Comparison of the photoredox properties of polyoxometallates and semiconducting particles
Anastasia Hiskia, Athanasios Mylonas and Elias Papaconstantinou*
Institute of Physical Chemistry, NCSR Demokritos, 153-10, Athens, Greece
First published on 28th November 2000
Abstract
We demonstrate that two large categories of compounds, namely, polyoxometallates (produced by acid condensation of mainly tungstates and/or molybdates) and aggregates of various metal oxides (formed by stirring, sonication etc) have similar properties. Emphasis is given to the photocatalytic properties of the two systems. The various steps in the photocatalytic cycles are described and several examples, from both systems, have been used to demonstrate that their photocatalytic performance is similar in terms of the overall mechanism of photodecomposition of organic compounds, the intermediate species involved and the final photodegradation products (i.e., CO2, H2O and inorganic anions).
 Anastasia Hiskia Anastasia Hiskia | Anastasia Hiskia was born in Athens, Greece in 1958. She received her Diploma in Chemistry (1981) from the University of Athens and a Doctorate Degree in Chemistry (1989) from the University of Athens in collaboration with NCSR Demokritos. She worked at the General Chemical State Lab., 1983–1995, in environmental chemistry. She joined the Catalytic–Photocatalytic Processes Lab. Institute of Physical Chemistry, NCSR Demokritos in 1995. Her research interests involve Advanced Oxidation Processes, polyoxometallates and environmental analysis. |
 Athanasios Mylonas Athanasios Mylonas | Athanasios Mylonas was born in Athens, Greece, in 1968. He received a Diploma in Chemistry from Patras University in 1990 and a Doctorate Degree in Chemistry from the National Technical University of Athens and NCSR Demokritos in 1996. Now he works as adjunct research scientist at NCSR Demokritos and as Director of Research and Development at Chemo Hellas. |
 Elias Papaconstantinou Elias Papaconstantinou | Elias Papaconstantinou holds a Diploma in Chemistry from the University of Athens, Greece, 1960. After a year as Research Fellow at the, then, NRC Demokritos, he moved to the USA and obtained a PhD degree in inorganic chemistry with M. T. Pope from Georgetown University, Washington DC, 1968. He was Postdoctoral Research Fellow (1968–1970) at Boston University on the photochemistry of inorganic complexes and Chief Research Chemist at the Electronic Chemicals Division, Philip A. Hunt Chemical Corporation, NJ, USA, 1980–1972. He joined the Institute of Physical Chemistry NCSR Demokritos in 1972 and was appointed Visiting Associate Professor at Boston University 1979–1980. He is currently Director of the Catalytic–Photocatalytic Processes Lab. His current research interests include redox catalytic processes, polyoxometallate, photocatalysis and environmental chemistry (Advanced Oxidation Processes). |
1 Introduction
Unlike CrO42− that only dimerizes upon lowering the pH of an aqueous solution, the corresponding MoO42− and WO42− anions form a variety of well defined metal oxygen clusters, the so-called polyoxometallates (POM),1,2 the history of which goes back to early 19th century.1,2 Other metal oxide anions such as vanadates, niobates and tantalates also undergo condensation reactions, but, generally the species formed are not well characterized.3Several categories of POM can be formed by careful selection of the ingredients and by adjustment of pH and temperature.2 We will mention a few characteristic groups of the great variety that exist: (a) acid condensation of pure MoO42− or WO42− produces the so-called isopoly compounds of the general formula MxOyq−. A typical example is W10O324− [Fig. 1(C)]. (b) If the original solution contains a heteroatom, A, like phosphorus, silicon, arsenic, iron or cobalt (about 60 elements can function as heteroatoms), acid condensation produces a heteropoly compound of the general formula AaMxOy q−, for instance, PW12O403− and P2W18O626− [Fig. 1(A, B)]. (c) Mixed heteropoly compounds that contain various ratios of molybdenum and tungsten arise from a solution that contains both MoO42− and WO42− and a heteroatom [Fig. 1(D)]. (d) By careful adjustment of pH, M–O moities can be removed from POM and can be replaced by mainly transition metals with a variety of ligands, thus forming the transition metal substituted POM (TMSP), for instance [PW11O39Mn(H2O)]6−, 1,2 [Fig. 1(E)].
