Towards artificial photosynthesis: ruthenium–manganese chemistry for energy production
Licheng Suna, Leif Hammarströmb, Björn Åkermarka and Stenbjörn Styringc
aDepartment of Organic Chemistry, Arrhenius Laboratory, Stockholm University, 106 91, Stockholm, Sweden
bDepartment of Physical Chemistry, Uppsala University, PO Box 532, 751 21, Uppsala, Sweden
cDepartment of Biochemistry, Centre for Chemistry and Chemical Engineering, University of Lund, PO Box 124, S-221 00, Lund, Sweden
First published on 28th November 2000
Abstract
The synthesis and characterisation of supramolecular model systems mimicking the light reactions on the donor side of Photosystem II (PSII) in green plants have been reviewed. In these systems, manganese complexes and tyrosine are electron donors, modelling the manganese cluster and tyrosineZ in PSII. The donors have been covalently linked to a photosensitizer, a ruthenium(II) tris-bipyridyl complex, that plays the role of the P680 chlorophylls in PSII. It has been demonstrated that, in the presence of an external electron acceptor in solution, the model systems can undergo an intermolecular electron transfer from the photoexcited state of RuII to an acceptor, followed by an intramolecular electron transfer from the coordinated Mn complexes or the tyrosine unit to the photogenerated RuIII. This leads to regeneration of the RuII and oxidation of the Mn complexes or generation of a tyrosine radical. The process closely mimics the primary reaction steps on the donor side of PSII.
 Licheng Sun Licheng Sun | Dr Licheng Sun (born 1962) obtained his PhD in Organic Chemistry at Dalian University of Technology in collaboration with the Chinese Academy of Sciences, China, in 1990. He did postdoctoral research on photochemistry with Helmut Görner at the Max-Planck-Institut für Strahlen Chemie, Germany. During 1993–1995 he was the Alexander von Humboldt Research Fellow with Harry Kurreck, Freie Universität Berlin, on porphyrin chemistry. In 1997, he became assistant professor at the Royal Institute of Technology (KTH), Sweden. He was appointed Docent (associate professor) in 1999. He leads the synthetic group at Stockholm University together with Professor Åkermark. His main interests are in supramolecular chemistry and artificial enzymes. |
 Leif Hammarström Leif Hammarström | Dr Leif Hammarström (born 1964) obtained his PhD 1995 in Physical Chemistry at Uppsala University, Sweden, working on electron transfer in lipid vesicles with Professor Mats Almgren. During his postdoctoral stay with Professor Vincenzo Balzani, Bologna, he studied the photochemistry and photophysics of transition metal complexes. After returning to Uppsala in 1997, he became assistant professor and formed his own group. In 1999, he was appointed Docent (associate professor). His main interests are in photochemistry and electron transfer reactions in supramolecular systems, studied by time-resolved optical spectroscopy. |
 Björn Åkermark Björn Åkermark | Professor Björn Åkermark (born 1933) obtained his PhD in 1967 in Organic Chemistry at the Royal Institute of Technology, where he also was appointed Associate Professor the same year. After a year as Visiting Scholar at Stanford, where he studied chemical nitrogen fixation with Professor Eugene Van Tamelen, he returned to the Royal Institute to work with cycloaddition reactions, photochemistry and homogenous metal catalysis. In 1980 he was appointed Professor of Organic Chemistry. He retired from this position in 1999 and moved to Stockholm university, where he is presently associated as research leader. His research interests are in homogenous catalysis and synthesis and function of supramolecular metal complexes. |
 Stenbjörn Styring Stenbjörn Styring | Professor Stenbjörn Styring (born 1951) obtained his PhD in Biochemistry at Göteborg, Sweden in 1985. During post-doctoral studies in Gif-sur-Yvette and Saclay with Anne-Lise Etienne and Bill Rutherford he turned to studies of Photosystem II. He returned to Stockholm in 1987 and continued studies on Photosystem II. During this period he also initiated studies of artificial photosynthesis. Since 1996 he has been Professor in Biochemistry at Lund University. His main interests are in spectroscopic studies of electron transfer reactions in Photosystem II and the development of artificial photosynthesis for fuel production. He has been the chairman of the Swedish Consortium for Artificial Photosynthesis since this started in 1995. |
1 Introduction
Life is dependent on energy conversion and energy balance. This holds for natural ecosystems as well as for our society. In nature, photosynthetic organisms convert the energy of solar light into chemical energy in the form of reduced organic compounds such as carbohydrates, fats, amino-acids etc. Since this energy source can be continuously replenished, it is used by all other organisms to develop and sustain life and the abundance of organic substrate is very often limiting in natural environments. The same holds in our modern society that, to a large extent, has reached its complexity and strength thanks to the employment of large amounts of energy. For a long time this was not a major concern but in the last decades it has become clear that our dominating energy sources are connected to socially or environmentally problematic effects. The use of fossil fuels (coal, oil, natural gas etc.) is often linked to very severe air pollution. Even more seriously, these energy sources are limited and some of them will be running out in the fairly near future. More recently the potential global warming caused by the net CO2 efflux due to combustion of fossil fuels has become a major concern. Nuclear power has a different risk profile, and seems, in its present shape, to be socially unacceptable in many countries. Other energy sources, such as wind power or hydroelectric power, have other recognised problems. Therefore, due to the increasing energy consumption in today’s society, it has become clear that new energy systems need to be based on sustainable and environmentally friendly energy sources.Solar energy emerges as one of the most promising sources for future sustainable production of fuel and electricity. One attractive way to harvest solar energy is to use the concepts of natural photosynthesis to build an artificial system. This system should convert solar energy into fuel, such as hydrogen, which could be stored in a chemical form. However, we are not aware of any man-made system which operates on the principles of the photosynthesis of green plants, thereby reaching nature’s high efficiency in the primary reactions.† Basic research is clearly needed in this area. In natural oxygen-producing photosynthesis, the solar energy is converted into chemical energy with water as electron source. The efficiency in the crucial steps is remarkably high. The construction of an artificial system that exactly mimics these reactions remains a great challenge to chemists. If a successful mimetic system could be made, much would be learnt at a fundamental level. Furthermore, and potentially very importantly, we would have new tools to better exploit solar energy for the generation of fuels. In this review we will describe our attempts to develop an artificial system that mimics one of the most vital parts of natural photosynthesis, the light-driven oxidation of water.
2 Photosystem II—the water oxidizing enzyme
Photosystem II (PSII) is the key enzyme in the light-driven reactions in photosynthesis of plants, algae and cyanobacteria.1 It is the only photosynthetic reaction centre (RC) that can use water as an electron source—thereby it provides the plants (and indirectly the entire biosphere) with an unlimited source of electrons. PSII carries out the general reactions: light absorption, energy transfer, charge separation, and charge stabilisation, that are common to all photosynthetic reaction centres. In addition, PSII has the ability to use light to drive electron transport from water to a quinone acceptor (plastoquinone) which is reduced in the reaction. The reduced plastoquinone subsequently carries the reducing equivalents further in the photosynthetic electron transfer chain. This provides reducing power to many biosynthetic reactions in the plants. As a waste product, water oxidation in PSII forms molecular oxygen which is the prerequisite for the existence of most higher life forms, placing the PSII reaction centre in a central position in the biosphere.In this review we will describe efforts to develop supermolecules mimicking the light driven oxidation of water accomplished by PSII. These efforts are inspired by the structure and function of the natural system and we will therefore describe, in considerable detail, the water oxidising reactions in PSII which 1) couple the one-photon charge separation to the four-electron oxidation of water and 2) reach potentials high enough to oxidize water (>0.9 V vs NHE) and handle these very oxidizing redox potentials in fragile biological structures.
The key player in water oxidation is a triad composed of a multimer of chlorophylls (named P680), a redox active amino acid (named tyrosineZ), and a Mn-cluster composed of 4 Mn ions of high valence (the oxygen evolving complex, OEC),2 see Fig. 1 and 2. PSII spans the thylakoid membrane in the chloroplasts and the water oxidising triad is located closely to one side of the membrane (Fig. 1). The Mn cluster also contains a Cl− and a Ca2+ ion which are tightly coupled to the Mn ions.
