Trinuclear CuIIMIICuII complexes of an oxamide/dioxime ligand and extension to a bimetallic magnetic compound
Nobuo
Fukita
a,
Masaaki
Ohba
a,
Takuya
Shiga
a,
Hisashi
Ōkawa
*a and
Yoshitami
Ajiro
b
aDepartment of Chemistry, Faculty of Sciences, Kyushu University, Hakozaki 6-10-1, Higashiku, Fukuoka, 812-8581, Japan
bDepartment of Physics, Faculty of Sciences, Kyushu University, Hakozaki 6-10-1, Higashiku, Fukuoka, 812-8581, Japan
First published on 7th December 2000
Abstract
N,N'-Bis[2-(hydroxyiminomethyl)phenyl]oxamide (H4L) provided trinuclear CuIIMIICuII complexes [M{Cu(HL)(DMF![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) )}2(DMF)2] (MII = Mn 1, Co 2, Ni 3 or Zn 4). The crystal structures of 1–4 have been determined by X-ray crystallography. They are isomorphous and have an oxamidate-bridged trinuclear CuIIMIICuII structure. The CuII resides in a pseudo-macrocyclic framework of (HL)3− comprised of an oxamidate and a hydrogen-bonded dioximate (
)}2(DMF)2] (MII = Mn 1, Co 2, Ni 3 or Zn 4). The crystal structures of 1–4 have been determined by X-ray crystallography. They are isomorphous and have an oxamidate-bridged trinuclear CuIIMIICuII structure. The CuII resides in a pseudo-macrocyclic framework of (HL)3− comprised of an oxamidate and a hydrogen-bonded dioximate (![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) N–O
N–O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ⋯H⋯O–N) groups to form a square-pyramidal structure {Cu(HL)(DMF)} together with a DMF molecule. Two {Cu(HL)(DMF)} entities co-ordinate to a MnII through the oxamidate oxygens to afford a cis octahedral environment about the metal together with two DMF oxygens. The CuII⋯MII intermetallic distance separated by the oxamidate bridge is 5.33–5.49 Å. In the case of 1 and 3 a significant antiferromagnetic interaction operates between the adjacent CuII and MII. The reaction of 1 with MnII in acetonitrile in the presence of KOH and 18-crown-6 formed Mn{Cu(L)}(H2O)4 that has a polymeric structure extended by the dioximate–MnII–dioximate linkage. It is a weak ferromagnet (TC = 5.5 K) exhibiting a weak antiferromagnetic interaction between the ferrimagnetic chains.
⋯H⋯O–N) groups to form a square-pyramidal structure {Cu(HL)(DMF)} together with a DMF molecule. Two {Cu(HL)(DMF)} entities co-ordinate to a MnII through the oxamidate oxygens to afford a cis octahedral environment about the metal together with two DMF oxygens. The CuII⋯MII intermetallic distance separated by the oxamidate bridge is 5.33–5.49 Å. In the case of 1 and 3 a significant antiferromagnetic interaction operates between the adjacent CuII and MII. The reaction of 1 with MnII in acetonitrile in the presence of KOH and 18-crown-6 formed Mn{Cu(L)}(H2O)4 that has a polymeric structure extended by the dioximate–MnII–dioximate linkage. It is a weak ferromagnet (TC = 5.5 K) exhibiting a weak antiferromagnetic interaction between the ferrimagnetic chains.
Introduction
The design of molecular-based magnetic materials is a current subject of many studies.1–4 The use of paramagnetic metal complexes as the constituents has great advantages over organic radical constituents because electron spin can be reserved to each metal center, 1-D to 3-D network structures can be constructed based on versatile stereochemistries of metal complexes, and the magnetic nature of complex-based magnets can be tuned by choice of metal ion and bridging group.5,6 For constructing complex-based network structures, ‘complex bridges’ are generally used that have two or more sites capable of acting as bridges to outer metal ions.The bridging function of oxamidate groups is well known.7–12 One of the present authors used N,N-bis(3-aminopropyl)oxamidatocopper(II) as a ‘complex ligand’ to obtain triangular tetranuclear MIICuII3 complexes with the MII at the center and CuII at the corners of the triangle.7 These and related complexes9 showed a significant antiferromagnetic interaction between the adjacent CuII and MII through the oxamidate bridge in cis arrangement. Oxamidate bridges in trans arrangement are similarly good magnetic mediators and have been used for providing bimetallic magnetic materials.9–12 The oximate group (N–O−) is another magnetic mediator between metal ions.13–22 In a trinuclear copper(II) complex derived from bis(dimethylglyoximato)cuprate(II) complete spin coupling occurs at room temperature through the dioximate bridge in cis arrangement.18 Significant antiferromagnetic interaction between dissimilar ions of CuII and MII through a dioximate bridge has been reported.19,22
In the context mentioned above, N,N'-bis[2-(hydroxyiminomethyl)phenyl]oxamide (Fig. 1), abbreviated as H4L, has been prepared in this work. Its mononuclear copper(II) complex [Cu(HL)]− is expected to act as a ‘complex bridge’ with its oxamidate and dioximate termini. Trinuclear CuIIMIICuII complexes [M{Cu(HL)(DMF)}2(DMF)2] (MII = Mn 1, Co 2, Ni 3 or Zn 4) have been prepared and characterized by X-ray crystallography and cryomagnetic studies. An extension of 1 into a polymeric compound Mn{Cu(L)}(H2O)4 exhibiting a magnetic phase transition at Tc = 5.5 K is reported.
