRef CAS; T. Wada, K. Tsuge and K. Tanaka, Angew. Chem., 2000, 39, 1479 CrossRef CAS.
- J. A. McCleverty, Prog. Inorg. Chem., 1969, 10, 49 Search PubMed.
- H. Oku, N. Ueyama and A. Nakamura, Inorg. Chem., 1995, 34, 3667 CrossRef CAS.
- E. S. Davies, R. L. Beddoes, D. Collison, A. Dinsmore, A. Docrat, J. A. Joule, C. R. Wilson and C. D. Garner, J. Chem. Soc., Dalton Trans., 1997, 3985 RSC.
- S. Boyde, C. D. Garner, J. A. Joule and J. A. Rowe, J. Chem. Soc., Chem. Commun., 1987, 800 RSC.
- A. Davison and R. H. Holm, Inorg. Synth., 1967, 10, 8 CAS.
- M. A. Greaney, C. L. Coyle, M. A. Harmer, A. Jordan and E. I. Stiefel, Inorg. Chem., 1989, 28, 912 CrossRef CAS.
- A. K. Bhattacharya and A. G. Hortmann, J. Org. Chem., 1974, 39, 95 CrossRef CAS.
- T. Norrby, A. Borje, B. Akermark, L. Hammarstrom, J. Alsins, K. Lashgari, R. Norrestam, J. Martensson and G. Stenhagen, Inorg. Chem., 1997, 36, 5850 CrossRef CAS.
- D. H. Gibson, B. A. Sleadd, M. S. Mashuta and J. F. Richardson, Organometallics, 1997, 6, 4421 CrossRef CAS.
-
TEXSAN, Single Crystal Analysis Software, Molecular Structure Corporation, Houston, TX, 1995.
Search PubMed.
- M. Kato and T. Ito, Inorg. Chem., 1985, 24, 504 CrossRef CAS.
- N. Kitajima, S. Hikichi, M. Tanaka and Y. Morooka, J. Am. Chem. Soc., 1993, 115, 5496 CrossRef CAS.
- K. Tanaka, Y. Kushi, K. Tsuge, K. Toyohara, T. Nishioka and K. Isobe, Inorg. Chem., 1998, 37, 120 CrossRef CAS.
- H. Tanaka, B.-C. Tzeng, H. Nagao, S.-M. Peng and K. Tanaka, Inorg. Chem., 1993, 32, 1508 CrossRef CAS.
- C. Mahadevan, M. Seshasayee and B. V. R. Murthy, Acta Crystallogr., Sect. C, 1983, 39, 1335 CrossRef.
- E. I. Stiefel, Z. Dori and H. B. Gray, J. Am. Chem. Soc., 1967, 89, 3353 CrossRef CAS.
- T. B. Hadda and H. L. Bozec, Inorg. Chim. Acta, 1993, 204, 103 CrossRef.
- S. C. Rasmussen, S. E. Ronco, D. A. Mlsna, M. A. Billadeau, W. T. Pennington, J. W. Kolis and J. D. Petersen, Inorg. Chem., 1995, 34, 821 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2001 |
Click here to see how this site uses Cookies. View our privacy policy here.