![Characteristic structures of POM. (A)
PW12O403−, Keggin structure; (B)
P2W18O626−,
Wells–Dawson structure; (C) The structure of the isopoly anion
W10O324−. They are composed of
WO6 octahedra sharing corners and edges. In
PW12O403− the heteroatom P is within
the central tetrahedron PO4.
P2W18O626− arises from
the Keggin anion, by removing three WO6 octahedra and joining
the two 9-tungsto half units. Also shown in this figure examples of: (D)
Mixed POM with Wells–Dawson structure, for instance,
P2W15Mo3O626−
in which three W atoms have been replaced by Mo atoms whose octahedra are
shown hatched. (E) TMSP with Keggin stucture, for instance
[PW11O39Mn(H2O)]6−. In
the shaded octahedron, −W–O moiety has been replaced by
–Mn–(H2O). Several ligands, besides H2O,
can coordinate in the position shown as solid circle. Oxidized and reduced
(by one and two electrons) spectra of
PW12O403− . The O→M CT band,
the intervalence electron transfer (M–M CT) and d–d
transitions, are indicated on spectra.2](/image/article/2001/CS/a905675k/a905675k-f1.gif) | ||
| Fig. 1 Characteristic structures of POM. (A) PW12O403−, Keggin structure; (B) P2W18O626−, Wells–Dawson structure; (C) The structure of the isopoly anion W10O324−. They are composed of WO6 octahedra sharing corners and edges. In PW12O403− the heteroatom P is within the central tetrahedron PO4. P2W18O626− arises from the Keggin anion, by removing three WO6 octahedra and joining the two 9-tungsto half units. Also shown in this figure examples of: (D) Mixed POM with Wells–Dawson structure, for instance, P2W15Mo3O626− in which three W atoms have been replaced by Mo atoms whose octahedra are shown hatched. (E) TMSP with Keggin stucture, for instance [PW11O39Mn(H2O)]6−. In the shaded octahedron, −W–O moiety has been replaced by –Mn–(H2O). Several ligands, besides H2O, can coordinate in the position shown as solid circle. Oxidized and reduced (by one and two electrons) spectra of PW12O403− . The O→M CT band, the intervalence electron transfer (M–M CT) and d–d transitions, are indicated on spectra.2 | ||
POM have well defined structures and properties and their size is typically a few nm. They have diverse chemistry with applications in the field of analytical chemistry, biochemistry and solid state devices and have been used as antiviral and antitumor reagents. However, their most important property is in the field of catalysis.4 POM, like metal oxides, participate in catalytic processes as oxygen and multielectron relays. Several books and review articles have already described the development of POM and the field is expanding rapidly. New, larger compounds have been formed that, besides their structural interest, have other promising prospects.5
Although condensation produces a few metal oxide anions, as mentioned above, aggregates of various sizes can be formed by stirring or sonication of a variety of metal oxides, such as TiO2 or WO3, in aqueous solution.6 Other methods such as chemical precipitation or controlled hydrolysis and subsequent polymerization can give materials of uniform size. Particulate systems are commonly distinguished by their size and fall into the two major categories of colloids and macroparticles. Macroparticles have a size that exceeds ca. 100 nm and form turbid suspensions, whereas colloids contain smaller particles that form clear solutions.7 In an excellent article Alivisatos described how the fundamental properties of these aggregates or clusters, vary, enormously, with size.8 Particles with a diameter over roughly 15 nm behave, generally, as bulk semiconductors (SC), whereas, smaller particles, of less than 5 nm have more or less molecular characteristics and are classified as quantum size clusters or quantum dots. Thus, POM, in SC terminology, may be considered ideal quantum size clusters, or quantum dots.8 Therefore, it is not surprising that these two systems exhibit overall similar behaviour.