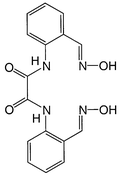 | ||
| Fig. 1 Chemical structure of H4L. | ||
Experimental
Physical measurements
Elemental analyses of C, H and N were obtained at the Service Center of Elemental Analysis of Kyushu University, metal analyses using a Shimadzu A-A 680 Atomic Absorption/Flame Emission Spectrophotometer. Infrared spectra were measured on KBr disks with a Perkin-Elmer Spectrum BX FT-IR system, Reflectance spectra on powdered samples with a Shimadzu UV-3100PC spectrophotometer using an integrating sphere attachment. Magnetic susceptibilities were measured using a Quantum Design MPMS 5XL or MPMS2 SQUID susceptometer in the temperature range 2–300 K. Calibrations were made with the standard Pd. Data were corrected for magnetization of the sample holder and capsule used. Diamagnetic corrections were made using Pascal’s constants.23 Magnetization studies were carried out with the same apparatus.Preparations
N,N'-Bis(2-formylphenyl)oxamide was prepared by the literature method.24 All chemicals were of reagent grade used as purchased.
'-Bis(2-formylphenyl)oxamide (2.96 g, 10 mmol) and hydroxylammonium chloride (1.39 g, 20 mmol) were added to ethanol–water (1∶1 in volume, 500 ml) and the mixture was refluxed for 3 hours. N,N'-Bis(2-formylphenyl)oxamide itself was insoluble in the mixed solvent but gradually dissolved to form a clear yellow solution. It was evaporated to dryness, and the resulting yellow residue treated with a saturated NaHCO3 solution (300 ml) and washed with water. Crystallization from hot ethanol gave pale yellow needles melting at 284 °C. The yield was 2.31 g (71%). Calc. for C8H7N2O2: C, 58.89; H, 4.32; N, 17.17%. Found: C, 58.91; H, 4.37; N, 17.12%. 1H NMR [d6-DMF]: δ 7.32 (t(2H), ring proton), 7.51(t(2H), ring proton), 7.61(d(4H), ring proton), 8.45(s(2H), ArCHN), 8.68(d(2H), ArNHC) and 12.46(s(2H), NOH). Selected FT-IR using KBr: ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) /cm−1 3540, 3272, 1691, 1578, 1315, 1293, 1007 and 750.
/cm−1 3540, 3272, 1691, 1578, 1315, 1293, 1007 and 750.
![[hair space]](https://www.rsc.org/images/entities/b_char_200a.gif) )}2(DMF)2] 1.
H4L (163 mg, 0.5 mmol) was dissolved in DMF (N,N-dimethylformamide) (50 ml) and an aqueous solution of NaOH (80 mg, 2 mmol) added. A DMF solution of copper(II) acetate monohydrate (100 mg, 0.5 mmol) was added dropwise and the mixture stirred for 30 minutes. A DMF solution of manganese(II) acetate tetrahydrate (61 mg, 0.25 mmol) was then added, and the mixture diffused with diethyl ether to form dark green crystals. The yield was 182 mg (65%). Calc. for C44H50Cu2MnN12O12: C, 47.14; H, 4.50; Cu, 11.34; Mn, 4.9; N, 14.99%. Found: C, 47.06; H, 4.63; Cu, 10.90; Mn, 4.6; N, 14.62%. Selected FT-IR using KBr: /cm−1 1664, 1639, 1603, 1569, 1558, 1337, 920 and 756. Visible spectrum on powdered sample: λ/nm 525 and 600.
)}2(DMF)2] 2.
This complex was obtained as dark green crystals similarly to 1, using cobalt(II) acetate tetrahydrate. The yield was 196 mg (70%). Calc. for C44H50CoCu2N12O12: C, 46.98; H, 4.48; Co, 5.2; Cu, 11.30; N, 14.94%. Found: C, 46.71; H, 4.65; Co, 5.0; Cu, 10.83; N, 14.62%. Selected FT-IR using KBr: /cm−1 2928, 1664, 1640, 1557, 1336, 920 and 756. Visible spectrum on powdered sample: λ/nm 515 and 600.
)}2(DMF)2] 3.
This complex was obtained as dark green crystals similarly to 1, using nickel(II) acetate tetrahydrate. The yield was 168 mg (60%). Calc. for C44H50Cu2N12NiO12: C, 46.99; H, 4.48; Cu, 11.30; Ni, 5.2; N, 14.94%. Found: C, 47.07; H, 4.71; Cu, 11.03; Ni, 5.5; N, 14.41%. Selected FT-IR using KBr: /cm−1 2928, 1663, 1636, 1555, 1341, 920 and 756. Visible spectrum on powdered sample: λ/nm 515, 600 and 965.
)}2(DMF)2] 4.
This complex was obtained as dark green crystals similarly to 1, using zinc(II) acetate tetrahydrate. The yield was 170 mg (60%). Calc. for C44H50Cu2N12O12Zn: C, 46.71; H, 4.45; Cu, 11.23; N, 14.86; Zn, 5.8%. Found: C, 46.80; H, 4.45; Cu, 11.43; N, 14.86; Zn, 6.3%. Selected FT-IR using KBr: /cm−1 2928, 1663, 1637, 1604, 1557, 1339, 920 and 756. Visible spectrum on powdered sample: λ/nm 515 and 600.
/cm−1 1615, 1562, 1333, 916 and 754. Visible spectrum on powdered sample: λ/nm 500 and 590.