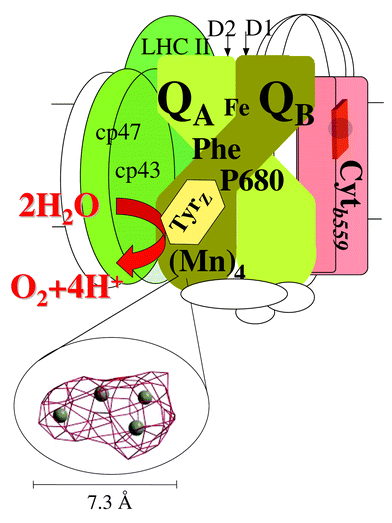 | ||
| Fig. 1 A schematic picture of the Photosystem II reaction centre with some of the central proteins in PSII. Most of the cofactors are bound to the D1 and D2 proteins. The reaction centre also contains a series of chlorophyll containing so-called antenna proteins (LHCII, CP43 and CP47 green in the figure; altogether an intact PSII complex in a normal plant contains about 200 chlorophyll molecules that are involved in the absorption and transfer of light energy). Water oxidation is accomplished by the triad composed of the primary donor P680, the tyrosineZ radical and the Mn4 cluster. Their approximate location close to each other on the lumenal side of PSII is shown in the figure. The distance between TyrZ and P680 is about 12 Å and the distance between the centre of TyrZ and the centre of the Mn cluster is 7–8 Å. The structure of the metal cluster is shown as 4 metal ions (Mn or Ca) that are drawn in the actually observed electron density map at 4.5 σ obtained from the 3.5 Å X-ray resolution of the PSII structure. The cluster structure has been generously provided by Drs Peter Orth, Athina Zeoni and Professor Wolfram Saenger (Berlin, Germany) prior to the publication of the structure. The density is shown at an approximately 45 degree angle compared to the membrane plane. | ||
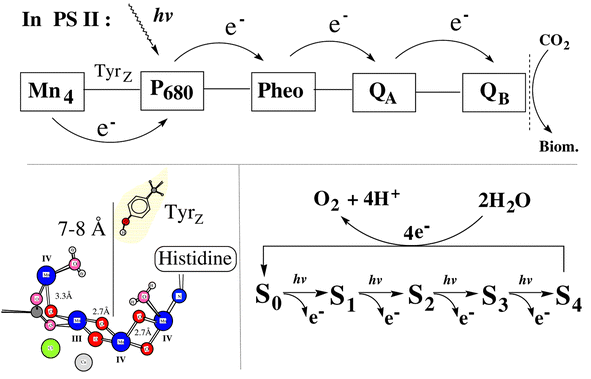 | ||
| Fig. 2 Schematic presentation of the electron transfer processes involved in the natural PSII reaction centre and a proposed structure for the Mn cluster. On the lower right we show the five oxidation states involved in the water oxidation cycle. The redox states are denoted S0 to S4 where S0 is the most reduced state. The structure of the Mn complex shown here is the so called ‘dangler’ model of the Mn4 cluster. This is derived from advanced simulations of EPR and ENDOR spectra from the S2 state and an imaginative, structural alignment of the outcome from the EPR simulations to the available EXAFS data. Some of the esential elements of the structure are the organisation of the two shorter Mn–Mn distances forming a core structure of 3 Mn ions which are close to each other while the fourth Mn ion is found 3.3 Å away from these. Two bound water molecules are also indicated but their exact location is arbitrary. The figure also shows the preferred assignemnt of the Mn valences in the S2 state as proposed from the EPR simulations. The structure has been kindly provided by Professor R. D. Britt (UC Davis, California) and coworkers prior to its publication.8 | ||
Upon the absorption of a photon, P680 is excited. It becomes strongly reducing and transfers an electron to the acceptor system (pheophytin and two quinones QA and QB) on the other side of the membrane. The oxidised primary donor, P680+, is one of nature’s most oxidising species and reaches a potential of +1.2 V vs NHE. P680+ is rapidly reduced by tyrosineZ which is located about 12 Å away. TyrZ has several functions. It is a very fast electron donor to P680+ (t1/2 = 20–200 nanoseconds, dependent on the S-state in which the Mn cluster is present before the electron transfer) and thereby prevents deleterious back reactions from the quinones on the acceptor side. In its reduced state, TyrZ is hydrogen bonded to a nearby histidine residue. However, upon oxidation, TyrZ gives rise to an EPR spectrum which originates from the tyrosine in its strongly oxidising (∼+0.95 V), neutral radical form. Thus, TyrZ is deprotonated upon its oxidation and the existence of the hydrogen bond to the histidine facilitates rapid deprotonation of the tyrosine, thereby promoting fast electron transfer to P680+. In situations when the hydrogen bond is not formed (at low pH, in mutants in the histidine, etc.) TyrZ is a slow, if at all functional, electron donor to P680+. The oxidised TyrZ radical is reduced by electrons which ultimately are derived from water. At present, it is much debated how the crucial hydrogen bond between TyrZ and the histidine is re-established.
In the past 4–5 years the water oxidation mechanism has become one of the most studied and debated problems in bio-energetic research. The main reason for this recent outburst of interest is a series of pioneering, innovative mechanistic and theoretical studies by G. T. Babcock and his co-workers. In this work they challenge the traditional view of how water is oxidized in PSII. The dominating view earlier was that TyrZ oxidises the Mn4-cluster in a stepwise manner by simple electron transfer reactions. Thereby, the Mn cluster would serve as a charge storing device that sequentially becomes more and more oxidizing as more electrons are withdrawn. Eventually the Mn4-cluster has reached its most oxidising state, the S4 state, which returns in a four-electron reduction to its most reduced state S0, simultaneously as two water molecules are oxidised to molecular oxygen (see Fig. 2 for the S-state formalism). During the catalytic cycle the protons are released from the OEC in a sequential, but not entirely elucidated manner. The function of TyrZ in these models was that of a pure electron transfer intermediate between the Mn-cluster and P680.
Babcock and co-workers observed that TyrZ was close enough to the Mn4-cluster that they might interact intimately during water oxidation.3 It was also noted that the available extra energy to drive the oxidative chemistry was similar and very small in each step in the S-cycle.4b Babcock also strongly argued that the OEC must be very conservative in its chemistry and that too large protein or substrate rearrangements must be avoided since about 200 water molecules per second can be oxidised by PSII when it works at full speed. Another argument they were first to apply to PSII chemistry was the concept of charge neutrality, i.e. that it is very costly to build up charge in a protein. From these arguments and supported by a series of theoretical studies Babcock and co-workers have formulated and fine-adjusted a mechanistic proposal in which TyrZ, after having been oxidized by P680+, retrieves not only an electron, but a hydrogen atom from water bound to the Mn complex.4 According to this mechanism, molecular oxygen is formed after four successive hydrogen atom transfers and the Mn cluster reverts to the S0 state. This mechanism is very attractive from an energetic point of view, because the reduction of (TyrZ)ox is charge neutral. A crucial consequence of this model is that protons are expelled from TyrZ immediately at its oxidation and that this proton release must reach the surface of the protein.4c Thus no protons are directly expelled from the Mn-cluster or Mn-coordinated substrate water, but instead the protons from substrate water are transferred to TyrZ upon its reduction. An elegant review on the function of TyrZ, covering most of these arguments, was recently presented.4c
Several of the proposals in the H-atom transfer mechanism are experimentally testable. In particular many groups have focused on the interaction between TyrZ and the hydrogen bonded histidine and it seems clear that this hydrogen bond (maybe in conjunction with another hydrogen bond) is crucial for the efficient functioning of the enzyme. However, at this moment it is not clear if protons are released from TyrZ to the medium via the histidine (as proposed by Babcock and co-workers) or if the proton release is mediated by a different pathway. This and other experimental ambiguities have manifested themselves in several other proposals for the oxidation of water in PSII (see for example ref. 5). In some of these, one or two of the S-state transitions involve pure electron transfer to TyrZ while the other transitions involve H-atom transfer. Thus, the field is very active and it is beyond the scope of this review to describe all the interesting proposals (many of these new proposals will be published in a special volume of Biochim. Biophys. Acta in the fall 2000).
Irrespective of mechanism, the Mn cluster cycles between five redox states during water oxidation. These are denoted S0–S4 where S4 is the most oxidised state. Molecular oxygen is released in the S4 → S0 transition, consequently S0 is 4 steps more reduced than the S4 state.
The structure, valence charges in the different S-states and the chemical function of the Mn cluster have been intensively studied for decades and we refer the reader to more specialised reviews to cover this topic.6 Structural and Mn valence information has mainly been obtained by EXAFS and EPR spectroscopy.7 These studies have established the presence of at least two di-μ-oxo bridged pairs of Mn distanced by 2.7 to 2.8 Å. It also seems clear that there is a longer distance between two of the Mn ions (3.3 Å) and much information indicates that the crucial cofactors Ca2+ and Cl− are found close to the Mn ions. The dominating opinion from these studies was for a long time that the 4 Mn ions form a C-shaped cluster held together by a system of oxo- and carboxylato-bridges although other structures than the C-shaped model were always pointed out as potential structures.7b An important consequence for organic chemists, who try to mimic the natural system by a synthetic approach, is that the Mn ions in PSII are organized in a non-symmetric environment.