It should be noted that despite determination of the Keggin and Wells–Dawson structures, mentioned below, many chemists, up until the Sixties, considered POM as inorganic polymers. A similarly unclear picture exists for the exact structure of metal oxide particulates. It is expected that these particles will resolve to definite structures, following the historical development of POM, but from the opposite direction, i.e., from larger to smaller sizes.
2 Properties
In the last twenty years or so, we have seen important developments in the photochemistry and photocatalysis of semiconductors and, to a lesser extent, POM. Each of the various observations and explanations given for one group appears to have a counterpart in the other group. Yet, there are generally, (and unintentionally), no cross references between the two groups. This matter has improved recently as discussed below.Some general points are worth mentioning. For instance:
(a) If one looks at the electronic absorption spectra of the oxidized and reduced WO3 colloids and at their electrochemical behaviour, it can be seen that they have a pattern characteristic of polyoxotungstates,9 as others have also noticed,10 (Fig. 2). The case of TiO2 is also similar.11. The overall resemblance to the spectra of POM (shown in Fig. 1) is apparent. The blue color of reduced POM is thus attributed to intra electron transfer between adjacent metal ions, M–M charge transfer (CT) bands2 and a somewhat similar explanation holds for the blue coloration of reduced WO3 and TiO2.
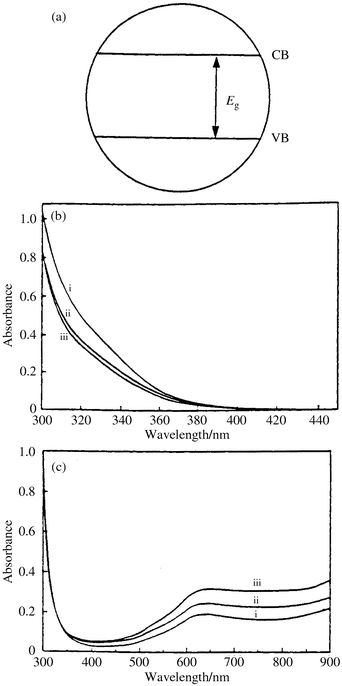 | ||
| Fig. 2 (a) A metal oxide particle, i.e., a SC, is usually represented as a circle with two bands: VB (valence band) and CB (conduction band), separated by the energy gap Eg; see text. (b) Absorption spectra of WO3 colloids (0.4 M) in water, with diameters 2–5 nm. The oxalic acid concentration that controls the size of the particles was i, 0.16, ii, 0.23 and iii, 0.31 M;9 (c) Absorption spectra of deaerated aqueous suspension of WO3 colloids (0.4 M) following UV photolysis (λ > 300 nm) for 6 min., in presence of various concentrations of oxalic acid i, 0.16, ii, 0.23 and iii, 0.31 M. (Reprinted from reference 9, with permission.) | ||
(b) The band gap in SC, Eg (see below), is a function of the size of the metal oxide particulates. The smaller the size, the higher the Eg.8 A similar trend is noticed with POM as exemplified by the threshold and peak absorptions of the spectra of Keggin (ionic size ca. 7.2 nm3) and Wells–Dawson (ionic size ca. 10.3 nm3) structures of phosphomolybdates and phosphotungstates, that have overall similar characteristics [Fig. 3(A and B)].2
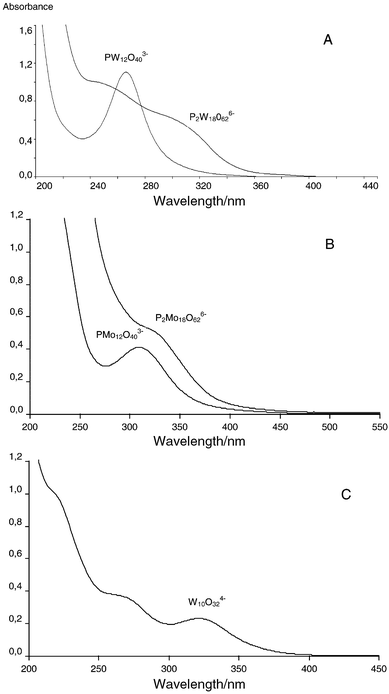 | ||
| Fig. 3 Comparison of the spectra of the oxidized forms of: (A) PW12O403− and P2W18O626−; (B) PMo12O403− and P2Mo18O626−; (C) W10O324−. POM, ca. 2 × 10−5 M, in 50∶50 CH3CN–H2O; 0.1 M HClO4. | ||
(c) It has been stated that accumulation of electrons on POM lowerss the efficiency of photoreaction. For instance, in the photooxidation of propan-2-ol to propanone by PW12O403−, the efficiency drops by an order of magnitude in going from the oxidized 12-tungstophosphate, PW12O403−, to the form PW12O404− that is reduced by one electron.12 An analogous statement has been made for SC (TiO2), namely, that accumulation of electrons on SC lowers the quantum yield13 by increasing the electron hole (h+ + e−) recombination rate (see below).