)}2(DMF)2] 1.
H4L (163 mg, 0.5 mmol) was dissolved in DMF (N,N-dimethylformamide) (50 ml) and an aqueous solution of NaOH (80 mg, 2 mmol) added. A DMF solution of copper(II) acetate monohydrate (100 mg, 0.5 mmol) was added dropwise and the mixture stirred for 30 minutes. A DMF solution of manganese(II) acetate tetrahydrate (61 mg, 0.25 mmol) was then added, and the mixture diffused with diethyl ether to form dark green crystals. The yield was 182 mg (65%). Calc. for C44H50Cu2MnN12O12: C, 47.14; H, 4.50; Cu, 11.34; Mn, 4.9; N, 14.99%. Found: C, 47.06; H, 4.63; Cu, 10.90; Mn, 4.6; N, 14.62%. Selected FT-IR using KBr: /cm−1 1664, 1639, 1603, 1569, 1558, 1337, 920 and 756. Visible spectrum on powdered sample: λ/nm 525 and 600.
)}2(DMF)2] 2.
This complex was obtained as dark green crystals similarly to 1, using cobalt(II) acetate tetrahydrate. The yield was 196 mg (70%). Calc. for C44H50CoCu2N12O12: C, 46.98; H, 4.48; Co, 5.2; Cu, 11.30; N, 14.94%. Found: C, 46.71; H, 4.65; Co, 5.0; Cu, 10.83; N, 14.62%. Selected FT-IR using KBr: /cm−1 2928, 1664, 1640, 1557, 1336, 920 and 756. Visible spectrum on powdered sample: λ/nm 515 and 600.
)}2(DMF)2] 3.
This complex was obtained as dark green crystals similarly to 1, using nickel(II) acetate tetrahydrate. The yield was 168 mg (60%). Calc. for C44H50Cu2N12NiO12: C, 46.99; H, 4.48; Cu, 11.30; Ni, 5.2; N, 14.94%. Found: C, 47.07; H, 4.71; Cu, 11.03; Ni, 5.5; N, 14.41%. Selected FT-IR using KBr: /cm−1 2928, 1663, 1636, 1555, 1341, 920 and 756. Visible spectrum on powdered sample: λ/nm 515, 600 and 965.
)}2(DMF)2] 4.
This complex was obtained as dark green crystals similarly to 1, using zinc(II) acetate tetrahydrate. The yield was 170 mg (60%). Calc. for C44H50Cu2N12O12Zn: C, 46.71; H, 4.45; Cu, 11.23; N, 14.86; Zn, 5.8%. Found: C, 46.80; H, 4.45; Cu, 11.43; N, 14.86; Zn, 6.3%. Selected FT-IR using KBr: /cm−1 2928, 1663, 1637, 1604, 1557, 1339, 920 and 756. Visible spectrum on powdered sample: λ/nm 515 and 600.
/cm−1 1615, 1562, 1333, 916 and 754. Visible spectrum on powdered sample: λ/nm 500 and 590.
Crystal structural analyses
Each single crystal of complexes 1–4 was mounted on a glass fiber and coated with epoxy resin. Intensities and lattice parameters were obtained on a Rigaku AFC-5S automated four-circle diffractometer with graphite-monochromated Mo-Kα radiation (λ = 0.71069 Å) for 2 and a Rigaku AFC-7R automated four-circle diffractometer with graphite-monochromated MoKα radiation (λ = 0.71069 Å) and 12 kW rotating anode generator for 1, 3 and 4. Cell constants and the orientation matrix for the data collection were obtained from 25 reflections and the ω–2θ scan mode was used for the intensity collections at 23 ± 1 °C. Pertinent crystallographic parameters are summarized in Table 1.| 1 | 2 | 3 | 4 | |
|---|---|---|---|---|
| Formula | C44H50Cu2MnN12O12 | C44H50CoCu2N12O12 | C44H50Cu2N12NiO12 | C44H50Cu2N12O12Zn |
| Formula weight | 1120.98 | 1124.98 | 1124.74 | 1131.42 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | C2/c (no. 15) | C2/c (no. 15) | C2/c (no. 15) | C2/c (no. 15) |
| a/Å | 24.904(4) | 24.903(5) | 24.86(1) | 24.910(4) |
| b/Å | 10.229(2) | 10.222(4) | 10.279(3) | 10.245(3) |
| c/Å | 19.581(2) | 19.569(6) | 19.373(5) | 19.459(3) |
| β/° | 103.93(1) | 103.87(2) | 104.86(3) | 104.72(1) |
| V/Å3 | 4841(1) | 4836(2) | 4784(2) | 4802(1) |
| Z | 4 | 4 | 4 | 4 |
| μ(Mo-Kα)/cm−1 | 12.12 | 12.85 | 13.45 | 14.48 |
| No. observations (I > 3.00σ(I)) |
2996 | 2831 | 1537 | 2082 |
| R | 0.043 | 0.049 | 0.063 | 0.049 |
| Rw | 0.051 | 0.058 | 0.068 | 0.050 |
Three standard reflections were monitored every 150 measurements. A linear correction factor was applied to the data to account for decay. Intensity data were corrected for Lorentz and polarization effects.
The structures were solved by the direct method and expanded using Fourier techniques. Non-hydrogen atoms were refined anisotropically. Hydrogen atoms were included in the structure analysis but not refined. Computation were carried out on an IRIS O2 computer using TEXSAN.25
CCDC reference number 186/2228.
