Catalytic enantioselective rearrangements and cycloadditions involving ylides from diazo compounds
David M. Hodgson*, Françoise Y. T. M. Pierard and Paul A. Stupple
Dyson Perrins Laboratory, Department of Chemistry, University of Oxford, South Parks Road, Oxford, UK OX1 3QY. E-mail: david.hodgson@chem.ox.ac.uk
First published on 27th November 2000
Abstract
This review summarizes the background and state of the art (as of June 2000) of catalytic enantioselective rearrangements and cycloadditions involving ylides from diazo compounds. The field is still in its infancy, as until a few years ago it was not considered likely that a catalyst, having decomposed a diazo compound to form an ylide, would still be able to exert an influence on subsequent transformations of the ylide. Recent developments have shown this not to be the case and a new era of synthetically important ylide transformations in organic chemistry has begun where appropriate metal–chiral (nonracemic) ligand combinations are starting to be developed to render these transformations enantioselective.
 David M. Hodgson David M. Hodgson | David Hodgson obtained his first degree in Chemistry at Bath University. After a PhD at Southampton University in the field of natural product synthesis (with Professor P. J. Parsons) and a research position at Schering, he was appointed in 1990 to a lecturership at Reading University. In 1995 he moved to the Dyson Perrins Laboratory at Oxford. He received a Glaxo Wellcome Award for Innovative Organic Chemistry in 1999 and Pfizer and AstraZeneca Awards in 2000. His research interests are broadly in the development and application of synthetic methods, particularly asymmetric generation and transformations of carbenoids. |
 Françoise Y. T. M. Pierard Françoise Y. T. M. Pierard | Françoise Pierard was born in Bastogne, Belgium in 1969. She studied Chemistry at the University of Louvain-la-Neuve in Belgium where she obtained her first degree. In 1998, she received her DPhil degree at the University of Louvain-la-Neuve in the field of [2 + 2] cycloadditions of olefins with α-aminoketene-iminium salts under the direction of Professor L. Ghosez. In 1998, she also joined the research group of Dr D. M. Hodgson working on asymmetric ylide cycloaddition chemistry. |
 Paul A. Stupple Paul A. Stupple | Paul Stupple studied Chemistry at Oxford University, where he obtained his first degree in 1996 at Hertford College and was awarded the Brian Bannister Prize for his Part II year of research with Dr J. Robertson on approaches to Roseophilin. As a Jubilee Scholar of St. Hugh’s College, he received his DPhil under the supervision of Dr D. M. Hodgson with a thesis entitled ‘Catalytic enantioselective carbonyl ylide formation–cycloaddition’. He is currently a Medicinal Research Chemist at Pfizer in Kent. |
1 Introduction
Ylides are normally reactive intermediates which are known to undergo a number of synthetically useful transformations (rearrangements and dipolar cycloadditions are considered in this review), to form stable products. Ylides can be viewed as species in which a positively charged heteroatom X (such as O, I, S, Se, N) is connected to an atom (carbon is considered in this review) possessing an unshared pair of electrons. An attractive method of ylide formation involves the reaction of heteroatom-substituted organic compounds via a lone pair on the heteroatom with carbenes (Scheme 1).1 | ||
| Scheme 1 | ||
The synthetic utility of free carbenes in such an ylide-forming process is limited partly by their methods of generation (thermally, photochemically or under basic conditions), and also by their high reactivity and lack of selectivity with functionalised organic compounds. It is often preferable to use metal-complexed carbenes (metallocarbenes) in which the metal, and the ligands with which it is usually associated, can potentially influence the reactivity of the carbene. Metallocarbenes are themselves often transient intermediates. One good way of generating metallocarbenes as intermediates is the reaction of a diazo (often an α-diazocarbonyl) compound with a metal (often copper or rhodium)–ligand system. The metal species used is believed to effect formation of the metallocarbene by acting as a Lewis acid and accepting electron density from the diazo carbon at a vacant coordination site on the metal (Scheme 2). This is then followed by back donation of electron density from the metal to the carbene carbon with concomitant loss of N2. As the intermediate metallocarbene can be considered to be electrophilic at carbon, it can accept electron density from a suitable donor XY with eventual loss of the metal–ligand complex which therefore functions as a catalyst.2
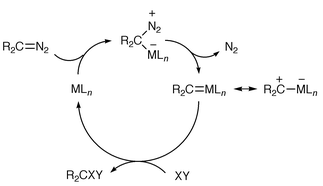 | ||
| Scheme 2 | ||
Specific metal–ligand combinations can allow the metallocarbene to
discriminate between potentially competing carbene reactions, such as
cyclopropanation and C–H insertion, and/or paths to different
stereoisomers by controlling the stereoselectivity of a reaction. If in
such a transformation achiral starting materials are used to generate a
product that contains a stereocentre, or stereocentres, then asymmetric
induction is possible with a metal–ligand catalyst where the ligand
is chiral (and nonracemic); this is because in the presence of such ligands
the transition states leading to the enantiomeric products become
diastereomeric. Significant progress has been made in transformations of
diazocarbonyl compounds involving enantioselective C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) C, C–H or
X–H (X = N, Si) insertions using chiral, non-racemic transition
metal-based catalysts. In such enantioselective insertions it is clear that
the metal–ligand system in an intermediate metallocarbene can
directly exert an influence on selectivity. The situation in a potentially
enantioselective ylide transformation is not so straightforward (Scheme 3).
C, C–H or
X–H (X = N, Si) insertions using chiral, non-racemic transition
metal-based catalysts. In such enantioselective insertions it is clear that
the metal–ligand system in an intermediate metallocarbene can
directly exert an influence on selectivity. The situation in a potentially
enantioselective ylide transformation is not so straightforward (Scheme 3).
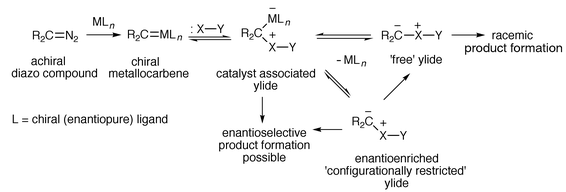 | ||
| Scheme 3 | ||
Once an ylide is formed, if the catalyst remains associated with the ylide during the subsequent transformation then asymmetric induction may be observed. But the catalyst could dissociate and therefore not be involved in the subsequent ylide tranformation. If the free ylide, formed after dissociation of the catalyst, is achiral then asymmetric induction would be very unlikely. However, if the free ylide is able to retain a chiral configuration during the subsequent transformation then the product may be formed enantioselectively.3,4 Recent developments have shown that catalytic asymmetric synthesis in a number of ylide transformations is possible and these will be reviewed in the following sections, along with background results which are relevant to the nature of the catalyst–ylide interaction and the potential reversibility of the steps indicated in Scheme 3.
2 Oxonium ylides
Ethereal oxonium ylides are highly reactive and short-lived intermediates compared to other onium ylides. Whilst oxonium ylides continue to elude spectroscopic identification, they are frequently proposed as intermediates in the reaction of ethers with a carbene or metallocarbene. Two major modes of rearrangement of oxonium ylides are [1,2] insertion (Stevens rearrangement) and [3,2] sigmatropic rearrangement (Scheme 4). With allylic (or propargylic) ether-derived oxonium ylides, symmetry-allowed5 [3,2] sigmatropic rearrangement is preferred over [1,2] insertion, and in many cases the ‘allylic transposition’ provides indirect evidence that the reaction has proceeded via an oxonium ylide, rather than direct C–O insertion (for an example see Scheme 14).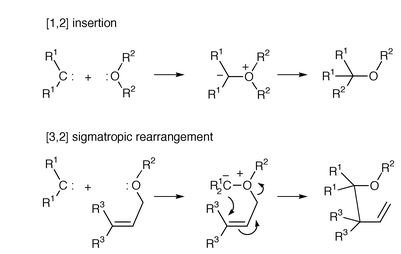 | ||
| Scheme 4 | ||
 | ||
| Scheme 14 | ||
The reactants and products in these processes may contain several ethereal oxygens which could form a number of different oxonium ylides, usually only one of which leads to the desired product. Reactions are often carefully designed to minimise, or eliminate the possibility of unwanted oxonium ylide formation by fulfilling some or all of the following criteria: the substrate is present in large excess and/or the carbene precursor is added slowly and/or the reaction is intramolecular and/or in the product ether the ethereal oxygen lone pair availability is attenuated by sterics and/or electronics relative to the substrate ether and therefore is less likely to form an ylide. However, even if undesired (catalyst-associated) oxonium ylide formation competes with, or is even preferred over, the desired ylide, this would not matter provided rearrangement of the undesired (catalyst-associated) oxonium ylide is slow relative to that of the desired ylide and relatively rapid reversal to ether and (metallo-)carbene occurs (by the Curtin–Hammett principle).