The situation changed in the summer 2000 when the first X-ray structure of PSII to 3.5 Å resolution was presented (P. Orth, W. Saenger and A. Zeoni, Berlin, Germany, personal communication). The structure was obtained from oxygen evolving crystals from the core of PSII from a cyanobacterium, Synechococcus vulcanus. There are many fascinating aspects of this first structure from PSII. Many of these are beyond the scope of this review. However, the most important feature was that the Mn cluster was observed for the first time (Fig. 1). At the available resolution it was possible to observe a large electron density which was located to the lumenal side of PSII. It was found close to the end of two membrane spanning helices in the D1 protein. One of these is known to contain TyrZ and already at this early stage it was possible to confirm the close distance between TyrZ and the Mn cluster. This distance was earlier determined as 7–8 Å from EPR investigations and the X-ray structure seemingly verifies the validity of these measurements. Fig. 1 shows the Mn cluster as seen by X-ray crystallography at 3.5 Å resolution (the X-ray density structure was kindly provided by P. Orth, W. Saenger and A. Zeoni, Berlin, Germany). Four metals are presently found in the electron density which is shaped a little like a ham. However, the distance between the metal ions, their identity and how the bonds are constructed cannot be deduced at this stage. Despite this limitation, there is little doubt that the new X-ray structure will make it possible to support one or a few of the proposed structures for the Mn cluster. At this stage, it is possible to rule out structures where TyrZ is directly linked to Mn via its phenolic proton. It also seems possible to rule out models where the Mn cluster is spread out forming for example two dimers of Mn which are quite distant (5 Å or more). Thus, there is little doubt that structural and mechanistic aspects of the OEC will become much clearer over the next few years.
There is also much biochemical knowledge about the structure of the Mn cluster. Most of the metal ligands are oxygens from carboxylic sidechains, the oxobridges, substrate water molecules and potentially the peptide backbone. At least two histidine residues are known to participate in Mn ligation. Thus, there are many more oxygen ligands than nitrogen ligands to Mn in the natural system. Close by, probably directly coordinated to the Mn cluster, we also find the two essential cofactors Cl− and Ca2+, and the latter is probably linked directly to Mn via a carboxylato bridge.
The best characterized redox state is S2 (Fig. 2) which gives rise to a set of EPR signals. These have been extensively studied by all possible EPR techniques and many groups have attempted to simulate the spectra obtained. Presently, one of the most successful EPR simulation studies8 suggests that the Mn cluster in the S2 state is in the Mn(III)(IV)3 state. In this study, the authors also attempted to couple the knowledge from EXAFS spectroscopy with the EPR simulation and this led them to propose a new structure (the ‘dangler’) for the Mn cluster (Fig. 2, lower left). In this structure, one Mn is more weakly coupled to the other three which form a stronger coupled trinuclear core. The authors also proposed which Mn ion was in which valence state (Fig. 2). Recently, a new EPR signal was discovered9 in the S0 state which is thought to contain at least one Mn ion in the Mn(II) state and this was corroborated by the EPR simulation study described above(Peloquin and coworkers).It is a common view that the S1 state involves Mn(III)2 Mn(IV)2 but the valence situation in S3 and S4 are less clear. Here the oxidative chemistry is likely to involve oxidised amino acids and/or partial oxidation of water.
Another step which is much debated, but where the experimental evidence is almost totally lacking, is that in which the oxygen–oxygen bond is formed. Here, most proposals are derived from inorganic manganese chemistry (V. L. Pecoraro, and W.-Y. Hsieh, 2000. The use of Complexes to Elucidate the Structure and Function of Manganese Redox Enzymes, 2000, see also the forth coming volume of Biochim. Biophys Acta and references therein) or from theoretical considerations.10 Several authors prefer Mn(V)-oxo states as the starting point for the oxygen–oxygen bond formation while others prefer peroxide-containing intermediates. Since experimental evidence is essentially lacking, much more work is clearly needed in this area and the synthetic chemists mimicking the natural system have few ideas to bring home. Instead, it is likely that the researchers studying the natural system have much to learn from the easier studied synthetic systems. It is noteworthy that Brudvig and co-workers recently reported a synthetic di-oxo-bridged Mn2(III/IV) complex that is able to evolve oxygen under strongly oxidising conditions with oxone.11
3 Artificial photosynthesis—fuel production from solar energy and water
Over the past twenty years much effort has been devoted to studies of photoinduced electron transfer (ET) reactions from chlorophyll and analogues to acceptors, e.g. to mimic the acceptor side of Photosystem II, for example, porphyrin(s)–quinone(s) or porphyrin–fullerene systems.12 In contrast, the regeneration of chlorophyll and analogues from their photo-oxidised forms by electron transfers from manganese or tyrosine, e.g. to mimic the donor side of Photosystem II, has received very limited attention. A few studies have been presented,11,13,14 which show that manganese complexes can catalyse the conversion of water into oxygen. In these studies, however, the manganese oxidation has not been coupled to photochemical charge separation. In 1994, we launched a project with the major goal of developing an artificial model system for PSII. More specifically, the aim was to construct a supramolecular system by using nature’s principles. Our hope is that such a system can be used to produce a sustainable and renewable fuel, e.g. hydrogen from water, in a similar way as nature produces biomass (see Fig. 3, top).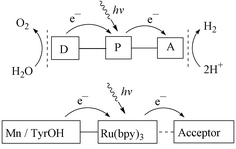 | ||
| Fig. 3 Schematic presentation of the electron transfer processes in an artificial photosynthetic system built on supramolecular Ru–Mn complexes. | ||
The primary charge separation in photosynthesis is the key reaction for life on earth in that it is the only reaction that converts solar energy into chemical energy. It is also remarkably efficient, for example almost all the energy in a photon of 680 nm is conserved in the charge separated state of Photosystem II (see below). However, the primary radical pair is short lived and will recombine within nanoseconds if the separated charges are not stabilised. This is accomplished by a series of charge stabilising reactions (Fig. 2) where the electron and electron hole are moved away from each other by a series of highly controlled reactions within the protein. In these reactions the organism ‘sacrifices’ some of the energy in the primary radical pair. The energy content in the final charge stabilised state in the PSII reaction centre is ∼50% of the photon energy at 680 nm. This seems wasteful at first glance, but the lifetime of the charge stabilised state is now long enough (ms to s) that complex chemistry is allowed both on the reducing and the oxidising side of the reaction centre.
Only ca. 1% of the solar energy hitting a field is converted to biomass by photosynthesis. This might seem poor. However, the primary light reactions are extremely efficient, converting a very large percentage of the absorbed light into chemical energy. The large losses that occur later in the life cycle are due to plant stress and inability to absorb light at all wavelengths. In addition, the converted energy is used by the plant to sustain life. The organism is not there to produce biomass, it is there to live!
When we design a system for artificial photosynthesis for fuel production it is the primary light processes, their efficiency and design we shall mimic. Here the potential is clearly very high and much is to be learnt from natural photosynthesis.
So far, we have focused our efforts on the synthesis, characterisation and light-induced electron transfer studies from manganese or tyrosine units to a photo-oxidised photosensitizer, to mimic the donor side of PSII (see Fig. 3, bottom). This is because there already exist many elegant mimics for the acceptor side, where a single electron is transferred between molecular components in triads, tetrads etc.12 In these systems, however, the electron recombines with the hole rather rapidly, usually on the ns–μs time scale. Therefore we have chosen to focus our synthetic and experimental efforts on the donor side and use simple bimolecular reactions on the acceptor side. Thus, we use methyl viologen or cobalt(III) pentamine chloride as external electron acceptors, this gives us ca. 100 μs (recombination half-life) to observe donor side reactions before the electron returns from the acceptor, which is better than with most triads and tetrads. We use ruthenium tris-bipyridyl complexes as photosensitizers to mimic the function of P680 in PSII, since they are stable, relatively easy to functionalise, and have favourable photophysical properties.
3.1 Ruthenium tris-bipyridyl complex covalently linked to monomeric manganese complexes
When we started the project, the first question was if it was at all possible to demonstrate an intramolecular electron transfer from a manganese complex to a photooxidized ruthenium tris-bipyridyl complex. To answer this question, a group of ruthenium tris-bipyridyl complexes were synthesised.15 Each of these complexes has a free ligand with variation in the bridging linkage and the distance between ruthenium and the ligand. Complexation of manganese(II) chloride with these compounds gave binuclear ruthenium–manganese complexes, 1–8.15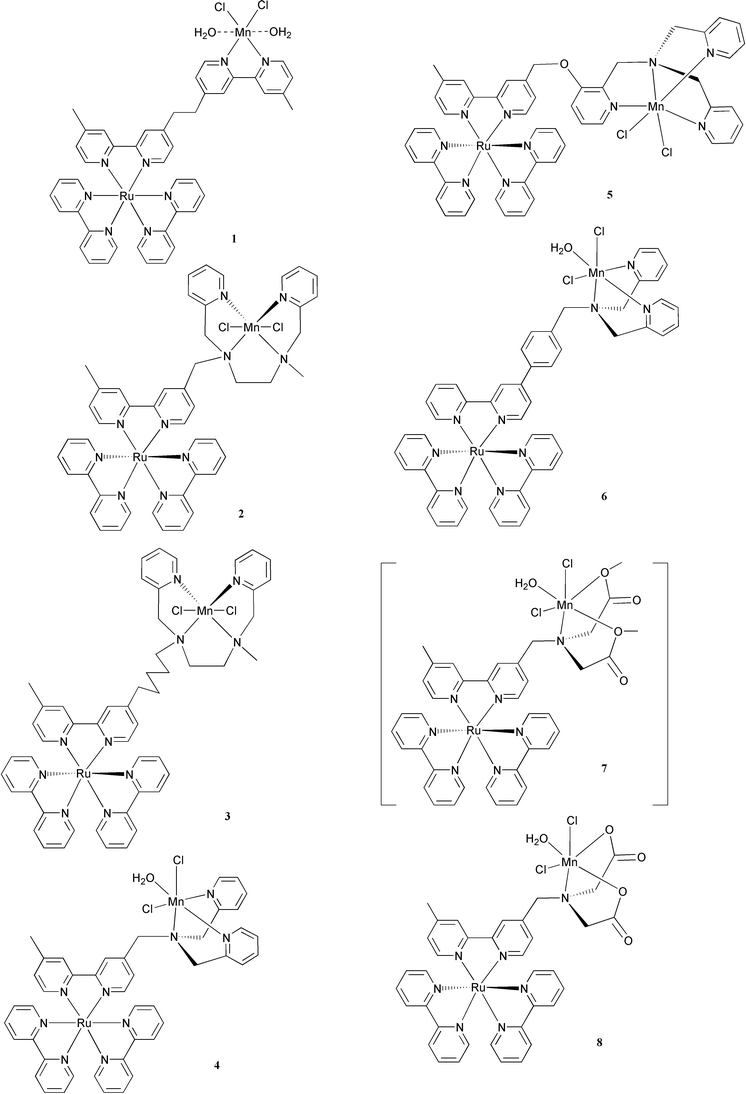
The intramolecular electron transfer from the co-ordinated Mn(II) to the photo-generated Ru(III) in acetonitrile solutions of the binuclear complexes 1–8 was studied with laser flash photolysis. The ruthenium excited state created by the laser flash transferred an electron to the external electron acceptor, methyl viologen (MV2+), forming the Ru(III) and MV+• species. Then, the Mn(II) was oxidised by intramolecular electron transfer to Ru(III), regenerating the photosensitiser before recombination of Ru(III) and MV+•.