See http://www.rsc.org/suppdata/dt/b0/b006613n/ for crystallographic files in .cif format.
Results and discussion
Synthesis and general properties
In spite of many efforts, all attempts to isolate a mononuclear copper(II) precursor complex of H4L were in vain. Thus, this was prepared in solution and treated with a second metal(II) ion to provide the trinuclear complexes [M{Cu(HL)(DMF)}2(DMF)2] 1–4. They show complicated IR bands in the region 2300–3000 cm−1, which are characteristic of the hydrogen-bonded –O–H⋯O– linkage of the dioximate group.26,27 The ν(CO) band of the oxamidate group is seen around 1635–1640 cm−1, which is low relative to that of H4L (1691 cm−1). This is in accord with the bridging function of the group as discussed below. An IR band around 1663 cm−1 is assigned to the ν(CO) mode of the co-ordinating DMF.
The reflectance spectrum of compound 1 (CuIIMnIICuII) shows two visible bands at 525 and 600 nm which are attributed to the d–d components of CuII: MnII in a high-spin state has no spin-allowed d–d band. Compound 4 (CuIIZnIICuII) also shows two visible bands at 515 and 600 nm. A similar visible spectrum was obtained for 2 (CuIICoIICuII). It is considered that the d–d bands of octahedral CoII are weak and concealed by those of CuII. In the case of 3 (CuIINiIICuII) an additional band is observed at 965 nm that is assigned to a d–d component of NiII.
Crystal structures
Complexes 1–4 are isostructural. An ORTEP28 drawing of 1 with the atom numbering scheme is given in Fig. 2. Selected bond distances and angles for 1–4 are summarized in Table 2.
| M = Mn (1) | Co (2) | Ni (3) | Zn (4) | |
|---|---|---|---|---|
| Symmetry operation: (*) −x, y, ½ − z. | ||||
| Cu–N(1) | 1.991(4) | 1.988(4) | 1.99(1) | 1.991(6) |
| Cu–N(2) | 1.998(4) | 1.999(4) | 1.99(1) | 2.009(6) |
| Cu–N(3) | 2.014(4) | 2.007(5) | 2.01(1) | 2.012(7) |
| Cu–N(4) | 1.976(4) | 1.989(5) | 1.99(1) | 1.987(6) |
| Cu–O(5) | 2.378(4) | 2.375(4) | 2.38(1) | 2.376(6) |
| M–O(1) | 2.174(4) | 2.173(4) | 2.052(9) | 2.103(5) |
| M–O(2) | 2.167(3) | 2.163(3) | 2.022(9) | 2.064(5) |
| M–O(6) | 2.159(4) | 2.159(5) | 2.04(1) | 2.089(6) |
| N(1)–Cu–N(2) | 84.2(1) | 83.8(2) | 84.0(5) | 84.0(2) |
| N(1)–Cu–N(3) | 89.0(2) | 89.0(2) | 89.5(5) | 89.2(3) |
| N(1)–Cu–N(4) | 172.7(1) | 172.7(2) | 171.0(5) | 171.7(3) |
| N(2)–Cu–N(3) | 164.1(1) | 164.1(2) | 164.6(4) | 164.7(3) |
| N(2)–Cu–N(4) | 90.7(1) | 91.1(2) | 90.4(5) | 90.3(3) |
| N(3)–Cu–N(4) | 94.6(2) | 94.7(2) | 94.1(5) | 94.9(3) |
| O(1)–M–O(1)* | 93.6(2) | 93.5(2) | 94.4(5) | 94.5(3) |
| O(1)–M–O(2) | 74.3(1) | 74.5(1) | 79.7(3) | 78.1(2) |
| O(1)–M–O(2)* | 97.7(1) | 97.7(1) | 96.0(3) | 95.8(2) |
| O(1)–M–O(6) | 91.3(1) | 91.2(2) | 90.8(4) | 90.7(2) |
| O(1)–M–O(6)* | 163.8(1) | 163.8(2) | 170.8(4) | 168.4(2) |
| O(2)–M–O(2)* | 168.6(2) | 168.8(2) | 173.6(6) | 171.2(3) |
| O(2)–M–O(6) | 98.5(1) | 98.4(2) | 92.3(4) | 95.4(2) |
| O(2)–M–O(6)* | 89.7(1) | 89.6(2) | 92.4(4) | 91.1(2) |
| O(6)–M–O(6)* | 88.3(2) | 88.5(3) | 85.1(7) | 86.1(4) |
![An ORTEP view of [Mn{Cu(HL)(DMF )}2(DMF )2] 1 with the atom numbering scheme.](/image/article/2001/DT/b006613n/b006613n-f2.gif) | ||
| Fig. 2 An ORTEP view of [Mn{Cu(HL)(DMF)}2(DMF)2] 1 with the atom numbering scheme.