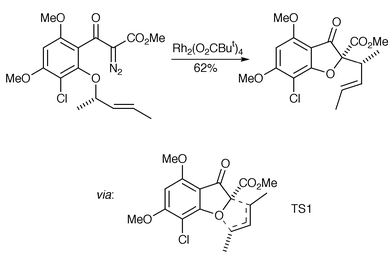
There are examples of oxonium ylide transformations in which it has been suggested that the (catalyst-associated) ylide formation is reversible. Pirrung and co-workers have reported an elegant total synthesis of (+)-griseofulvin where the key step involves an oxonium ylide formation–[3,2] rearrangement transformation and which serves to illustrate some important selectivity issues (Scheme 5).6
 | ||
| Scheme 5 | ||
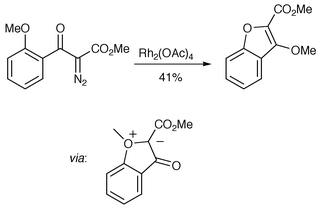
The stereochemistry of this process can be understood in terms of a transition state model (TS1) that resembles an oxabicyclo[3.3.0]octane ring system with the key, stereochemistry-defining methyl group located on the convex face (possible involvement of the catalyst in this transition state is not shown). No other stereoisomers or regioisomers were detected in the crude reaction mixture. This raised an important issue of selectivity as there are two ortho-alkoxy groups which could potentially form oxonium ylides. To discern the possible fate of an O-methyl ylide, a simpler diazoether was examined which gave a benzofuran, presumablyarising from formal [1,4] migration after oxonium ylide formation (Scheme 6).
 | ||
| Scheme 6 | ||
In the case of the (+)-griseofulvin synthesis (Scheme 5), no product derived from the O-methyl ylide could be found. It was therefore concluded that the system is under Curtin–Hammett control and that the product profile is a result of the symmetry-allowed [3,2] rearrangement having a lower activation barrier than [1,4] migration. Any (catalyst-associated) O-methyl ylide derived from the starting diazo compound must reform the (metallo)carbene. Although the [1,4] rearrangement is also a symmetry-allowed process {[σ2s + ω2s + π2s]}, the distance between the migration origin and terminus in the O-methyl ylides under consideration here (e.g., Scheme 6) means that the process from (catalyst-associated) ylide to benzofuran cannot be a single-step pericyclic reaction.
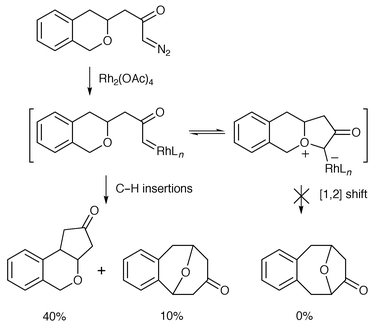
In a related study, West and co-workers also explained product profiles on the basis of oxonium ylide formation being reversible (Scheme 7).7 It was suggested that a [1,2] insertion from the intermediate (catalyst-associated) ylide was slow (a pericyclic process would be strongly disfavoured according to the Woodward–Hoffmann rules), which allowed the metallocarbene to reform and undergo more facile C–H insertion reactions.
 | ||
| Scheme 7 | ||
2.1 [1,2] Insertion (Stevens rearrangement)
In 1966 Nozaki et al. reported the reaction of racemic 2-phenyl oxetane and methyl diazoacetate with a chiral (nonracemic) ligand-containing copper chelate which furnished a mixture (75%) of cis and trans-tetrahydrofurans (Scheme 8).8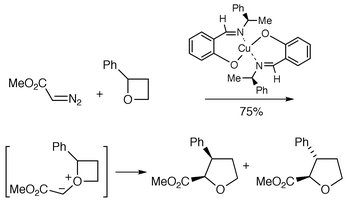 | ||
| Scheme 8 | ||
Both tetrahydrofurans were optically active, indicating that an enantiomer differentiation process had occurred. The reaction was suggested as proceeding through an intermediate oxonium ylide which underwent regioselective insertion into the phenyl-substituted C–O bond to generate the ring-expanded tetrahydrofurans. Although the enantiomeric excesses were not reported for this transformation, the work and the co-reported asymmetric cyclopropanation (6% ee) was a landmark study because it showed, for the first time outside the area of polymer chemistry, that homogeneous organometallic catalysis could be made asymmetric by using chiral ligands attached to the metal.
One of the first suggestions that an oxonium ylide transformation was proceeding via a metal-complexed intermediate was made by Roskamp and Johnson in 1986.9 It was found that after transition metal catalysed decomposition of a diazo-containing precursor, the rearrangement pathway of an intermediate oxonium ylide was sensitive to the nature of the catalyst used (Scheme 9).
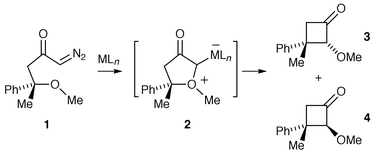 | ||
| Scheme 9 | ||
(S)-Diazoketone 1 was treated with three different catalysts and the ratio of diastereomeric cyclobutanones 3 and 4 obtained was found to be as follows: Rh2(OAc)4, 3∶1; Cu(acac)2, 1∶6 and RhCl(PPh3)3, 1∶10. It was established that cyclobutanone 3 was formed with retention of configuration at the quaternary centre. Roskamp and Johnson concluded that the most likely explanation for the results involves the oxonium ylide rearrangement proceeding via a catalyst-complexed intermediate 2. Since the ethereal oxygen lone pairs in diazoketone 1 are diastereotopic, up to four diastereomeric catalyst-associated oxonium ylides could be formed, the ratio of which could be influenced by the catalyst used. Although this work demonstrates that a catalyst-complexed ylide could be the product-forming intermediate, the results obtained do not prove this. If the catalyst subsequently dissociates and the onium centre remains configurationally stable, then the potentially differing reactivity of the two diastereoisomeric metal-free oxonium ylides could account for the varying product ratios. However, Clark in his studies of [3,2] sigmatropic rearrangements of oxonium ylides (Section 2.2) suggested that it is unlikely the configuration at oxygen in an oxonium ylide would be preserved prior to a subsequent rearrangement.10 This was proposed as, although the rates of inversion of oxonium ylides have not been determined, it is known that the barriers to inversion of analogous oxonium ions are low.11
Whilst the early work by Nozaki et al. paved the way for intensive investigations in asymmetric cyclopropanation and, subsequently, asymmetric C–H insertion chemistry, asymmetric oxonium ylide transformations were not further investigated until the early 1990s. At this time Katsuki et al. reported a more detailed investigation of the oxetane ring-expansion process.12 These workers first studied the reaction of excess racemic 2-phenyl oxetane (2 equivalents) under conditions which they had previously used for asymmetric cyclopropanations with high enantiomeric excesses (ees), that is with tert-butyl diazoacetate using a copper catalyst with a C2-symmetric reduced bipyrindine ligand (Scheme 10).