In the case of 1, the first order rate constant for electron transfer from the Mn(II) was 1.8 × 105 s−1.15,16 This was the first time a manganese complex was used as an intramolecular electron donor to a photo-oxidised photosensitizer. As expected, the distance between manganese and ruthenium was important for the intramolecular electron transfer rate (Table 1), and in general it should be short to ensure a rapid reaction. However, when the distance was decreased as in case of 2, the lifetime of the Ru excited state was about two orders of magnitude shorter than in 1 due to rapid quenching by the manganese (see Table 1). In fact, for this series of complexes, there was an exponential decrease in quenching rate constant with increasing distance (see Fig. 4). This may be expected for quenching via an exchange energy transfer mechanism‡ ,17 if the excited state energies of the manganese complexes are similar. Complex 6, where the ligand is conjugated, is an exception where the quenching is strong although the metal–metal distance is quite long. We could show that the lowest excited state in this compound was delocalised over the bipyridine phenyl groups, close to the manganese,18 while the lowest excited state of the other complexes was presumably localised on the unsubstituted, remote bipyridines. By changing spacer and ligand structure, we can to some extent avoid too rapid quenching, although this is difficult when the Ru–Mn distance is short. Rapid quenching results in a short lifetime of the excited state, i.e. a rapid decay to the ground state, and inefficient electron transfer to the external acceptors. On the other hand, a long Ru–Mn distance leads to slow electron transfer from the manganese to the photo-oxidised ruthenium.
| Com- pound | Calculated distancea (Ru–Mn)/Å | Excited state lifetime,bτ/ns | Electron transfer rate constant,ckET/s−1 |
|---|---|---|---|
| a The Ru–Mn distance was estimated from the most extended conformation determined by molecular mechanics calculation.b The excited state lifetimes of the ruthenium complexes were measured in acetonitrile solution at room temperature.c Intramolecular electron transfer from the Mn or the tyrosine moiety to photogenerated Ru(III), data were measured by ns flash photolysis in acetonitrile solution at room temperature in the presence of methyl viologen as acceptor.d In aqueous solution at pH = 7. | |||
| 1 | 13 | 260 | 1.8 × 105 |
| 2 | 9 | 7 | >2 × 107 |
| 3 | ≈14 | 300 | ≈1 × 105 |
| 4 | 9 | 2 | 1.3 × 106 |
| 5 | ≈13 | 120 | ≈1 × 105 |
| 6 | 14 | 23 | 1.6 × 106 |
| 7 | 9 | 19 | — |
| 8 | 9 | — | — |
| 9 | — | 1150 | 5 × 104d |
| 11 | — | 1350 | >1 × 107 |
| 11a | 16 | 110 (72%), 880 (18%), 35 (10%) | >1 × 107 |
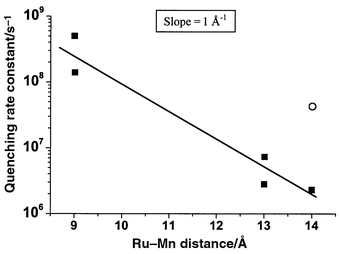 | ||
| Fig. 4 The rate constant for quenching of the Ru excited state by manganese in the binuclear complexes 1–6, plotted as a function of Ru–Mn distance. The line is an exponential fit to the data. The point for complex 6 (open circle) is excluded from the fit because the excited state of Ru is differently localised as compared to the other complexes (see text). | ||
It is interesting to note that the singlet excited state of a porphyrin was also rapidly quenched by a Mn(II) complex linked to the porphyrin periphery.19 Thus, we suggest that quenching of the excited P680 chlorophylls in PSII by the manganese cluster could also be a potential problem, since Mn(II) is involved at least in the fully reduced S0 state of the OEC. It seems that nature has solved the problem by keeping the manganese cluster at a large distance from P680, using tyrosineZ as a redox intermediate to maintain a rapid chain of electron transfer reactions. Thus, to avoid the quenching problem using principles from nature, we decided to increase the complexity in our system by introducing a redox intermediate between the manganese and ruthenium complexes.
A second reason to introduce an intermediate is that the manganese complexes will probably never be fast electron donors. From the temperature dependence of the intramolecular electron transfer in our Ru–Mn complexes, we obtained the activation free energy (ΔG#) from eqn. (1), and by using eqn. (2) we calculated the reorganisation energy (λ) for each complex:20
| kET = A exp(−ΔG#/RT) | (1 ) |
| ΔG# = (ΔG0 + λ)/4λ | (2) |
| A = 4π2Hrp2(4πλ RT)−1/2 | (3) |
A third reason for using an intermediate donor is a particular problem in multiple electron transfer reactions. For water oxidation to occur, several oxidising equivalents must be accumulated in the manganese complex, one equivalent for each photon that is absorbed by the system. In order to be of interest for water oxidation, the Mn potential in the single oxidation steps must be at least around +0.8 V vs. NHE. However, the excited Ru-complex is a strong reductant, E1/2 = −0.8 V, that may reduce any redox couple with a potential more positive than −0.8 V. Therefore, when the next photon is captured by the Ru complex, there is a large driving force for the reduction of the oxidised manganese complex by the capture of an electron from the Ru excited state. This is an energy-wasting back reaction that obviously cannot be avoided by manipulations of the redox potentials. Instead it has to be solved by making this backward reaction slow compared to the forward electron transfer from the Ru excited state. In the next section we describe the development of systems with an intermediate donor.
3.2 A ruthenium tris-bipyridyl complex covalently linked to L-tyrosine
As described in the Introduction, the electron transfer between the manganese cluster and P680 in PSII is mediated by the phenolic group of tyrosineZ. To solve our quenching and ET rate problems in the simple binuclear Ru–Mn model systems, discussed in the previous section, we sought to mimic nature more closely by introducing a tyrosine group in our systems. Thus, we synthesised complex 9, in which a L-tyrosine ethyl ester was covalently linked to the ruthenium tris-bipyridyl complex by an amide bond.21 Time-resolved emission measurements showed that the excited state of the Ru moiety was indeed not affected by the tyrosine unit in 9 at pH < 10. In deoxygenated aqueous solution at pH 7, the lifetime of the excited state of 9 was 370 ns. This is the same as for a reference compound where tyrosine was replaced by alanine, which has no interaction with the excited state of the ruthenium complex.Flash photolysis of the complex 9 in aqueous solution in the presence of a sacrificial electron acceptor, cobalt(III) pentamine chloride [Co(NH3)5Cl]2+, led to the formation of [Co(H2O)6]2+ and Ru(III). The latter was reduced by intramolecular ET from the tyrosine unit, and a neutral tyrosine radical 9′ was generated. This was detected by direct observation of a transient absorption at 410 nm, where tyrosine radicals are known to absorb (see Figs. 5 and 6). This result was further established by time-resolved EPR measurements of 9 in aqueous solution in the presence of [Co(NH3)5Cl]2+. After a laser flash, an EPR signal was observed with a g-value of 2.0045 (Fig. 9, inset), which is typical for a neutral tyrosine radical. Photoinduced electron transfer from a phenol type ligand has also recently been reported by Wieghardt and coworkers.22 Using the complex 10 they were able to show that on irradiation with visible light in the presence of a methyl viologen acceptor, the ruthenium(III) complex was first formed. Ruthenium(II) was then rapidly regenerated by intramolecular electron transfer from one phenolic group (kET > 5 × 107 s−1), giving a phenoxyl radical. This radical was quite long lived, half life ca. 20 s, which may be compared to 0.1 s for the simple tyrosine complex 9.