| ||
The asymmetric unit consists of half of the [Mn{Cu(HL)(DMF)}2(DMF)2]: the Mn exists on the mirror plane. The Cu resides in the N4 cavity of the ligand with two oxamidate nitrogens and two oxime nitrogens. The geometry about the Cu is square pyramidal with N(1), N(2), N(3) and N(4) of (HL)3− on the basal plane and O(5) of DMF at the apex. The Cu–N bond distances range from 1.976(4) to 2.014(4) Å. The axial Cu–O(5) bond distance is 2.378(4) Å which is elongated due to the Jahn–Teller effect of the d9 electronic configuration. The Cu is displaced by 0.179 Å from the basal least-squares plane toward O(5). One proton of the oxime group is deprotonated to form a N–O–H⋯O–N hydrogen bond. The N(3)–O(3)–H(6), N(4)–O(4)–H(6) and O(3)–H(6)–O(4) angles are 98.4, 102.0 and 156.9°, respectively. The O(3)⋯O(4) separation is short (2.381(6) Å), suggesting that the hydrogen bond is very strong. The oxamidatomanganese entity forms a good coplane, but the [Cu(HL)]− molecule is not coplanar. The least-squares plane defined by Cu, N(1), N(2), N(3) and N(4) and that defined by N(1), N(2), C(15), C(16), O(1), O(2) and Mn are bent at the N(1)⋯N(2) edge with a dihedral angle of 21.02° (see Fig. 2). The MnII has a cis-octahedral geometry with four oxamidate oxygens, O(1), O(2), O(1)* and O(2)*, and two DMF oxygens, O(6) and O(6)* (* indicates the symmetry operation of −x, y, ½ − z). The Mn–O bond distances range from 2.159(4) to 2.174(4) Å. The CuII⋯MnII separation is 5.491(1) Å.
The {Cu(HL)}− parts in complexes 1–4 are essentially similar. Some noticeable differences in the structures are seen in the geometry around the MII. The average M–O bond distance decreases in the order: 1 (2.167) ≈ 2 (2.165) > 4 (2.085) > 3 (2.038 Å). Notably, 1 and 2 have the same M–O bond distance in spite of the markedly differing ionic radius between MnII (0.97) and CoII (0.89 Å).29 The M–O bond distance of 2–4 decreases with decreasing ionic radius of the MII. The M⋯Cu intermetallic distances separated by the oxamidate bridge are in a similar order: 1 (5.491(1)) ≈ 2 (5.490(2)) > 4 (5.385(1)) > 3 (5.334(3) Å).
Magnetic properties
The room-temperature magnetic moment of complex 1 is 6.20 μB (per molecule) that is slightly smaller than the spin-only value (6.40 μB) expected for two CuII (S = 1/2) and one MnII (S = 5/2). The effective magnetic moment decreased with decreasing temperature to a near plateau value of 3.90 μB below 20 K (Fig. 3). The plateau moment is close to the spin-only value for ST = 3/2 (3.87 μB) resulting from the antiferromagnetic spin coupling in the CuII–MnII–CuII system. The interaction between the terminal CuII can be ignored because complex 4 (CuZnCu) shows little temperature dependence of magnetic moment (1.78 μB at 300 K and 1.76 μB at 10 K). Based on the spin Hamiltonian H = −2JSMn(SCu1 + SCu2), the magnetic susceptibility expression for the CuII–MnII–CuII system is given by eqn. (1),30 where N is Avogadro’s number, g the Lande g factor, β the Bohr magneton, k the Boltzmann constant, J the exchange integral, T the absolute temperature, θ the Weiss constant and Nα the temperature-independent paramagnetism. The same g factor is applied for four different spin states, (S = 7/2, S' = 1), (S = 5/2, S' = 1), (S = 3/2, S' = 1) and (S = 5/2, S' = 0), defined by S = SMn + SCu1 + SCu2 and S' = SCu1 + SCu2. A good magnetic simulation is obtained by eqn. (1), using the best-fit parameters J = −14 cm−1, g = 2.07, and Nα = 120 × 10−6 cm3 mol−1 and θ = −0.1 K (see Fig. 3). The discrepancy factor defined as R(χ) = [∑(χobs − χcalc)2/∑(χobs)2]1/2 was 2.99 × 10−3.|
χ
m = {Ng2β2/4k(T − θ)}[84exp(5J/kT) + 35exp(−2J/kT) + 10exp(−7J/kT) + 35]/[4exp(5J/kT) + 3exp(−2J/kT) + 2exp(−7J/kT) + 3] + Nα
| (1) |
![χ
m
vs.
T and μeffvs.T plots for [Mn{Cu(HL)(DMF )}2(DMF )2] 1.](/image/article/2001/DT/b006613n/b006613n-f3.gif) | ||
| Fig. 3
χ
m
vs.
T and μeffvs.T plots for [Mn{Cu(HL)(DMF)}2(DMF)2] 1.
| ||
Oxamato-bridged CuIIMnIICuII complexes with a similar trinuclear core structure are known.31 It is noted that their exchange integrals (−14.7 to −16.9 cm−1) are comparable to that observed for 1 (J = −14 cm−1). Comparable exchange integrals (−11.7 to −18.3 cm−1) have also been reported for other complexes with CuII and MnII combined by oxamido or oxamato bridges.10,32,33
The μeffvs.T curve of complex 2 is given in Fig. 4. The effective magnetic moment at room temperature is 5.78 μB that is larger than the spin-only value (4.24 μB) expected for two CuII (S = 1/2) and one CoII (S = 3/2). The magnetic moment slightly increased with decreasing temperature to a maximum of 5.88 μB near 245 K and then continuously decreased to 1.52 μB at 2 K. Such magnetic behavior is probably due to a large orbital contribution arising from the 4T1g ground term of CoII.34 An antiferromagnetic interaction may operate between the adjacent CuII and CoII, but the exchange integral could not be evaluated because of the orbital contribution from the CoII.
![χ
m
vs.
T plots for [Co{Cu(HL)(DMF )}2(DMF )2] 2 and χmvs.T and μeffvs.T plots for [Ni{Cu(HL)(DMF )}2(DMF )2] 3.](/image/article/2001/DT/b006613n/b006613n-f4.gif) | ||
| Fig. 4
χ
m
vs.