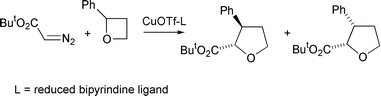 | ||
| Scheme 10 | ||
Surprisingly, the recovered oxetane was found to be essentially racemic which implies that no kinetic resolution of the two oxetane enantiomers had occurred. The cis- and trans- ring-expanded tetrahydrofurans were each formed in good ee (∼80%) and the predominant absolute configuration at the ester-substituted stereocentre (C-2) in both tetrahydrofurans was the same. Further studies with the individual enantiomers of 2-phenyloxetane established that with the (R,R)-reduced bipyrindine ligand, (R)-2-phenyloxetane led predominantly to the trans-(2S,3R)-disubstituted tetrahydrofuran, whereas (S)-2-phenyloxetane gave predominantly the cis-(2S,3S)-disubstituted tetrahydrofuran. These results indicate the same face of the metallocarbene is approached by both oxetane enantiomers and this determines the common absolute configuration at C-2 in the predominant cis- and trans-ring expanded tetrahydrofurans. It is suggested that this common approach is to the Si face of the intermediate metallocarbene because this minimises developing steric interactions between the ester group and the C-7 substituent of the ligand (Scheme 11).
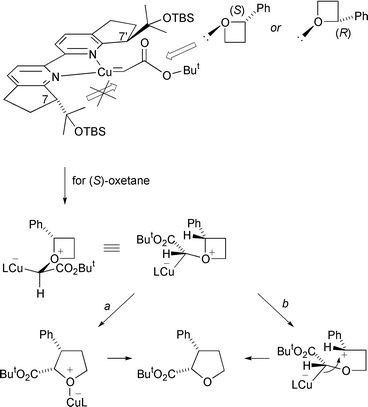 | ||
| Scheme 11 | ||
The detailed mechanism leading from the catalyst-complexed oxonium ylide to the ring-expanded tetrahydrofurans is not clear. A concerted rearrangement via the catalyst-free oxonium ylide is strongly disfavoured as orbital symmetry requirements would demand a geometrically difficult and energetically unfavourable backside attack on the PhC–O σ* orbital; moreover this would require inversion at C-2 of the oxetane, whereas retention is predominantly observed. However, by analogy to a suggestion from Woodward and Hoffmann concerning Stevens rearrangement of an ammonium ylide,5 a symmetry-allowed process may be envisaged if the metal is strongly associated with the carbanionoid centre at the outset of the reaction and with the original oxetane oxygen atom after the ring expansion has occurred (Scheme 11, route a). In this analysis, inversion occurs at oxygen and at the carbanionoid carbon and retention occurs at the migrating oxetane carbon, and an orbital of the metal is involved such that the process is suprafacial on the C–Cu component and antarafacial on the C–O component {a [σ2s + σ2a] rearrangement}. Whilst involvement of the metal provides an attractive solution to how a concerted rearrangement may be possible, one may question if the strength of association of the metal with the ethereal oxygen as the rearrangement proceeds is enough to make a pericyclic process plausible. Any stepwise mechanism requires final C–O bond formation to be faster than C–CHPh bond rotation in order to be consistent with the observed minimal loss of stereochemical information at C-2 in the oxetane. Dissociation of the catalyst from the metal-complexed oxonium ylide could occur before, during, or after C–O bond cleavage, or during C–O bond formation. To be consistent with the experimental results a stepwise process that begins with dissociation of the catalyst to give the free oxonium ylide would require that the subsequent C–O bond cleavage and C–O bond formation steps were faster than ButO2CHC–O bond rotation. An alternative stepwise sequence from the catalyst-complexed oxonium ylide to tetrahydrofuran starts with (rate-determining) C–O bond cleavage, followed by C–O bond formation as the catalyst dissociates (Scheme 11, route b); this provides a more reasonable explanation as to why the catalyst is able to exert stereochemical control during formation of C-2 in the tetrahydrofurans. This sequence also implies that C–O homolysis (to produce a singlet radical pair) is less likely than C–O heterolysis,13 since homolysis additionally requires loss of the catalyst prior to radical recombination.
Doyle has recently devised an alternative strategy to chiral oxonium ylides based on desymmetrisation of 1,3-dioxanes (Scheme 12).14
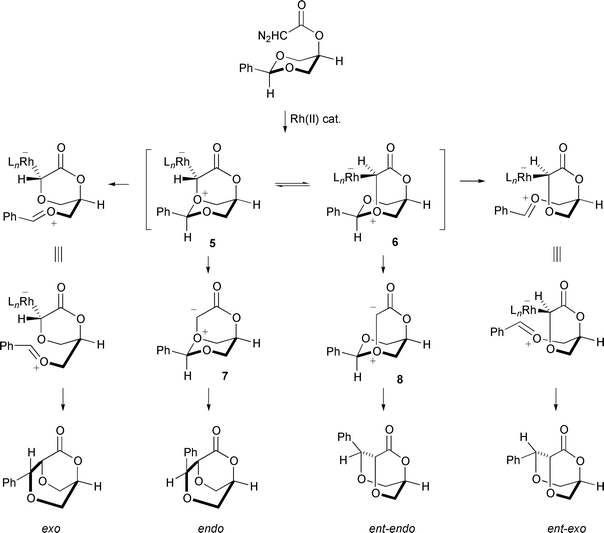 | ||
| Scheme 12 | ||
In this rearrangement the intermediate metallocarbene discriminates enantiotopic ethereal oxygen atoms; variable amounts of direct C–H insertion giving a fused butyrolactone (not shown in Scheme 12) were also observed. In the example shown in Scheme 12, catalyst-dependent ee was observed for the oxonium ylide-derived exo product, but the ee of the endo- diastereomer was essentially not catalyst-dependent, and the ratio of exo- to endo-diastereomer was catalyst-dependent. The exo-isomer could be derived from rapidly equilibrating catalyst-associated ylides (5 and 6) where rate-determining C–O bond cleavage is catalyst-dependent. Because the α-carbon in the intermediate lactone remains stereogenic, a transition state might be preferred for ring closure (Scheme 12) in which CH2–O+ bond rotation has occurred. The endo-isomer could be derived solely from the enantiomeric catalyst-free ylides (7 and 8) where catalyst dissociation rates are different for the two possible catalyst-associated ylides (which are in rapid equilibrium relative to catalyst dissociation), but the ratio of the two rates is relatively independent of the individual ligands.
2.2 [3,2] Sigmatropic rearrangement
In 1992 McKervey and co-workers reported the first examples of catalytic enantioselective oxonium ylide formation–[3,2] sigmatropic rearrangement. A novel chiral Rh(II) catalyst was employed [Rh2(S-binaphtholphosphate)2(O2C OH)2], which resulted in enantioselectivities of up to 30% ee after [3,2] sigmatropic rearrangement of the intermediate oxonium ylide derived from 9 (Scheme 13). Further improvement of enantiocontrol (up to 60% ee) was achieved when a range of chiral carboxylate Rh(II) catalysts were screened in the transformation from precursor 10 (Scheme 13).15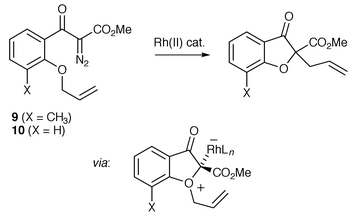 | ||
| Scheme 13 | ||
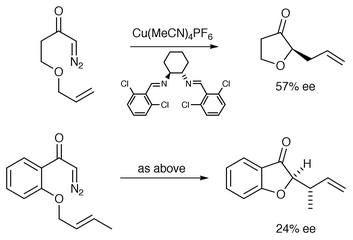
Following Roskamp and Johnson’s observations of catalyst-dependent diastereoselectivity in a Stevens rearrangement, in 1992 Clark reported analogous observations in [3,2] sigmatropic rearrangements.10 In subsequent studies copper(II) hexafluoroacetylacetonate emerged as a (pre)catalyst of choice for these rearrangements as it minimised competing C–H insertion. It was suggested that using such an electrophilic catalyst would increase the rate of insertion into the ethereal oxygen lone pair and it might also reduce the propensity to reform the original metallocarbene–tethered ether and/or reduce the energy difference between the catalyst-bound ylide species and the transition states for rearrangement. More recently, Clark and co-workers have reported enantioselective rearrangements using a copper catalyst in combination with a C2-symmetric diimine (up to 57% ee, Scheme 14).16 The presence of a phenyl ring in the tether gave slightly lower ees. The origin of the asymmetric induction was discussed as either being due to rearrangement via a catalyst-free ylide in which the energy barrier to inversion of configuration at the sp3 oxonium centre of the ylide must be significantly greater than the transition state energy for the rearrangement reaction, or rearrangement through the catalyst-bound ylide. In the latter case the stereochemical outcome of the reaction is dictated by the stereogenic centre(s) established during initial attack on the carbenoid, and the environment created by the chiral ligands attached to copper.