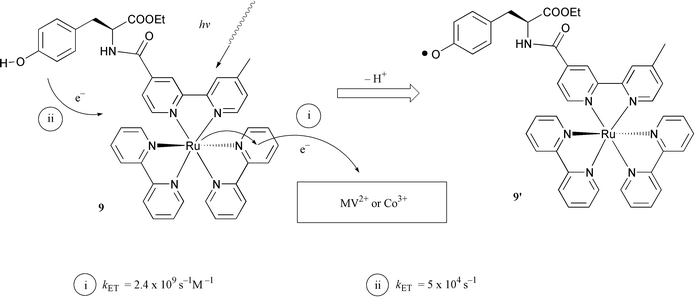 | ||
| Fig. 5 Photoinduced electron transfer reactions and the generation of a tyrosine radical 9′ at room temperature. Methyl viologen was used as electron acceptor in acetonitrile solution of 9; cobalt(III)pentamine chloride was used as the electron acceptor in experiments in aqueous solution with 9. | ||
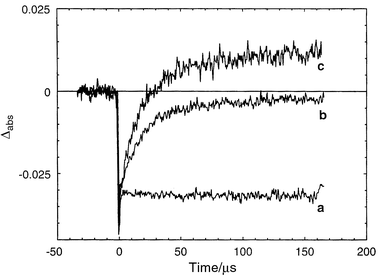 | ||
| Fig. 6 Transient absorption following a laser flash of 9, or its Ru–Ala analogue, with cobalt(III)pentamine chloride as acceptor:21 (a) trace at 450 nm for Ru–Ala; (b) trace at 450 nm for 9; (c) trace at 410 nm for 9. No regeneration of the Ru(II) ground state was observed for the Ru–Ala complex (trace a). In 9, however, the Ru(II) was regenerated (trace b) by electron transfer from the linked tyrosine. The tyrosine radical gives a positive absorption at 410 nm at the end of the reaction (trace c, the signal in this trace is magnified by a factor 2.2, to help visualise the identical kinetics at 450 and 410 nm). | ||
It seemed interesting to see if the tyrosyl radical 9′, which has a redox potential of +0.95 V vs. NHE, could oxidize an artificial manganese complex.
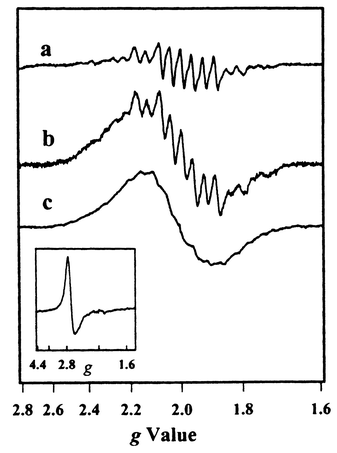
A suitable Mn(III/III) dimer was available through a collaboration with Professor Jean-Jacques Girerd (Orsay).23 The Mn(III/III) dimer complex (see Fig. 7) does not give rise to any EPR signal.24 When complex 9 was mixed with the Mn(III/III) dimer complex in aqueous solution in the presence of [Co(NH3)5Cl]2+, and the solution was subjected to laser flashes, a multiline signal was observed (see Fig. 8). This signal was assigned to the Mn dimer in the Mn(III/IV) state. The decay of the flash induced tyrosine radical in the absence and presence of the Mn(III/III) dimer was studied by transient EPR measurements (Fig. 9). These revealed that the tyrosine radical decayed faster in the presence of the Mn dimer which shows that the Mn dimer was oxidised by the tyrosine radical, and not simply by the Ru(III). The redox potentials of the Mn dimer, the tyrosine unit and the photosensitizer in compound 9 are comparable to those in PSII. Thus this system is the first model to mimic the electron donor ‘triad’ in PSII.
 | ||
| Fig. 7 In aqueous solution of 9 in the presence of cobalt(III) pentamine chloride as the electron acceptor, the Mn(III,III) dimer complex was photooxidized to Mn(III,IV) at room temperature. The oxidation occurred via the photogenerated tyrosine radical 9′. This system is the first redox active ‘triad’ which mimics the corresponding redox components in natural PS II both functionally, in a one-electron transfer sequence, and where also the redox potentials involved are very similar. | ||
 | ||
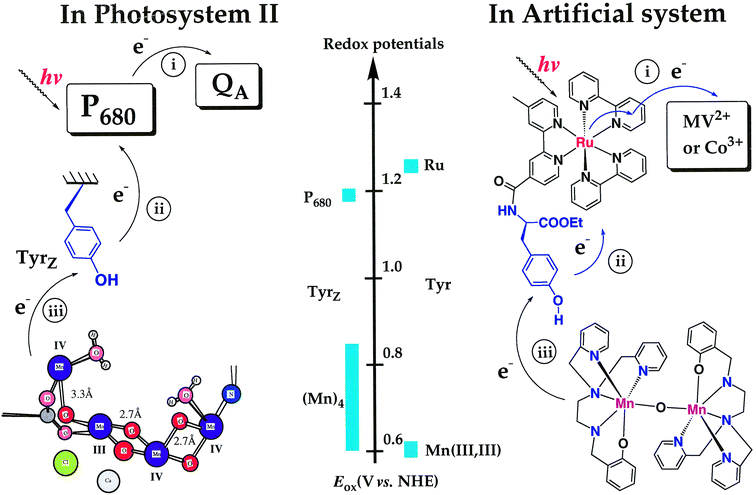
| Fig. 8 EPR spectra of different oxidised forms of the Mn(III/III) dimer.23 (a) EPR spectrum of the Mn(III/IV) state formed, as the result of 10 flashes given to a solution of 9 and Mn(III/III) dimer in acetonitrile in the presence of MV2+. (b) EPR spectrum recorded after oxidation of Mn(III/III) dimer by ca. 1 equivalent Ru(III)(bpy)3(PF6)3 in the dark. (c) EPR spectrum recorded after 25 flashes were given to solution of Mn(III/III) dimer and Ru(bpy)3 in the presence of MV2+. The inset shows the easily distinguishable EPR spectrum of Ru3+. The broad, featureless signal in (b) and (c) suggests a further oxidation of parts of the Mn(III/IV) species by the Ru(III)(bpy)3. This is in contrast to the milder oxidation by the tyrosyl radical in 9 that results in a clean Mn(III/IV) spectrum shown in (a). | ||
 | ||
| Fig. 9 Time-resolved EPR measurements of the induction and decay of the tyrosyl radical signal (see inset) from complex 9,23 after single laser flashes in water solution at pH 7.0, in the presence of the sacrificial electron acceptor cobalt(III)pentamine chloride. Flash-induced induction and decay of the tyrosyl radical in complex 9 alone (a) and in the presence of Mn(III/III) dimer at 0.3 mM (b) and 0.6 mM (c) concentration, respectively. | ||
A further biomimetic feature of the system is that the Mn(III,III) complex protects the tyrosine against photodegradation, which in fact also gives further evidence for the occurrence of reaction step (3). The tyrosine radical undergoes irreversible degradation reactions, if it is not reduced rapidly enough. Because the irreversible acceptor [Co(NH3)5Cl]2+ was used in our experiments, the electron never returned to the oxidised tyrosine. Therefore the initial tyrosine EPR signal decreased in magnitude with increasing number of flashes given to the same sample due to photodegradation. However, when the Mn(III,III) was added and could regenerate the tyrosine, a radical signal of the same magnitude was observed after every flash, until the Mn(III,III) had been consumed. An analogous protective behaviour is observed in PSII. In reaction centres depleted of manganese, irreversible reactions of the oxidised tyrosineZ radical are important pathways for what is known as donor side photo-inhibition.25 However, when Mn(II) is added to the PSII particles, the photodegradation is prevented.