(d) Appreciable negative charge can be built up on metal oxide particulates in the absence of an acceptor. This is also true for POM, whose characteristic property is that they undergo multiple, stepwise, ‘reversible’ (from a chemical point of view) reductions without decomposition.2
(e) Accumulation of charge on SC drives the redox potential to more negative values, i.e., the redox potential is a function of the charge density.14 This result was demonstrated, a long time ago for POM with the Keggin structure by Pope and Varga, (Fig. 4).15
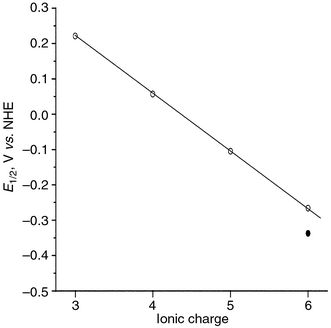 | ||
| Fig. 4 Half-wave potentials for one-electron reduction vs. charge of the anions with Keggin structure: PW12O403−, SiW12O404−, FeW12O405−, CoW12O406−, H2W12O406− ●. (Reprinted from reference 15, with permission.) | ||
(f) The rate-determining step in photocatalytic processes with metal oxide particulates was reported to be the removal of the electrons from the SC.13 Again, a similar statement has been made for POM, namely, that the rate-determining step in the photocatalytic cycle is the regeneration (reoxidation) of the catalyst.12
(g) Both systems, i.e., polyoxotungstates and TiO2 particulates, are effective photocatalysts for the mineralization to CO2, H2O and inorganic anions of a great variety of organic pollutants (see below).
Thus, as mentioned earlier, the two systems have advanced independently, yet they exhibit great similarities, as is now quite obvious.
As always, the generalities break down when we get down to specifics. For POM the well defined properties, such as solubility, stability,1 excited state life times,16 can be associated with the structure. Thus, for example, W10O324− with a structure other than the Keggin and Wells–Dawson structure (Fig. 1), does not follow the rule of size, having ca. 450 and 320 nm for the threshold and peak absorptions, respectively [Fig. 3(C)].
No such specific properties can be noted for SC clusters for lack of structural data until now. In POM electrons are trapped in reasonably well understood molecular orbitals, so that reduced POM can be isolated, studied and prevented from undergoing further reactions. No such treatment is possible, so far, with band gap illuminated SC. For instance, most commercial SC samples have catalytic activity that varies from batch to batch. In SC oxidations and reductions are not easily separated and cannot be stoichiometrically controlled.
3 Photochemical behaviour
The points listed above are some general thoughts and observations scattered in the literature, referring to either of the systems under discussion. It is the similarities in behaviour that we will focus upon, providing an overall picture of the parallel photochemical behaviour of these two categories of compounds in their reactions with organic substrates in aqueous solutions, as presented in the literature.We will mainly consider the behaviour of TiO2 and WO3 as SC and that of polyoxotungstates as POM. A somewhat similar approach to the overall characteristics of POM and SC, but not to their specific photochemical behaviour, has been reported by Gomez-Romero;10 a comparison of several POM and semiconducting materials in CH3CN has also been presented by Hill and Chambers.17 Maldotti et al. have mentioned the similarity between the behaviour of photoexcited WO3 and TiO2 with that of POM.18 We will present a few key points of the several that substantiate each case.