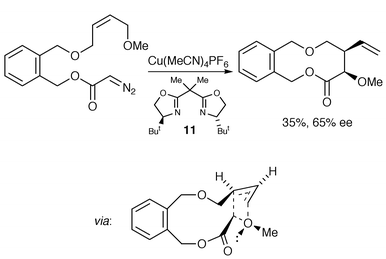
In 1998 Doyle et al. reported an example of an enantioselective intramolecular oxonium ylide formation–[3,2] sigmatropic rearrangement to form a ten-membered ring in 65% ee (Scheme 15, possible involvement of the catalyst in the transition state is not shown).17
 | ||
| Scheme 15 | ||
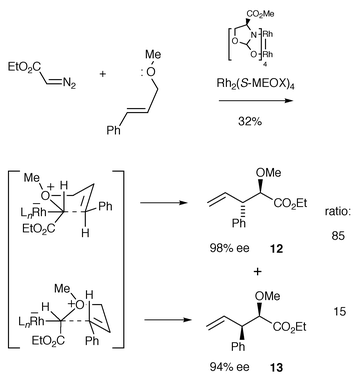
Doyle also reported a study of an enantioselective intermolecular oxonium ylide formation–[3,2] sigmatropic rearrangement—remarkably in up to 98% ee, although yields were low (Scheme 16).17 As in the previous oxonium ylide studies, one could consider that the asymmetric induction arises from configurationally stable (enantiomeric) free ylide intermediates. However, the observation of significant catalyst-dependent diastereoselectivity [using Rh2(OAc)4 gave 17∶83, 12 : 13] implicates a catalyst-associated ylide in the product-forming step. Unlike in Roskamp and Johnson’s study and Clark’s early studies, an achiral starting material was used in Doyle’s work. Catalyst decomplexation from the dipolar complexes shown in Scheme 16 would give a common intermediate, which would presumably not show catalyst-dependent diastereoselectivity. Therefore, these results are consistent with the oxonium ylide transformation proceeding via a catalyst-complexed intermediate. It was suggested that the rearrangement occurs during catalyst dissociation in what is formally a backside displacement reaction that involves inversion of configuration (Scheme 16, for similar suggestions see Schemes 11 and 12).
 | ||
| Scheme 16 | ||
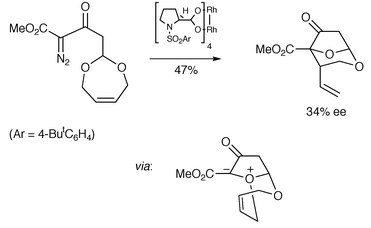
Calter and Sugathapala have reported an asymmetric approach to the core of the zaragozic acids based on desymmetrisation of cyclic unsaturated acetals (Scheme 17).18 The enantiodiscriminating event is similar to that examined by Doyle (Scheme 12), in that the chiral metallocarbene discriminates between enantiotopic oxygen atoms.
 | ||
| Scheme 17 | ||
3 Iodonium, sulfonium and selenonium ylides
If one accepts that direct carbene insertion into the C–I bond of allyl iodide is unreasonable compared with iodonium ylide formation, then the clearest evidence that a catalyst-associated onium species is involved in an ylide rearrangement has been obtained by Doyle et al.17 Reaction of ethyl diazoacetate with allyl iodide in the presence of chiral catalysts led to an α-iodoester in up to 69% ee (Scheme 18). In this case, a catalyst free but ‘configurationally constrained’ ylide cannot be considered as the origin of the enantioselectivity because the catalyst-free iodonium ylide is achiral by virtue of the iodine lone pairs.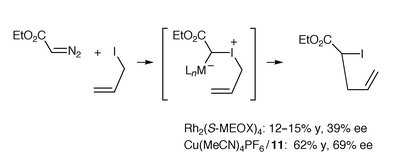 | ||
| Scheme 18 | ||
Compared with the ephemeral nature of oxonium ylides, sulfonium ylides (non-allylic) can be much more stable. Indeed there are several examples of X-ray structure determinations of sulfonium ylides.1,2 In 1995 Uemura et al. reported the first examples of catalytic asymmetric [3,2] sigmatropic rearrangements of allylic sulfur and selenonium ylides derived from trans-cinnamyl phenyl sulfide (and related selenides) with ethyl diazoacetate in the presence of a copper(I)–bisoxazoline catalyst or Rh2(S-MEPY)4 in up to 20% ee for the sulfide and up to 41% ee with the corresponding selenide (e.g., Scheme 19).19
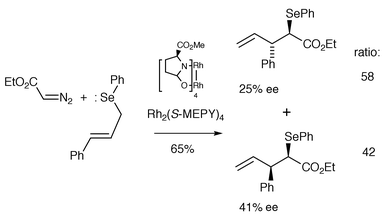 | ||
| Scheme 19 | ||
Higher enantioselectivities (up to 64%) have been achieved by Katsuki and co-workers with trans-cinnamyl phenyl sulfide using chiral cobalt(III)–salen catalysts and the sterically larger tert-butyl diazoacetate (Scheme 20).20
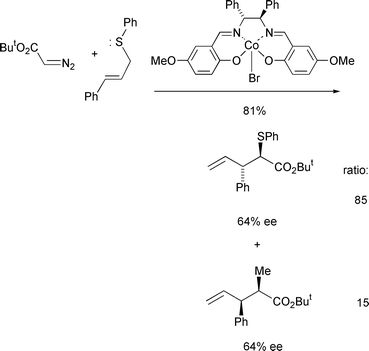 | ||
| Scheme 20 | ||
Where comparisons could be made between different catalysts for the same substrate, essentially catalyst-independent diastereoselectivity was observed in both Uemura’s and Katsuki’s studies. These results tend to suggest that rearrangement occurs mainly from a catalyst-free ylide. It is generally considered that configurational integrity at sulfur or selenium is maintained during [3,2] sigmatropic rearrangements of these ylides.19 That is, the energy barrier to (formal) pyramidal inversion at the sulfur or selenium atom of the ylide is greater than the barrier for rearrangement. The enantioselectivity in these reactions is then principally decided during differentiation of the enantiotopic lone pair electrons on the sulfur or selenium atom by the chiral metallocarbene. However, the structure of the ylide can influence the chirality transfer process, because the structure will determine the size of the energy differences between competing transition states for the rearrangement. So the level of enantioselectivity in the discrimination of the enantiotopic lone pairs on sulfur or selenium will not necessarily correlate directly to the ee(s) of the sigmatropic rearrangement product(s). Furthermore, if diastereomers form, their ees will not necessarily be identical. This can be seen for example, in Uemura’s study (up to 22% difference in ee between diastereomers),19 and in Katsuki’s calculated heats of formation of the transition structures leading to the four possible isomers in the sigmatropic rearrangment of an (R)-trans sulfur ylide, where the energy differences between the structures leading to the enantiomers of each diastereomer are not the same (Scheme 21).20
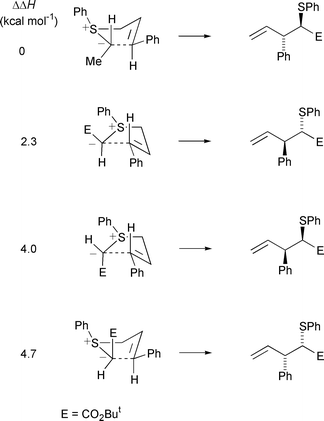 | ||
| Scheme 21 | ||
In 1999 Aggarwal and co-workers reported low ees from rearrangement of allylic sulfur ylides derived from trimethylsilyldiazomethane (Scheme 22).21 Interestingly however, catalyst-dependent diastereoselectivity was observed (ranging from 90∶10 to 49∶51, for 14 : 15) which (as in Doyle’s work with oxonium ylides)17 implies catalyst association in the ylide rearrangement step.