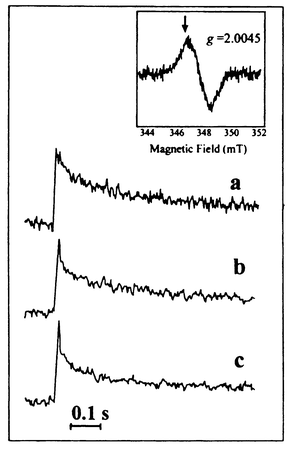
The pH dependence of the electron transfer rate constant in the Ru–tyrosine compound is shown in Fig. 10a.29 In Fig. 10b, the corresponding results for electron transfer from tyrosineZ to the oxidised chlorophylls of P680+ in manganese-depleted PSII are shown.26 There is a remarkable similarity between the two sets of data. The results for Ru–tyrosine showed29 that electron transfer and deprotonation are not consecutive processes, but instead a concerted reaction, with a common transition state. The OH bond of the tyrosine is then an important part of the reaction coordinate for electron transfer. When the OH bond is extended, reaching the transition state, electron transfer may occur, after which the proton dissociates. Since the reaction goes directly from Ru(III)–TyrOH to Ru(II)–TyrO•, without intermediates, the free energy change of the reaction follows that determined electrochemically. Thus, at pH < 10, the rate constant follows the dependence on pH (driving force) expected from conventional Marcus theory [eqn. (1)–(3)]30 as shown in Fig. 10a. However, the reorganisation energy for electron transfer [eqn. (1)–(2)] is unusually high, ca. 1.8 eV, because of the stretching of the tyrosine OH bond. Above pH = 10, when the reduced tyrosine is already deprotonated, the reorganisation energy is ca. 0.9 eV which is normal for a pure electron transfer reaction in a high polarity solvent. Consequently, the activation energy is lower [eqn. (2)] and the reaction becomes much faster.
 | ||
| Fig. 10 The rate constant for electron transfer from tyrosine to the photosensitiser as a function of pH (a) in an artificial Tyr–Ru complex 9.29 The solid line is a theoretical, Marcus-type function for the pH-dependence of the electron transfer rate constant; (b) in Mn-depleted PSII particles. Dashed lines are linear fits as guidance for the eye. | ||
Returning to the data for PSII (Fig. 10b) the rate, the pH-dependence, driving force and activation energy around pH = 6 are all nearly identical to the corresponding data for the Ru–tyrosine complex. This strongly suggests that the electron transfer and deprotonation are concerted in this case also, which explains the high activation (and reorganisation) energy. This is in contrast to what is currently suggested in the literature.27 Furthermore, the comparison in Fig 10 supports a model26 in which the tyrosineZ proton is lost to the bulk at pH < 7.6, but where it is hydrogen bonded to a nearby histidine (His 190) at higher pH. The histidine pKa is 7.6 and the hydrogen bond is therefore not formed at lower pH values. However, at pH > 7.6, the hydrogen bond weakens the tyrosineZ OH-bond strength and thus lowers the reorganisation energy. The reaction is then faster, and also pH-independent, since the proton is transferred within the hydrogen bond. However, it is not as fast as when the tyrosine is completely deprotonated, which obviously removes the reorganisation energy contribution from the OH bond completely.
To summarise, the detailed kinetic study of the Ru–tyrosine system leads to conclusions which help our understanding of PSII reactions. We propose that measurements of the reorganisation energy may reveal the protonation state of tyrosineZ, and that other hydrogen bonds may be detected, for example in PSII mutants where the histidine has been exchanged. For our artificial systems, our results suggest that hydrogen bonds to the tyrosine should be introduced in order to make the electron transfer faster.
3.3 A ruthenium tris-bipyridyl complex covalently linked to L-tyrosine with hydrogen-bonding substituents
From emission measurements of compound 9, we know that the tyrosine unit does not quench the excited state of the ruthenium moiety. Consequently, there is enough time for the desired electron transfer reactions to occur. However, as discussed above, the ET rate from tyrosine to the photogenerated Ru(III) is low, kET is ca. 105 s−1 at neutral pH. Our results and comparison with PSII prompted us to construct a complex in which the tyrosine is hydrogen bonded, in analogy with the proposed hydrogen bond between tyrosineZ and His190. This would be expected to increase the electron transfer rate significantly.Thus, we have synthesised complex 11,31 see structure in Fig. 11, in which two dpa-arms are connected to the ortho-positions of the phenol group of L-tyrosine. The synthetic routes for complex 11 as well as complexes 9 and 11a are summarised in Scheme 1. The dpa-arms in 11 form a strong hydrogen bond to the phenol group, evidenced by 1H NMR and NOESY spectroscopy. Again, the substituted tyrosine unit does not quench the excited state of ruthenium, actually complex 11 displayed a single exponential decay in acetonitrile with a lifetime τ = 1350 ns.
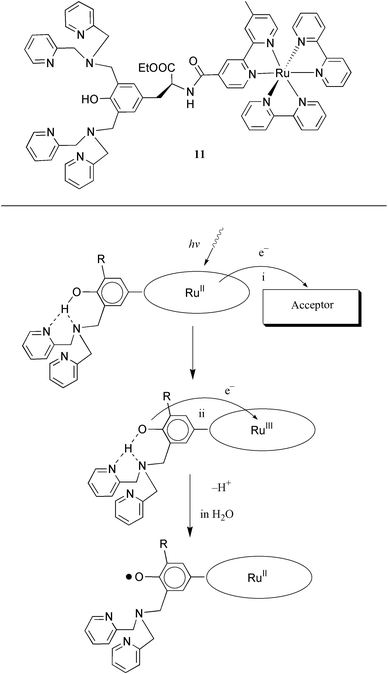 | ||
| Fig. 11 A proposed mechanism for the electron transfer from a dpa substituted tyrosine unit to the photogenerated Ru(III) at room temperature. The dpa arm forms a hydrogen bond with the tyrosine unit, and this hydrogen bond promotes the intramolecular electron transfer. In water, the result is a deprotonated, neutral tyrosine radical. In acetonitrile solution, EPR measurements show that the radical formed is a cationic tyrosine species. | ||
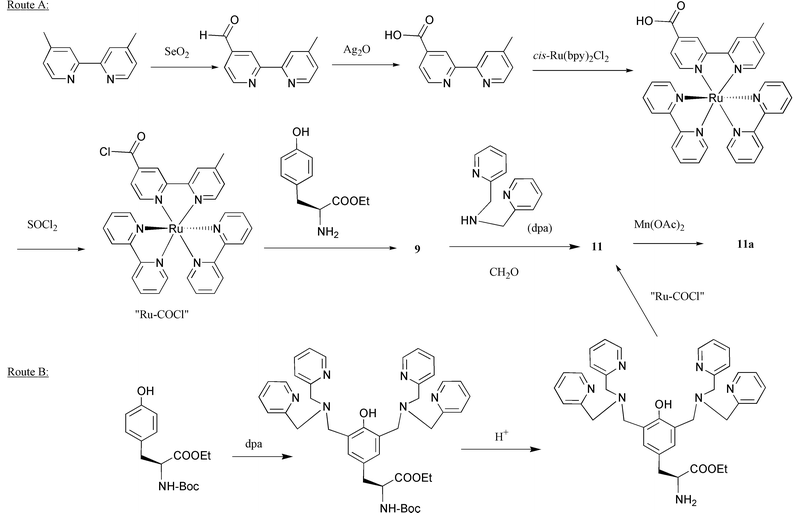 | ||
| Scheme 1 The general routes to summarise the synthetic procedures of complex 9, 11 and 11a. | ||
Flash photolysis and EPR measurements on 11 in water, in the presence of an electron acceptor (methyl viologen, MV2+, or cobaltpentamine chloride, [Co(NH3)5Cl]2+) showed that an intermolecular electron transfer from the excited state of Ru(II) in 11 to the electron acceptor took place, forming Ru(III) and the methyl viologen radical MV+• or [Co(H2O)6]2+. This was followed by intramolecular electron transfer from the substituted tyrosine moiety to the photogenerated Ru(III), regenerating Ru(II) and forming a tyrosyl radical; see the scheme in Fig. 11.31 The electron transfer process is intramolecular with a rate constant kET > 1 × 107 s−1, which is at least two orders of magnitude greater than in the similar compound 9, in which no dpa-arm is attached to the tyrosine unit. Therefore the hydrogen bonding between the substituted tyrosine and the dpa-arms in 11 is proposed to be responsible for the fast electron transfer. This interaction mimics the proposed His190 and tyrosineZ interaction on the donor side of PSII. The intramolecular electron transfer was so rapid that the overall rate was limited by the initial quenching of the Ru excited state by the acceptor. Although favourable from a functional point of view, this precluded an investigation of the dependence of the electron transfer rate on pH and temperature. We are currently investigating other complexes with a hydrogen bonded tyrosine in order to test and expand our model for the proton coupled electron transfer.