T plots for [Co{Cu(HL)(DMF)}2(DMF)2] 2 and χmvs.T and μeffvs.T plots for [Ni{Cu(HL)(DMF)}2(DMF)2] 3.
| ||
The μeffvs.T and χmvs.T curves of complex 3 are shown in Fig. 4. The effective magnetic moment at room temperature is 3.38 μB, which is small relative to the spin-only value (3.74 μB) expected for two CuII (S = 1/2) and one NiII (S = 1). The magnetic moment decreased with decreasing temperature to 0.82 μB at 2 K. The magnetic behavior indicates significant antiferromagnetic interaction between the adjacent CuII and NiII through the oxamidate bridge. The magnetic susceptibility expression for the CuII–NiII–CuII system is given by eqn. (2),30 based on the Heisenberg model H = −2JSNi(SCu1 + SCu2). Magnetic simulations with this equation gave a poor fitting in the low temperature region below 60 K. A sharp increase in χm below 10 K suggests a secondary contribution such as an intermolecular interaction or contamination with a paramagnetic impurity. Thus, magnetic simulations were carried out using the modified expression eqn. (2'), where ρ is the fraction of paramagnetic impurity. The paramagnetic impurity is presumed to be a nickel(II) species because no copper(II) complex was isolated from H4L. As seen in Fig. 4, a tolerable magnetic simulation is achieved using J = −48 cm−1, g = 2.06, Nα = 400 × 10−6 cm3 mol−1, and ρ = 0.08. The discrepancy factor R(χ) was 6.98 × 10−2.
|
χ
m = {2Ng2β2/kT}[5exp(4J/kT) + 1 + exp(2J/kT)]/[5exp(4J/kT) + 3 + exp(−2J/kT) + 3exp(2J/kT)] + Nα
| (2) |
|
χ
m = (1 − ρ){2Ng2β2/k(T − θ))}[5exp(4J/kT) + 1 + exp(2J/kT)]/[5exp(4J/kT) + 3 + exp(−2J/kT) + 3exp(2J/kT)] + (2Ng2β2ρ/3kT) + Nα
| (2') |
Extension to an ordered network
The trinuclear complexes 1–4 are expected to combine another metal ion through the dioximate bridge to give a polymeric compound. This is possible only when the trinuclear CuMCu core does not cause metal scrambling in the reaction with a third metal ion. In order to avoid such a problem, in this work 1 (CuMnCu) was treated with MnII. It is considered that MnII prefers the dioximate oxygens to the N4 cavity of L4−.In our first attempt using LiOH as the base in CH3CN, complex 1 formed a stable dilithium salt that had little reactivity toward MnII. Thus, KOH was used fully to deprotonate the dioxime proton and 18-crown-6 added to eliminate potassium ion by complexation. To the resulting solution was added manganese(II) perchlorate hexahydrate, precipitating crystalline Mn{Cu(L)}(H2O)45. The synthesis is shown in Scheme 1.
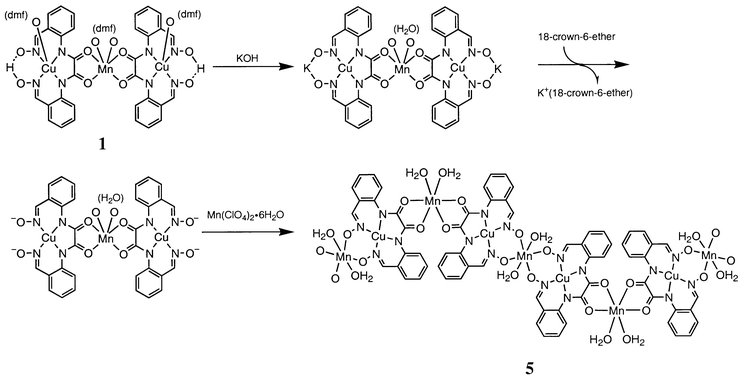 | ||
| Scheme 1 Synthesis of Mn{Cu(L)}(H2O)45 from complex 1. | ||
Compound 5 shows no IR vibration in the region 2300–3000 cm−1, indicating that the dioxime part is fully deprotonated and involved in polymeric structure formation. Another notable feature is the lack of the ν(CO) vibration of DMF and the appearance of a ν(OH) mode around 3500 cm−1. This means that the DMF molecules in 1 are replaced with water molecules in 5. In fact, analytical data for 5 indicate the presence of four water molecules instead of DMF. The reflectance spectrum of 5 resembles that of 1 and shows two visible bands at 500 and 590 nm. Together with the crystallographic result for 1, the most likely structure of 5 is as in Scheme 1.
The cryomagnetic properties of complex 5 were studied in the temperature range 2–300 K. The χmvs.T and μeffvs.T plots are given in Fig. 5. The effective magnetic moment at room temperature is 5.99 μB, which is slightly smaller than the spin-only value (6.16 μB) expected for uncoupled CuII (S = 1/2) and MnII (S = 5/2) ions. The magnetic moment decreased with decreasing temperature to a minimum value of 5.04 μB near 60 K. This is close to the spin-only value for ST = 2 (4.90 μB) arising from antiferromagnetic spin coupling between CuII (S = 1/2) and MnII (S = 5/2). This fact implies a strong antiferromangetic interaction between CuII and MnII through the cis dioximate bridge in spite of a large intermetallic distance (ca. 3.65–3.75 Å18). With further decrease in temperature, the magnetic moment of 5 increased to a maximum value of 17.80 μB at 5 K and then decreased below this temperature. The cryomagnetic behavior suggests that 5 is a weak ferromagnet exhibiting a weak antiferromagnetic interaction between the ferrimagnetic chains.