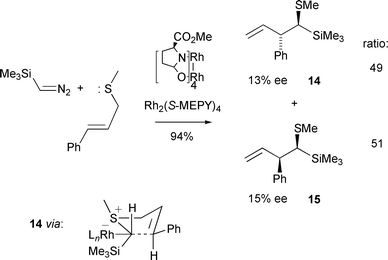 | ||
| Scheme 22 | ||
More recently McMillen has found that increasing the steric demands of an allyl sulfide (from allyl phenyl sulfide to allyl 2,6-dimethylphenyl sulfide) increased ee (from 14% to 52%) in the resulting homoallylic sulfide following treatment with ethyl diazoacetate and a copper(I)–bisoxazoline catalyst.22
4 Carbonyl ylides
Carbonyl ylides are usually non-isolable reactive intermediates whose principal synthetic uses are in 1,3-dipolar cycloadditions. Of the various methods for carbonyl ylide formation, the interaction of a carbene or metallocarbene with the oxygen atom of a carbonyl group is particularly attractive because of its apparent simplicity (Scheme 23).1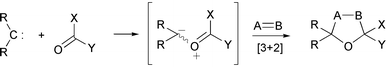 | ||
| Scheme 23 | ||
One of the first suggestions that a carbonyl ylide transformation could proceed via a metal-complexed species was made by Landgrebe et al. in 1989.23 A series of metal-catalysed reactions of ethyl diazoacetate with β-ketoesters were studied and the main products, at least with Rh2(OAc)4, were found to be enol ethers, which were suggested as arising from an intermediate carbonyl ylide 16 (possible association of the catalyst is not shown) followed by [1,4] sigmatropic hydrogen-shift (Scheme 24).
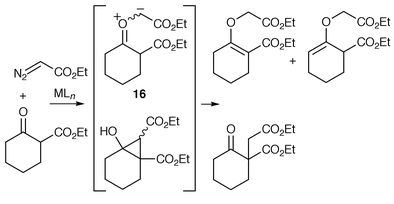 | ||
| Scheme 24 | ||
Using CuCl, significant amounts of ketodiesters were also isolated, possibly formed by cyclopropanation of the enol form of the starting material followed by ring opening. Interestingly the ratio of regioisomeric enol ethers was found to be catalyst-dependent, which led Landgrebe to conclude that ‘it seems inescapable that the catalyst must be present during the [1,4] sigmatropic shift of hydrogen’.23 However, catalyst-independent ratios of isomeric enol ethers and no cyclopropanation-derived products were obtained from 2-methyl- and 2-phenyl-cycloalkanones. It is therefore conceivable that the catalyst-dependent enol ether ratios result from competition between carbonyl ylide formation {with [1,4] hydrogen shift from the catalyst-free ylide} and direct OH insertion of the enol which vary according to the metallocarbene and β-ketoester used; because cyclopropanation and direct OH insertion could be competing reaction pathways from the enol, this may also be a catalyst-dependent factor which influences the ratio of enol ethers.
Landgrebe and co-workers have also suggested that carbonyl ylide formation can be irreversible.24 This suggestion was based on the observation that treatment of an excess equimolar mixture of the ketones shown in Scheme 25 with ethyl diazoacetate and CuCl furnished a 1∶1 mixture of the deuterated enol and non-deuterated enol (yield not stated). This shows that the [1,4] hydrogen-shift in the intermediate carbonyl ylide is intramolecular. Furthermore, formation of equal amounts of both products seems to provide evidence that the carbonyl ylide formation is not appreciably reversible. If the 1,3-dipole formation was reversible, i.e., the metallocarbene could reform by dissociation of the ketone from the carbonyl ylide, one may expect to have isolated the non-deuterated enol as the major product, due to a kinetic isotope effect in the [1,4] shift.
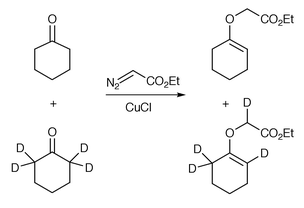 | ||
| Scheme 25 | ||
Davies et al. have reported a highly selective approach towards asymmetric cyclopropanation which utilises α-hydroxy esters or α-hydroxy lactones as chiral auxiliaries (Scheme 26).25
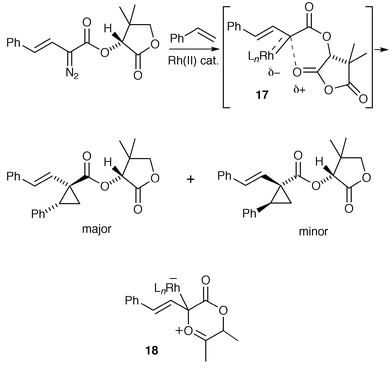 | ||
| Scheme 26 | ||
Davies proposed that one of the key interactions which determines the extent of asymmetric induction is between the carbonyl of the auxiliary and the rhodium-bound carbene, generating a relatively rigid intermediate. If this interaction is interpreted in an alternative way, it can be viewed as a catalyst-complexed carbonyl ylide. This was emphasised by the observation of competition between cyclopropanation and carbonyl ylide cycloaddition with styrene when a ketone functionality was used in the auxiliary, rather than an ester–lactone. The ester–lactone-derived intermediates may resemble metallocarbenes with a weak association with the auxiliary carbonyl (e.g., 17), which is sufficient, however, to orient the chiral auxiliary predominantly in one conformation which favours efficient asymmetric induction. The intermediate formed by interaction with a ketone may resemble more a catalyst-complexed carbonyl ylide (e.g., 18) (or even the catalyst-free carbonyl ylide), which could undergo cyclopropanation or cycloaddition (for a possibly related example see Scheme 28). Double stereodifferentiation was observed when the reaction shown in Scheme 26 was carried out using chiral Rh(II) catalysts derived from the enantiomers of mandelic acid; these results suggest that, at least with a lactone auxiliary, the catalyst participates in the cyclopropanation.
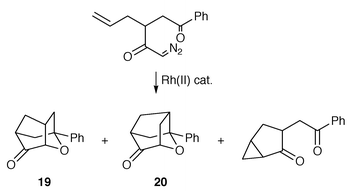
A study which provides more direct support for the involvement of a Rh(II) catalyst in a carbonyl ylide reaction was reported by Padwa and co-workers in 1992.26 A series of competition experiments were carried out in order to establish trends in metallocarbene selectivity between cycloaddition and cyclopropanation with respect to the nature of the ligands used in the Rh(II) catalyst (Scheme 27).