3.4 A ruthenium tris-bipyridyl complex covalently linked to a Mn dimer via L-tyrosine
In order to oxidise water, it is necessary to store four oxidation equivalents in a Mn complex. This might be hard to achieve with a mononuclear complex. In PSII, the operating unit is a manganese cluster consisting of four manganese ions, but provided the right ligands are coordinated to manganese, a Mn dimer might be sufficient. In fact, the complex 11 was prepared with this idea in mind, and a reaction with manganese(II) acetate gave the dinuclear complex 11a, see Fig. 12.28 The two alternative synthetic sequences are shown in Scheme 1. In the first, the tyrosine complex 9 was prepared by reacting the acid chloride of ruthenium tris-bipyridine ‘Ru-COCl’ with L-tyrosine ethyl ester. The dpa side arms were then introduced by reaction with formaldehyde and dipicolylamine (dpa) to give 11 which was transformed into the manganese complex 11a. In the second sequence, a protected tyrosine derivative was reacted with formaldehyde and dpa, then deprotected and reacted with ‘Ru-COCl’ to give 11 which was again converted to the dinuclear manganese complex 11a. Both these sequences demonstrate the sturdiness of the Ru(II)–tris-bipyridine system, which can often be used as a convenient synthetic building block.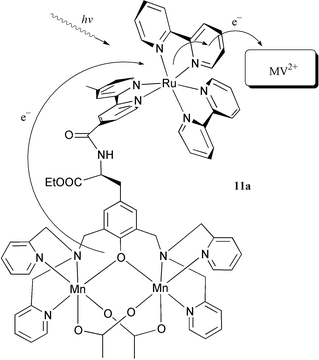 | ||
| Fig. 12 An intramolecular electron transfer from a Mn(II,II) dimer to a photooxidised Ru(III) centre in acetonitrile solution of the trinuclear complex 11a in the presence of methyl viologen as electron acceptor. | ||
Excitation of complex 11a in acetonitrile solution showed that, despite the presence of the Mn-dimer, the emission lifetime was 110 ns. This is long enough for efficient bimolecular electron transfer to acceptors such as methyl viologen(MV•2PF6) and [Co(NH3)5Cl]2+. In flash photolysis experiments with 11a in acetonitrile in the presence of MV2+, the excited state of the ruthenium complex 11a was quenched by intermolecular electron transfer to MV2+, forming the charge separated products Ru(III) and MV+•. The Ru(III) was immediately reduced back to Ru(II) by fast (kET > 1× 107 s−1) intramolecular electron transfer from the manganese(II/II) moiety to give Mn(II/III). This means that we have in hand a system with an efficient recovery of the photosensitiser after the photoinduced oxidation and also efficient oxidation of a manganese cluster in the recovery process. In a similar approach, Wieghardt and coworkers have prepared the complex 10a, which contains a linear cluster of three manganese(II) ions.22 On irradiation of 10a in the presence of an acceptor, a ruthenium(III) species was formed, which was rapidly (kET > 5 × 107 s−1 ) reduced to ruthenium(II) by intramolecular electron transfer from the manganese cluster. The performance of the model systems 11a and 10a is thus clearly approaching that of natural PSII. Recently, work in progress carries this similarity even further.32 In these experiments, we have been able to show that by repeated flashes in the presence of a [Co(NH3)5Cl]2+ acceptor the manganese cluster of 11a can be oxidised from the Mn(II/II) state to the Mn(III/IV) state in a 3-electron oxidation as shown by EPR experiments. This multi-step electron transfer process thus closely models the 4-electron cycle in PSII.
4 Future outlook
We are presently extending our work on Ru–Mn2 complexes. A modification of the Mn dimer ligand in 11 has been made, in which a substituted phenol group was introduced into the dpa arm to replace one of picolyl groups (see structure of 12).33 Preliminary results showed that the ligand in 12 can coordinate two manganese ions, forming a Mn(III,III) dimer complex which is shown in 12a.34 Introduction of the anionic phenolate into the ligand is expected to stabilise the Mn dimer in its higher valence states. It should also prevent dissociation of the Mn dimer in aqueous solution (which is a problem with dpa-based systems), and thus increase the possibility of storing more oxidising equivalents in the Mn complex, which will be required to oxidise water to oxygen.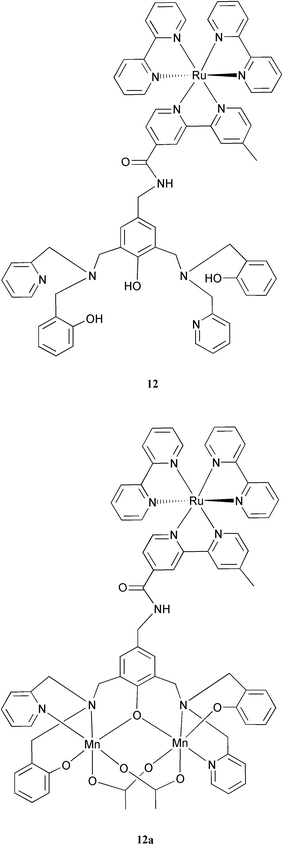
In all electron transfer experiments which were discussed above, an external electron acceptor (methyl viologen, MV2+, or cobalt pentamine chloride, [Co(NH3)5Cl]2+) was used. The external electron acceptors have a disadvantage in these systems, because the electron transfer rate from the excited state of ruthenium to the acceptor is limited by diffusion due to the bimolecular reaction. This requires that the lifetime of the excited state of ruthenium should not be too short. Also, if faster reactions from the donors should be resolved, the initial electron transfer to the acceptor must not be rate limiting. One way to solve this problem is to link an electron acceptor covalently to the photosensitizer, but this will sometimes make the synthetic procedure very difficult. Alternatively, we can use a nanostructured semiconductor like TiO2 as electron acceptor,35 by binding the photosensitizer to the TiO2 surface. It is likely that the use of TiO2 as the electron acceptor will facilitate the construction of an artificial photosynthetic device. We are confident that in a future which is not too distant, solar energy will be converted into fuel (hydrogen) by such artificial photosynthetic devices.
5 Concluding remarks
In this review we have described the present level in a research project intended to develop artificial photosynthesis as a means to convert solar energy into a fuel.Our strategy is to design and synthesise multifunctional supramolecular complexes to achieve light-driven oxidation of water. Our complexes are designed using principles from the PSII reaction centre. In PSII, a triad composed of a multimer of chlorphyll molecules (P680), a tyrosine residue (TyrZ) and a tetranuclear Mn complex are the active redox components in light-driven water oxidation.
We have described how we approach our goal stepwise, by synthesis and characterisation of gradually more complex compounds. Our most recent supramolcular complexes contain analogues to all of the redox components on the donor side in PSII. Instead of P680 we use Ru tris-bipyridine complexes which are similar to P680 in terms of oxidative potential and which have a rich chemistry to use in synthetic attempts. Our complexes also contain an incorporated Mn complex and we have managed to synthesize molecules containing one Ru and two Mn ions. In addition we have introduced a tyrosine link between the Ru centre and the Mn dimer.
At present we have been able to oxidise a coupled Mn(II)–Mn(II) dimer in three steps to the Mn(III)–Mn(IV) level by light-driven intramolecular electron transfer to the photo-oxidized Ru centre. Similar to the situation in PSII, it is highly likely that incorporation of an intervening redox active link (in both cases a tyrosine) is crucial to achieve multistep electron transfer from the Mn cluster to the Ru centre.
Although we have not yet been able to demonstrate catalytic water oxidation we hope we have been able to convey to the reader the exciting photochemistry these complexes are able to perform. Already in the first family of compounds we have accomplished many features that will be necessary to achieve artificial photosynthesis. Our next goal, is to try to design a new generation of Ru–Mn supermolecules in which we have included our knowledge about the limitations of our present compounds. This is an exciting and difficult task but we are convinced that it will soon be possible to expand the photochemistry in Ru–Mn complexes to reach the level of catalytic, light driven water oxidation. In our view, this would be the key step in our attempts to produce a system for the production of hydrogen from water in a true artificial photosynthesis system.
The path to a technological device is very long and winding and it may well be that other supermolecules than Ru–Mn based systems will eventually prove best. However, our strong belief is that the Ru–Mn track is well worth pursuing and that it might well be that catalytic photosynthetic water oxidation in a supramolecular system is not very far away.
Artificial photosynthesis is a vision that attracts much interest in our society. It is clear that this can not be accomplished without enormous research efforts. These must include concerted, collaborative effort of scientists from many disciplines including synthetic chemistry, physical and theoretical chemistry and biochemistry. Later, engineering and material sciences must also be included. It is also clear to us that many different approaches have to be tested before a final system is reached and in this review we have described our own attempt, the Ru–Mn track, which we initiated as a collaborative effort between our groups a few years ago.
6 Acknowledgements
We thank our many earlier and present coworkers in the Swedish Consortium for Artificial Photosynthesis. This work was financially supported by grants from the Knut and Alice Wallenberg Foundation, the Swedish National Energy Administration, the Swedish Natural Science Research Council (NFR), the Swedish Research Council for Engineering Sciences (TFR), the European TMR Program (TMR Network CT96-0031) and the Nordic Energy Research Program. We also thank our colleagues and coworkers in the TMR program.7 References
- (a) J. Barber and B. Andersson, Nature, 1994, 370, 31 CrossRef CAS; (b) B. Andersson and S. Styring, in Current Topics in Bioenergetics, ed. C. P. Lee, Academic Press, San Diego, 1991, vol. 16, pp. 1–81 Search PubMed; (c) G. Renger, Physiol. Plant, 1997, 100, 828 CrossRef CAS.
- (a) B. A. Diner and G. T. Babcock, in Oxygenic Photosynthesis: The Light Reactions, ed. D. R. Ort and C. F. Yocum, Kluwer Academic Publishers, Dordrecht, The Netherlands, 1996, p. 213 Search PubMed; (b) R. D. Britt, in Oxygenic Photosynthesis: The Light Reactions, ed. D. R.Ort and C. F. Yocum, Kluwer Academic Publishers, Dordrecht, The Netherlands, 1996, p. 137 Search PubMed; (c) R. J. Debus, Biochim. Biophys. Acta, 1992, 1102, 269 CAS.
- C. Tommos, X.-S. Tang, K. Warncke, C. W. Hoganson, S. Styring, J. McCracken, B. A. Diner and G. T. Babcock, J. Am. Chem. Soc., 1995, 117, 10325 CrossRef CAS.