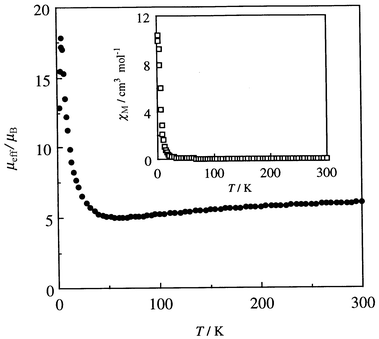 | ||
| Fig. 5 χ m vs. T and μeffvs.T plots for Mn{Cu(L)}(H2O)45. | ||
The field-cooled magnetization (FCM) under an applied field of 5 G increased rapidly below 9 K to a maximum at 5.2 K, decreased to a minimum at 4.2 K, and then increased to 67 cm3 G mol−1 at 2.0 K (Fig. 6). When the applied field was switched off at 2 K a remnant magnetization of 34 cm3 G mol−1 remained that decreased upon warming and vanished at ≈8 K. The zero-field-cooled magnetization (ZFCM) under an applied field of 5 G showed a maximum at 5.5 K. From these studies the magnetic phase transition temperature (TC) was determined to be 5.5 K. The ZFCM curve shows another phase transition at 3.6 K, but its origin was not studied.
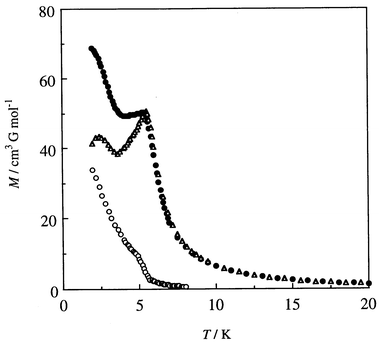 | ||
| Fig. 6 Field-cooled magnetization (FCM) (●) under 5 G, zero-field-cooled magnetization (ZFCM) (○), and remnant magnetization (RM) (△) for Mn{Cu(L)}(H2O)45. | ||
The field dependences of magnetization measured at 2 K are given in Fig. 7. The magnetization increased sharply with applied field to demonstrate a magnetic ordering in the bulk. The magnetization at 50 kG is 3.78 NμB adding support to antiferromagnetic coupling between the adjacent CuII (S = 1/2) and MnII (S = 5/2). An expansion in the field of 0–500 G is given in the insert where the field-dependence curve shows a break around 150 G. This means a phase transition from a weak ferromagnet to a ferromagnet with applied magnetic field. The hysteresis curve of 5 was determined at 2 K in the applied field of −1000 to +1000 G (see Fig. 8). It shows a remnant magnetization of 672 cm3 G mol−1 and a coercive force of 30 G. The hysteresis curve shows a break due to the phase transition near 150 G.
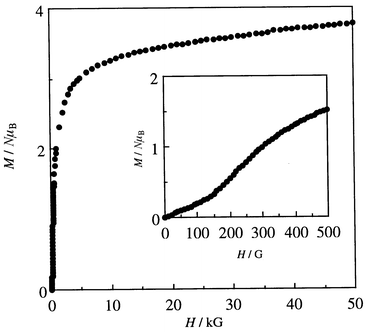 | ||
| Fig. 7 Field dependences of magnetization for Mn{Cu(L)}(H2O)45 (determined at 2 K). | ||
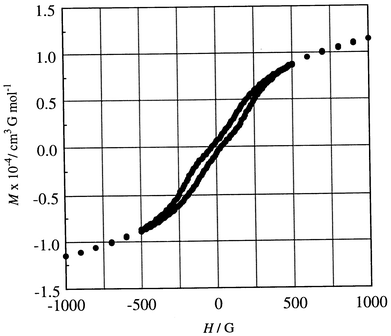 | ||
| Fig. 8 Hysteresis curve of Mn{Cu(L)}(H2O)45. | ||
The magnetic phase transition is further supported by magnetization studies under different magnetic fields (Fig. 9). The M vs. T curves at 50, 100 and 200 G show a break around 5 K, whereas the curves at 250 and 300 G show no such a break. Thus, the weak antiferromagnetic interaction between the ferrimagnetic chains is overcome by the weak applied field.
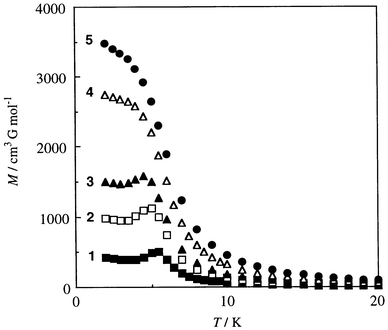 | ||
| Fig. 9 M vs. T curves of Mn{Cu(L)}(H2O)45 under different applied fields (measured at 2 K): (1) 50, (2) 100, (3) 200, (4) 250, (5) 300 G. | ||
In conclusion the oxamide/dioxime ligand is promising for providing complex-based magnetic materials. This work illustrates a stepwise synthesis of magnetic materials using a ‘complex bridge’.
Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research (No. 09440231), Scientific Research on Priority Area ‘Metal-assembled Complexes’ (No. 10149106) and an International Scientific Research Program (No. 09044093) from the Ministry of Education, Science and Culture, Japan.References
-
Proceedings of the NATO Advanced Research Workshop on Magnetic Molecular Materials, Kluwer, Dordrecht, 1990.