 | ||
| Scheme 27 | ||
Although the chemoselectivity profile was found to be insensitive to the catalyst used, a regiochemical crossover in the carbonyl ylide cycloaddition was observed on changing the catalyst. The major regioisomer formed using Rh2(OAc)4 or Rh2(cap)4 (cap = caprolactamate) was found to be 19; however, 20 was the predominant cycloadduct formed under Rh2(tfa)4 (tfa = trifluoroacetate) catalysis. Padwa originally proposed two explanations to account for these observations. One possibility is that immediately after dipole generation, the Rh2(tfa)4 catalyst coordinates with the nearby olefinic π-bond. This complexation could affect the product partitioning by lowering the LUMOalkene energy and the cycloaddition then becoming controlled by the HOMOdipole, instead of cycloaddition under LUMOdipole control with the more electron-rich free alkene. Alternatively, the Rh2(tfa)4 catalyst may still be coordinated with the dipole and this catalyst-complexed species could then undergo a subsequent cycloaddition with a different regiochemical profile to that of the free carbonyl ylide. The first option cannot be discounted, since it has been shown that Rh2(tfa)4 can coordinate to a range of olefins. Conversely, the UV absorption spectrum of Rh2(OAc)4 remains unchanged on exposure to the same set of alkenes, suggesting there is no interaction between Rh2(OAc)4 and alkenes in solution. The second proposal should be extended to include the possibility that Rh2(OAc)4 or Rh2(cap)4 can remain associated to the carbonyl ylide, despite presumably being less electrophilic than Rh2(tfa)4. Pirrung and Morehead have found that in reactions under Rh2(tfa)4 catalysis, the free carbene is more likely to be released, due to very weak Rh–carbene back bonding.27
In contrast with Landgrebe (Scheme 25),24 Padwa et al. have provided a set of results which were suggested as indicating reversibility of carbonyl ylide formation.28 Unsaturated diazotrione 21 under Rh(II) catalysis failed to undergo intramolecular cyclisation–cycloaddition; cyclopropanation was found to be the only observed reaction pathway (Scheme 28).
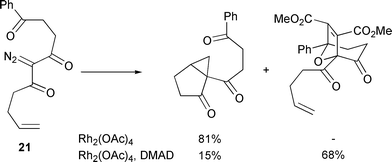 | ||
| Scheme 28 | ||
The isolation of a product derived from cyclopropanation suggested that
perhaps carbonyl ylide formation was somehow disfavoured in this particular
instance. To investigate this hypothesis the transformation was repeated in
the presence of dimethylacetylene dicarboxylate (DMAD). Interestingly, this
reaction furnished the cycloadduct derived from intermolecular carbonyl
ylide cycloaddition as the major product (82∶18,
cycloadduct∶cyclopropane). This observation was rationalised by
suggesting that in both cases the (catalyst-complexed) carbonyl ylide
formed, but is unable to undergo cycloaddition to the tethered
alkene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) —possibly because of the carbonyl group in the
tether resulting in the two-plane orientation approach required for
cycloaddition not being easily achievable. In the absence of DMAD, it was
proposed that this intermediate reverted to the rhodium metallocarbene
which then underwent intramolecular cyclopropanation. Although this
interpretation is consistent with the observed results, it could also be
possible that cycloaddition and cyclopropanation are proceeding
via a common intermediate, which is not necessarily formed
reversibly. Since variation of the catalyst had no effect on the ratio
cycloadduct∶cyclopropane, this intermediate is most straightforwardly
assigned as the catalyst-free carbonyl ylide.
—possibly because of the carbonyl group in the
tether resulting in the two-plane orientation approach required for
cycloaddition not being easily achievable. In the absence of DMAD, it was
proposed that this intermediate reverted to the rhodium metallocarbene
which then underwent intramolecular cyclopropanation. Although this
interpretation is consistent with the observed results, it could also be
possible that cycloaddition and cyclopropanation are proceeding
via a common intermediate, which is not necessarily formed
reversibly. Since variation of the catalyst had no effect on the ratio
cycloadduct∶cyclopropane, this intermediate is most straightforwardly
assigned as the catalyst-free carbonyl ylide.
Doyle et al. have reported a study of the stereocontrol in a series of intermolecular carbonyl ylide formation–intermolecular cycloaddition transformations. The results indicate (and were analysed on the basis) that some of the reactions proceed via catalyst-associated ylides, although concerted cycloadditions were not considered from these intermediates.29 Dioxolane products were formed using aromatic aldehydes, which were employed both to form a carbonyl ylide by reaction with a metallocarbene, and also to function as dipolarophile (Scheme 29).
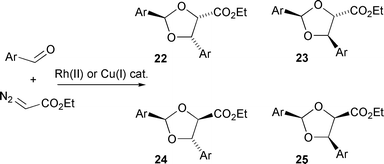 | ||
| Scheme 29 | ||
It was found that when using 4-methoxybenzaldehyde or benzaldehyde, the dioxolane isomers formed were 22 and 23 and the ratio of these dioxolanes (52∶48) was independent of the catalyst used in their formation, implying the absence of the catalyst in the cycloaddition step. For each of the aromatic aldehydes molecular modelling investigations were carried out on the four isomeric catalyst-free carbonyl ylides that potentially can be formed. On the basis of these modelling studies, it was proposed that cycloadditions of carbonyl ylides formed from 4-methoxybenzaldehyde and benzaldehyde occur through the thermodynamically most stable ‘sickle’-shaped ylide 27 (Scheme 30).
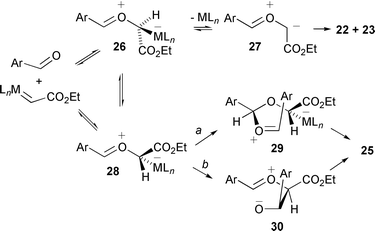 | ||
| Scheme 30 | ||
For 4-methoxybenzaldehyde and benzaldehyde, interconversion between the
catalyst-associated ylides 26 and 28 (either by C–O
rotation, and/or dissociation–recombination) followed by catalyst
dissociation from 26 gives ylide 27; for 28
Doyle has suggested there may be a stereoelectronic barrier to catalyst
dissociation, due to lack of lone pair donation from O+ to
σ* C–CO2Et and/or repulsion between the
lone pair of O+ and oxygen lone pairs of CO2Et.
Alternatively, any of the other three catalyst-free ylides isomeric to
27 that may be formed isomerise to 27 before
cycloaddition. Interestingly, when the transformation was carried out using
4-nitrobenzaldehyde all four possible dioxolanes
22–25 were obtained, and the product profile was
sensitive to the catalyst used—with the
thermodynamically least stable all-cis dioxolane 25
predominating when catalysts with more electron-withdrawing ligands were
used [% of 25 in dioxolane mixture: 16% using
Rh2(cap)4; 54% using Rh2(OAc)4;
45% with Rh2(perfluorobutyrate)4; 70% with
Cu(MeCN)4PF6]. In a subsequent report by Doyle,
aromatic aldehydes with less-powerful electron-withdrawing substituents in
the para position were also found to produce some of the
all-cis dioxolane 25, although as a relatively smaller
proportion of the dioxolane product mixture.30 In 1998 Doyle and Forbes reported in a review
that using Rh2(S-MEOX)4 with
4-nitrobenzaldehyde gave dioxolane 25 in 32% yield and 28% ee,
along with a 26% yield of isomeric dioxolanes.3 The observation of enantioselectivity supports the
suggestion of dioxolane 25 being formed, at least in part,
via a catalyst-complexed intermediate. Because the
all-cis dioxolane 25 is only seen in the
catalyst-dependent reactions, the ‘w’-shaped
catalyst-associated ylide 28 is implicated as an intermediate
leading to 25. A stepwise mechanism was proposed for reaction
through the catalyst-complexed ylide 28 (Scheme 30, path a), which involves
nucleophilic attack on such an intermediate by the aldehyde to give
29. The orientation of the incoming aldehyde, via
complexation through the less accessible lone pair on the aldehyde O, was
explained as arising from minimisation of steric effects that allows the Ar
group to avoid interaction with the protruding H on the carbenic carbon.
Catalyst decomplexation and ring-closure then give dioxolane 25 as
the major product. An alternative stepwise mechanism to that proposed by
Doyle involves initial formation of the carbon–carbon bond between
the catalyst-complexed carbon of 28 and the aldehyde with
concomitant catalyst dissociation to give 30 (Scheme 30, path b), followed by
cyclisation. This could explain why reaction occurs through the
catalyst-complexed intermediate when using more electrophilic aldehydes
such as 4-nitrobenzaldehyde.