- (a) C. W. Hoganson and G. T. Babcock, Science, 1997, 277, 1953 CrossRef CAS; (b) C. Tommos and G. T. Babcock, Acc. Chem. Res., 1998, 31, 18 CrossRef CAS; (c) C. Tommos and G. T. Babcock, Biochim. Biophys. Acta, 2000, 1458, 199 CrossRef CAS.
- M. Hauman and W. Junge, Biochim. Biophys. Acta, 1999, 1411, 86.
- W. Ruettinger and G. C. Dismukes, Chem. Rev., 1997, 97, 1 CrossRef CAS.
- (a) V. K. Yachandra, V. J. De Rose, M. J. Latimer, I. Mukerji, K. Sauer and M. P. Klein, Science, 1993, 260, 675 CrossRef CAS; (b) V. K. Yachandra, K. Sauer and M. P. Klein, Chem. Rev., 1996, 96, 2927 CrossRef CAS.
- J. M. Peloquin, K. A. Campbell, D. W. Randall, M. A. Enavchik, V. L. Pecoraro, W. H. Armstrong and R. D. Britt, J. Am. Chem. Soc, 2000, submitted. Search PubMed.
- (a) K. A. Århling, S. Petersson and S. Styring, Biochemistry, 1997, 36, 13148 CrossRef; (b) J. Messinger, J. H. Robblee, W. O. Yu, K. Sauer, V. K. Yachandra and M. P. Klein, J. Am. Chem. Soc., 1997, 119, 11349 CrossRef CAS.
- P. E. M. Siegbahn and R. H. Crabtree, J. Am. Chem. Soc., 1999, 121, 117 CrossRef CAS.
- (a) J. Limburg, J. S. Vrettos, L. M. Liable-Sands, A. L. Rheingold, R. Crabtree and G. W. Brudvig, Science, 1999, 283, 1524 CrossRef CAS; (b) J. Limburg, V. A. Szalai and G. W. Brudvig, J. Chem. Soc., Dalton Trans., 1999, 1353, and references therein. Search PubMed.
- (a) H. Imahori and Y. Sakata, Eur. J. Org. Chem, 1999, 2445 CrossRef CAS; (b) H. Kurreck and M. Huber, Angew. Chem. Int. Ed. Engl., 1995, 34, 849 CrossRef CAS; (c) J.-P. Sauvage, J.-P. Collin, J.-C. Chambron, S. Guillerez, C. Coudret, V. Balzani, F. Barigelletti, L. De Cola and L. Flamigni, Chem. Rev., 1994, 94, 993 CrossRef CAS; (d) M. R. Wasielewski, Chem. Rev., 1992, 92, 435 CrossRef CAS.
- Y. Naruta, M.-A. Sasayama and T. Sasaki, Angew. Chem. Int. Ed. Engl., 1994, 33, 1839 CrossRef.
- (a) M. Watkinson, A. Whiting and C. A. McAuliffe, J. Chem. Soc., Chem. Commun., 1994, 2141 RSC; (b) F. M. Ashmawy, C. A. McAuliffe, R. V. Parish and J. Tames, J. Chem. Soc. Dalton Trans., 1995, 1391 RSC; (c) N. Aurangzeb, C. E. Hulme, C. A. McAuliffe, R. G. Pritchard, M. Watkinson, M. Bermejo, A. Garcia-Deibe, M. Rey, J. Sanmartin and A. Sousa, J. Chem. Soc., Chem. Commun., 1994, 1153 RSC.
- (a) L. Sun, L. Hammarström, T. Norrby, H. Berglund, R. Davidov, M. Andersson, A Börje, P. Korall, C. Philouze, M. Almgren, S. Styring and B. Åkermark, Chem. Comm., 1997, 607 RSC; (b) L. Sun, H. Berglund, R. Davidov, T. Norrby, L. Hammarström, P. Korall, A. Börje, C. Philouze, K. Berg, A. Tran, M. Andersson, G. Stenhagen, J. Mårtensson, M. Almgren, S. Styring and B. Åkermark, J. Am. Chem. Soc., 1997, 119, 6996 CrossRef CAS; (c) L. Hammarström, L. Sun, B. Åkermark and S. Styring, Biochim. Biophys. Acta, 1998, 1365, 193 CAS; (d) K. E. Berg, A. Tran, M. K. Raymond, M. Abrahamsson, J. Wolny, S. Redon, M. Andersson, L. Sun, S. Styring, L. Hammarström, H. Toftlund and B. Åkermark, Eur. J. Inorg. Chem., 2000, in press. Search PubMed.
- H. Berglund-Baudin, L. Sun, R. Davidov, M. Sundahl, S. Styring, B. Åkermark, M. Almgren and L. Hammarström, J. Phys. Chem. A, 1998, 102, 2512 CrossRef CAS.
- (a) D. L. Dexter, J. Chem. Phys., 1953, 21, 836 CrossRef CAS; (b) G. L. Closs, M. D. Johnson, J. R. Miller and P. Piotrowiak, J. Am. Chem. Soc., 1989, 111, 3751 CrossRef CAS.
- Abrahamsson et al., manuscript in preparation..
- L. Sun, L. Hammarström, et al., manuscript in preparation..
- R. A. Marcus and S. Sutin, Biochim. Biophys. Acta, 1985, 811, 265 CAS.
- A. Magnusson, H. Berglund, P. Korall, L. Hammarström, B. Åkermark, S. Styring and L. Sun, J. Am. Chem. Soc., 1997, 119, 10720 CrossRef CAS.
- (a) D. Burdinski, K. Wieghardt and S. Steenken, J. Am. Chem. Soc., 1999, 121, 10781 CrossRef CAS; (b) D. Burdinski, E. Bothe and K. Wieghardt, Inorg. Chem., 2000, 39, 105 CrossRef.
- A. Magnuson, Y. Frapart, M. Abrahamsson, O. Horner, B. Åkermark, L. Sun, J.-J. Girerd, L. Hammarström and S. Styring, J. Am. Chem. Soc., 1999, 121, 89 CrossRef CAS.
- O. Horner, E. Anxolabéhére-Mallart, M.-F. Charlot, L. Tchertanov, J. Guilhem, T. Mattioli, A. Boussac and J.-J. Girerd, Inorg. Chem., 1999, 38, 1222 CrossRef CAS.
- C. Jegerschöld and S. Styring, Biochemistry, 1996, 35, 7794 CrossRef CAS.
- R. Ahlbrink, M. Haumann, D. Cherepanov, O. Bögerhausen, A. Mulkidjanian and W. Junge, Biochemistry, 1998, 37, 1131 CrossRef CAS.
- (a) D. A. Force, D. W. Randall and R. D. Britt, Biochemistry, 1997, 36, 12062 CrossRef CAS; (b) B. A. Diner, D. A. Force, D. W. Randall and R. D. Britt, Biochemistry, 1998, 37, 17931 CrossRef CAS; (c) M. Haumann, A. Mulkidianian and W. Junge, Biochemistry, 1999, 38, 1258 CrossRef CAS.
- L. Sun, M. K. Raymond, A. Magnuson, D. LeGourriérec, M. Tamm, M. Abrahamsson, J. Mårtensson, G. Stenhagen, L. Hammarström, S. Styring and B. Åkermark, J. Inorg. Biochem., 1999, 78, 15 CrossRef CAS Preliminary accounts of this work have been presented as invited lectures at: EUCHEM Conference, Artificial Photosynthesis, May 1998, Sigtuna, Sweden; Fourth Nordic Congress on Photosynthesis, Nov. 1998, Naantali, Finland; EBEC July, 1998, Göteborg, Sweden..
- M. Sjödin, S. Styring, B. Åkermark, L. Sun and L. Hammarström, J. Am. Chem. Soc., 2000, 122, 3932 CrossRef.
- R. A. Marcus, Angew. Chem. Int. Ed. Engl., 1993, 32, 1111 CrossRef.
- L. Sun, M. Burkitt, M. Tamm, M. K. Raymond, M. Abrahamsson, D. LeGourriérec, Y. Frapart, A. Magnuson, P. Huang-Kenéz, P. Brandt, A. Tran, L. Hammarström, S. Styring and B. Åkermark, J. Am. Chem. Soc., 1999, 121, 6834 CrossRef CAS.
- P. Huang Kenéz, A. Magnuson, M. Abrahamsson, R. Lomoth, M. Tamm, B. van Rotterdam, L. Sun, J. Park, L. Hammarström, B. Åkermark and S. Styring, manuscript submitted. Search PubMed.
- Tran, A. et al., manuscript in preparation..
- L. Sun, J. Zheng, et al., manuscript in preparation..
- A. Hagfeldt and M. Grätzel, Chem. Rev., 1995, 95, 49 CrossRef CAS and references therein..
Footnotes |
| † Light-induced water splitting has been achieved using heterogeneous systems (metals and semi-conductors; see e.g. O. Khaselev and J. A. Turner, Science, 1998, 280, 425, and references therein). The mechanistic understanding is still limited, however, and the results are not directly related to homogeneous, molecular catalysts in biological or biomimetic systems. |
| ‡ The mechanism was probably Dexter-type energy transfer, since alternative mechanisms could be ruled out; see ref. 28. |
| This journal is © The Royal Society of Chemistry 2001 |