Search PubMed
.
-
O. Kahn
, Molecular Magnetism, VCH, Weinhem, 1993;
Search PubMed
- O. Kahn, Adv. Inorg. Chem., 1995, 43, 179 Search PubMed
-
M. M. Turnbull,
T. Sugimoto
and L. K. Thompson,
ACS Symp. Ser.,
1996, 644.
Search PubMed
- H. Tamaki, Z. J. Zhong, N. Matsumoto, S. Kida, M. Kiokawa, N. Achiwa, Y. Hashimoto and H. Ōkawa, J. Am. Chem. Soc., 1992, 114, 6974 CrossRef
- H. Ōkawa, M. Mitsumi, M. Ohba, M. Kodera and N. Matsumoto, Bull. Chem. Soc. Jpn., 1994, 67, 2139 CAS
- H. Ōkawa, Y. Kawahara, M. Mikuriya and S. Kida, Bull. Chem. Soc. Jpn., 1980, 53, 549 CAS
- K. Nonoyama, H. Ojima, K. Ohkiand and M. Nonoyama, Inorg. Chim. Acta, 1980, 41, 155 CrossRef CAS
- A. Bencini, M. Di Vaira, A. C. Fabretti, D. Gatteschi and C. Zanchini, Inorg. Chem., 1984, 23, 1620 CrossRef CAS
- Y. Pei, O. Kahn, K. Nakatani, E. Codjovi, C. Mathoniere and J. Sletten, J. Am. Chem. Soc., 1991, 113, 6558 CrossRef CAS
- Y. Pei, O. Kahn, J. Sletten, J. P. Renard, R. Georges, J. C. Gianduzzo, J. Curely and Q. Xu, Inorg. Chem., 1988, 27, 47 CrossRef CAS
- Y. Pei, K. Nakatani, O. Kahn, J. Sletten and J. P. Renard, Inorg. Chem., 1989, 28, 3170 CrossRef CAS
- R. Beckett, R. Colton, B. F. Hoskins, R. L. Martin and D. G. Vince, Aust. J. Chem., 1969, 22, 2527 CAS
- J. A. Bertrand, J. H. Smith and P. Garyeller, Inorg. Chem., 1974, 13, 1649 CrossRef CAS
- Y. Agnus, R. Louis, R. Jesser and R. Weiss, Inorg. Nucl. Chem. Lett., 1976, 12, 455 Search PubMed
- A. Chakravorty, Coord. Chem. Rev., 1974, 13, 1 CrossRef CAS
- G. A. Nicholson, C. R. Lazarus and B. J. McCormick, Inorg. Chem., 1980, 19, 192 CrossRef CAS
- D. Luneau, H. Oshio, H. Ōkawa and S. Kida, Chem. Lett., 1989, 443 CAS
- Z. J. Zhong, H. Ōkawa, N. Matsumoto, H. Sakiyama and S. Kida, J. Chem. Soc., Dalton Trans., 1991, 497 RSC
- R. Ruiz, M. Julve, J. Faus, F. Lloret, M. C. Muñoz, Y. Journaux and C. Bois, Inorg. Chem., 1997, 36, 3434 CrossRef CAS
- E. Colacio, J. M. Dominguez-Vera, A. Escuer, R. Kivekäs and A. Romerosa, Inorg. Chem., 1994, 33, 3914 CrossRef CAS
- F. Birkelbach, M. Winter, U. Flörke, H.-J. Haupt, C. Butzlaff, M. Lengen, E. Bill, A. X. Trautwein, K. Wieghardt and P. Chaudhuri, Inorg. Chem., 1994, 33, 3990 CrossRef CAS
-
E. A. Boudreaux
and L. N. Mulay
, Theory and Applications of Molecular Paramagnetism, Wiley, New York, 1976, pp. 491–494.
Search PubMed
- W. L. F. Armarego and R. E. Wilette, J. Chem. Soc., 1965, 1258 RSC
-
TEXSAN, Crystal Structure Analysis Package, Molecular Structure Corporation, Houston, TX, 1985 and 1992.
Search PubMed
- R. Blinc and D. Hadzi, J. Chem. Soc., 1958, 4536 RSC
- K. Burger, I. Ruff and F. Ruff, J. Inorg. Nucl. Chem., 1965, 27, 179 Search PubMed
-
C. K. Johnson
, ORTEP, Report 3794, Oak Ridge National Laboratory, Oak Ridge, TN, 1965.
Search PubMed
- D. E. Shannon and C. T. Prewitt, Acta Crystallogr., Sect. B, 1969, 25, 925 CrossRef CAS
- E. Sinn and C. M. Harris, Coord. Chem. Rev., 1969, 4, 391 CrossRef
- J.-P. Costes, J.-P. Laurent, J. M. M. Sanchez, J. S. Varela, M. Ahlgren and M. Sundberg, Inorg. Chem., 1997, 36, 4641 CrossRef CAS
- Y. Pei, Y. Journaux and O. Kahn, Inorg. Chem., 1988, 27, 399 CrossRef CAS
- L. Mörgenstern-Badarau and H. H. Wickman, Inorg. Chem., 1985, 24, 1889 CrossRef
-
F. E. Mabbs
and D. J. Machin
, Magnetism and Transition Metal Complexes, Chapman and Hall, London, 1973.
Search PubMed
| This journal is © The Royal Society of Chemistry 2001 |