Doyle extended his study to include a three-component system, in which DMAD was also present, thus allowing the free or catalyst-complexed carbonyl ylide to undergo competitive cycloaddition with two dipolarophiles. When 4-methoxybenzaldehyde was used, the trans dihydrofuran 31 was isolated as the sole product (Scheme 31); this is consistent with the cycloaddition taking place through free ylide 27.
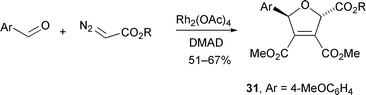 | ||
| Scheme 31 | ||
Interestingly, when 4-nitrobenzaldehyde and DMAD were used with Rh2(OAc)4 the full complement of products (total yield 30%) was obtained: both cis- and trans-dihydrofurans (30% and 43% of total) and all four dioxolanes 22–25 (27% of total). As in the reaction in the absence of DMAD, the major dioxolane product isolated (82% of dioxolane mixture) was the thermodynamically least stable dioxolane 25. When the concentration of 4-nitrobenzaldehyde was doubled, the ratio of dihydrofurans to dioxolanes changed from 73∶27 to 45∶55; however, the cis–trans dihydrofuran ratio and the ratio of dioxolanes were both unchanged. The most straightforward explanation for these observations is that both 4-nitrobenzaldehyde and DMAD compete for the catalyst-associated ylides 26 and 28 which interconvert more rapidly than any cycloaddition processes occur.
Hodgson and co-workers reported the first examples of enantioselective carbonyl ylide cycloaddition (up to 53% ee) using unsaturated α-diazo-β-keto esters in 1997 (e.g., Scheme 32).31
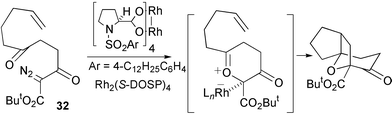 | ||
| Scheme 32 | ||
Because the catalyst-free carbonyl ylide would be achiral, the observation of enantioselectivity provides unambiguous evidence for an enantioselective ylide transformation taking place via a catalyst-complexed intermediate (for a generalised analysis of the process see Scheme 33).
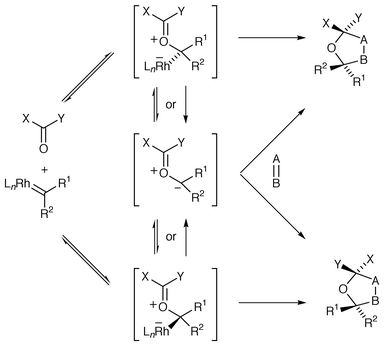 | ||
| Scheme 33 | ||
Attack of the metallocarbene by the tethered carbonyl would give
initially a catalyst-complexed ylide species where the catalyst is attached
to the originally carbenic carbon. Assuming that during the ensuing
cycloaddition the catalyst remains associated with the
CO+–C− part of the ylide, rather
than ligation with a carbonyl group or carbonyl groups, then the chiral
catalyst can only be associated with either face of a single carbonyl
ylide, since the ylide is part of a six-membered ring. It is then suggested
that cycloaddition occurs on the opposite face of the ylide to the catalyst
as the catalyst dissociates. In this case the cycloaddition is likely to be
a concerted process because the dipolarophile is a simple unpolarised
alkene. If one also assumes for the moment that no cycloaddition occurs
competitively from the catalyst-free ylide, then two suggestions for the
origin of the enantioselectivity are as follows. If the two
catalyst-associated ylide isomers do not interconvert within the timescale
of the cycloaddition, then the enantioselectivity is governed by the
preference of the tethered carbonyl to cyclise to the Re or
Si face of the metallocarbene under the influence of the chiral
ligands of the catalyst. Alternatively, interconversion between the two
catalyst-associated ylide isomers via a dissociation (to the
acyclic metallocarbene)–recombination mechanism could be faster than
the rate(s) of cycloaddition. This latter case describes a
Curtin–Hammett situation with the relative proportions of the two
catalyst-associated ylides being inconsequential and the enantioselectivity
being determined by the difference in the free energies of the activation
barriers (ΔΔG#) of the two
catalyst-associated ylide isomers for cycloaddition. Regardless of which of
these two processes operates, enantioselectivity could be affected if the
catalyst dissociates from the ylide prior to, or competitively with,
cycloaddition from the catalyst-associated ylide. If catalyst dissociation
is reversible and is fast compared with the rates of catalyst-associated
and catalyst-free cycloadditions then the relative rates of these
cycloadditions will also be an important factor influencing the level of
asymmetric induction observed.
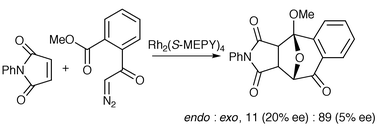
In 1998 Suga, Ishida and Ibata reported a study of catalyst stereocontrol in intermolecular cycloaddtion of an ‘aromatic’ carbonyl ylide with N-substituted maleimides (Scheme 34).32
 | ||
| Scheme 34 | ||
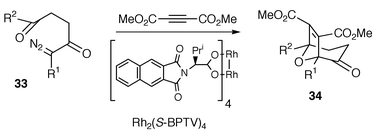
Interestingly, copper-based catalysts favoured the endo (with respect to the ylide-containing ring) cycloadduct (endo∶exo, up to 94 : 6 with N-methyl maleimide and 20% CuOTf), whereas rhodium catalysts favoured the exo adduct. Low levels of enantioselectivity were observed with N-phenyl maleimide and a CuOTf–bisoxazoline catalyst or Rh2(S-MEPY)4. More recently, Hashimoto and co-workers observed significant asymmetric induction using α-diazo ketones with DMAD as the dipolarophile, where enantioselectivities up to 92% were reported (Scheme 35, R1 = H, R2 = Ph).33
 | ||
| Scheme 35 | ||
In Hashimoto’s work high levels of enantioselectivity were consistently maintained with electron-donating substituents at the para position on the benzene ring (R1 = H, R2 = 4-MeOC6H4 or 4-MeC6H4), whereas a slight drop in enantioselectivities was observed by the introduction of electron-withdrawing groups on the benzene ring (Cl or CF3 at the 4-position) as well as with R2 = alkyl substituents. The procedure was also found to allow for variation of the tether length, with cycloadducts via five- and seven-membered cyclic carbonyl ylide intermediates being formed in 68% and 80% ee, respectively. Hashimoto’s observations of good to excellent asymmetric induction may be contrasted with the isolation of a racemic cycloadduct by Hodgson,34 when the intramolecular cycloaddition precursor 32 was exposed to the same catalyst–solvent combination as used in Hashimoto’s study. Furthermore, cycloadduct 34 (R1 = CO2Et, R2 = Me) was obtained in only 33% ee under the same conditions in the reaction of α-diazo-β-ketoester 33 (R1 = CO2Et, R2 = Me) with DMAD34 [α-diazoketone 33 (R1 = H, R2 = Me) gave cycloadduct in 80% ee].33 These latter results indicate that ee is rather sensitive to variation in the electronic structure of the dipole. Hodgson and co-workers have also shown that rhodium phosphate catalysts can be superior to the more commonly utilised carboxylate and carboximidate analogues in asymmetric transformations of diazocarbonyl compounds by observing enantioselectivities of up to 90% in the cycloaddition of 32 using a hydrocarbon soluble variant of Rh2(binaphtholphosphate)4.34
5 Azomethine ylides
Jacobsen and co-workers have reported the enantioselective formation of aziridines from the reaction of a metallocarbene with imines (Scheme 36).35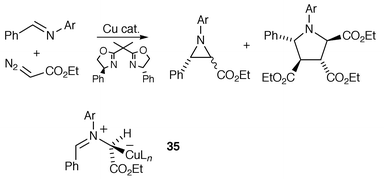 | ||
| Scheme 36 | ||
Asymmetric induction (up to 67% ee) was achieved in the aziridinations by using a Cu(I) catalyst–bisoxazoline combination. Pyrrolidines were often obtained as by-products in these reactions, and were likely formed by [3 + 2] cycloaddition of an azomethine ylide intermediate and diethyl fumarate (generated from ethyl diazoacetate). Jacobsen proposed a mechanism for the reaction which involves initial nucleophilic attack of the metallocarbene by the imine nitrogen lone-pair. The resulting catalyst-complexed azomethine ylide 35 was suggested as either cyclising, effecting enantioselective aziridine formation, or undergoing (reversible) catalyst decomplexation to give free ylide from which cyclisation could afford a racemic aziridine. Jacobsen’s results are therefore consistent with the possibility of aziridination taking place via a catalyst-complexed ylide. As the pyrrolidine was obtained as the racemate, it was considered as arising solely from the free ylide, although non-enantioselective cycloaddition of the catalyst-associated ylide 35 cannot be ruled out. It is also possible that the catalyst-complexed azomethine ylide 35 is in rapid (relative to aziridination or cycloaddition) equilibrium with the metallocarbene and imine, and aziridine formation could then occur, at least in part, by a direct insertion process from the metallocarbene, analogous to alkene cyclopropanation.
6 Concluding remarks
It is obvious from the preceding sections that a catalyst can exert significant influence on ylide-based rearrangements and cycloadditions using diazo compounds. However, in many cases the origin of that influence is currently unclear. In [3,2] sigmatropic rearrangements, the observations by Doyle and Aggarwal of catalyst-dependent diastereoselectivities provide evidence that catalysts can remain associated with chalcogen ylides during the rearrangement process. These results suggest the exciting possibility that further development of chiral catalysts will ultimately allow a range of these transformations to be carried out to deliver products not only in high ee, but also with enhanced de over the non-catalyst influenced process. Similarly, in [3 + 2] cycloadditions, Hodgson’s, Ibata’s and Hashimoto’s results suggest that efficient catalyst control over enantioselectivity and diastereoselectivity can eventually be developed, although major challenges clearly lie ahead in developing catalysts that are effective with various ylide types and substitution patterns and different dipolarophiles. Despite the encouraging and significant progress that has been made so far in all of these fields, future developments are likely to depend on gaining a greater understanding of the mechanisms of these fascinating and synthetically important processes.7 Acknowledgements
We thank the EPSRC (Research grant GR/L98022), AstraZeneca and GlaxoWellcome for supporting our work in the field of enantioselective ylide transformations.8 References
- A. Padwa and S. F. Hornbuckle, Chem. Rev., 1991, 91, 263 CrossRef CAS.
- M. P. Doyle, M. A. McKervey and T. Ye, Modern Catalytic Methods for Organic Synthesis with Diazo Compounds, Wiley, New York, 1998. Search PubMed.
- M. P. Doyle and D. C. Forbes, Chem. Rev., 1998, 98, 911 CrossRef CAS.
- A.-H. Li, L.-X. Dai and V. K. Aggarwal, Chem. Rev., 1997, 97, 2341 CrossRef CAS.
- R. B. Woodward and R. Hoffmann, Angew. Chem., Int. Ed. Engl., 1969, 8, 781 CrossRef CAS.
- M. C. Pirrung, W. L. Brown, S. Rege and P. Laughton, J. Am. Chem. Soc., 1991, 113, 8561 CrossRef CAS.
- F. G. West, T. H. Eberlein and R. W. Tester, J. Chem. Soc., Perkin Trans. 1, 1993, 2857 RSC.
- H. Nozaki, H. Takaya, S. Moriuti and R. Noyori, Tetrahedron, 1968, 24, 3655 CrossRef CAS and references cited therein..
- E. J. Roskamp and C. R. Johnson, J. Am. Chem. Soc., 1986, 108, 6062 CrossRef CAS.
- J. S. Clark, Tetrahedron Lett., 1992, 33, 6193 CrossRef CAS.
- J. B. Lambert and D. H. Johnson, J. Am. Chem. Soc., 1968, 90, 1349 CrossRef CAS.
- K. Ito, M. Yoshitake and T. Katsuki, Heterocycles, 1996, 42, 305 Search PubMed.
- M. P. Doyle, in Comprehensive Organometallic Chemistry II, eds. E. W. Abel, F. G. A. Stone and G. Wilkinson, Pergamon Press, Oxford, 1995, Vol. 12, p. 421. Search PubMed.
- M. P. Doyle, D. G. Ene, D. C. Forbes and J. S. Tedrow, Tetrahedron Lett., 1997, 38, 4365.
- N. Pierson, C. Fernandez-Garcia and M. A. McKervey, Tetrahedron Lett., 1997, 38, 4705 CrossRef CAS and references cited therein..
- J. S. Clark, M. Fretwell, G. A. Whitlock, C. J. Burns and D. N. A. Fox, Tetrahedron Lett., 1998, 39, 97 CrossRef CAS.
- M. P. Doyle, D. C. Forbes, M. M. Vasbinder and C. S. Peterson, J. Am. Chem. Soc., 1998, 120, 7653 CrossRef CAS.
- M. A. Calter and P. M. Sugathapala, Tetrahedron Lett., 1998, 39, 8813 CrossRef CAS.
- Y. Nishibayashi, K. Ohe and S. Uemura, J. Chem. Soc., Chem. Commun., 1995, 1245 RSC and references cited therein..
- T. Fukuda, R. Irie and T. Katsuki, Tetrahedron, 1999, 55, 649 CrossRef CAS.
- V. K. Aggarwal, M. Ferrara, R. Hainz and S. E. Spey, Tetrahedron Lett., 1999, 40, 8923 CrossRef CAS.
- D. W. McMillen, N. Varga, B. Ann Reed and C. King, J. Org. Chem., 2000, 65, 2532 CrossRef CAS.
- A. C. Lottes, J. A. Landgrebe and K. Larsen, Tetrahedron Lett., 1989, 30, 4093 CrossRef CAS.
- A. C. Lottes, J. A. Landgrebe and K. Larsen, Tetrahedron Lett., 1989, 30, 4089 CrossRef CAS.
- H. M. L. Davies, N. J. S. Huby, W. R. Cantrell, Jr. and J. L. Olive, J. Am. Chem. Soc., 1993, 115, 9468 CrossRef CAS.
- A. Padwa, D. J. Austin and S. F. Hornbuckle, J. Org. Chem., 1996, 61, 63 CrossRef CAS and references cited therein..
- M. C. Pirrung and A. T. Morehead, Jr., J. Am. Chem. Soc., 1994, 116, 8991 CrossRef CAS.
- A. Padwa, S. F. Hornbuckle, G. E. Fryxell and Z. J. Zhang, J. Org. Chem., 1992, 57, 5747 CrossRef CAS.
- M. P. Doyle, D. C. Forbes, M. N. Protopopova, S. A. Stanley, M. M. Vasbinder and K. R. Xavier, J. Org. Chem., 1997, 62, 7210 CrossRef CAS.
- M. P. Doyle, D. C. Forbes and K. R. Xavier, Russ. Chem. Bull., 1998, 47, 932 Search PubMed.
- D. M. Hodgson, P. A. Stupple and C. Johnstone, Tetrahedron Lett., 1997, 38, 6471 CrossRef CAS.
- H. Suga, H. Ishida and T. Ibata, Tetrahedron Lett., 1998, 39, 3165 CrossRef CAS.
- S. Kitagaki, A. Masahiro, O. Kataoka, K. Matsuno, C. Umeda, N. Watanabe and S. Hashimoto, J. Am. Chem. Soc., 1999, 121, 1417 CrossRef CAS see also: S. Kitagaki, M. Yasugahira, M. Anada, M. Nakajima and S. Hashimoto, Tetrahedron Lett., 2000, 41, 5931 CrossRef CAS.
- D. M. Hodgson, P. A. Stupple and C. Johnstone, Chem. Commun., 1999, 2185 RSC.
- K. B. Hansen, N. S. Finney and E. N. Jacobsen, Angew. Chem., Int. Ed. Engl., 1995, 34, 676 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2001 |