The following steps are encountered in the photochemistry and photocatalytic processes undergone by polyoxotungstates and metal oxide particulates.
3.1 Preassociation of catalysts with substrate
Several observations suggest the formation of a preassociated complex or a preassociated equilibrium of the catalyst with the organic substrate in aqueous solution,| M + S ↔ M···S | (1) |
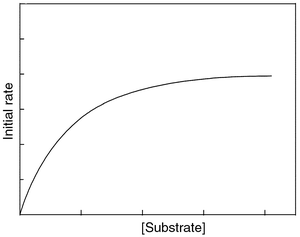 | ||
| Fig. 5 Typical plot showing the variation in the initial rate of the photoredox reaction as a function of increased concentration of organic substrate, for a certain concentration of catalyst (metal oxide particulate or POM). | ||
3.2 Excitation of the catalysts
In metal oxide particulates atomic and molecular orbitals coalesce to form energy bands. Impinging light λ > Eg (Eg, the band gap energy of semiconductor) leads to electron hole separation (Fig. 2a).By analogy, excitation into the O→M CT band of POM (near-visible
and UV region), results in electron hole separation, which can be
represented as
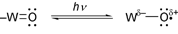
![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) O represents a tungsten–oxygen
bond in polyoxotungstates). Thus, for both systems the excited state can be
described as
O represents a tungsten–oxygen
bond in polyoxotungstates). Thus, for both systems the excited state can be
described as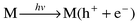 | (2) |
3.3 Photolysis in deaerated solutions
Excitation renders these systems powerful oxidizing reagents, able to oxidize a great variety of organic compounds. In the absence of electron trapping reagents (mainly dioxygen), electrons accumulate on metal oxide particulates, and POM, leading to the gradual development of a blue color, especially in the case of POM (Fig. 6). Noteworthy are the similarities in the photolysis spectra of deaerated aqueous solutions of PW12O403− (Fig. 6)12 and aqueous suspensions of WO3 colloids in the presence of organic substrates,9 (Fig. 2c).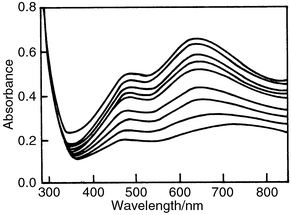 | ||
| Fig. 6 Gradual development of a blue color, indicating the successive formation of one-electron (750 nm) and two-electron (650 nm) reduction products of PW12O403−, 0.1 mM; propan-2-ol, 2 mM in 0.1 M HClO4; deaeration with Ar. Photolysis at 254 nm. | ||
3.4 Formation of OH radicals upon photolysis of catalysts in aqueous solution
Reaction of the excited catalyst with H2O (i.e. oxidative hole trapping) creates “surface” bound OH radicals, according to the following equation.| M(e− + h+) + H2O ↔ M(e−) + OH + H+ | (3) |
The following observations substantiate the formation of OH radicals: (a) OH adducts (hydroxylation products) have been detected in photolysis experiments with aromatic hydrocarbons in the presence of the POM21,24 and TiO2.25,26 Thus, for example, photolysis of phenol, whether in the presence of TiO2 or PW12O403− yields hydroquinone, catechol and 1,2,4-trihydroxybenzene (pyrogallol).23 In principle, the hydroxylated product may originate from the hydrolysis of cation radicals produced from direct hole trapping by the organic adsorbate;23 this subject is considered below. (b) ESR trapping experiments had detected OH radicals upon photolysis of POM18,27 and SC.28 (c) The generation of OH radicals is also suggested by the fact that the excited state potentials of most POM and SC are more positive than the reaction given in eqn. (4).
| OH + H+ + e− → H2O E = 1.90 V vs. NHE | (4) |
Fig. 7 shows the ground state and excited state potentials of some POM together with those of characteristic SC for comparison. The excited state potential of POM was calculated from the equation: [excited state potential of POM (V vs. NHE)] = [ground state reduction potential of POM (V vs. NHE) + 1240/(O–O) transition energy], where (O–O) transition energy is, roughly, the threshold absorption (nm) of the O→M CT band in the near-visible area.
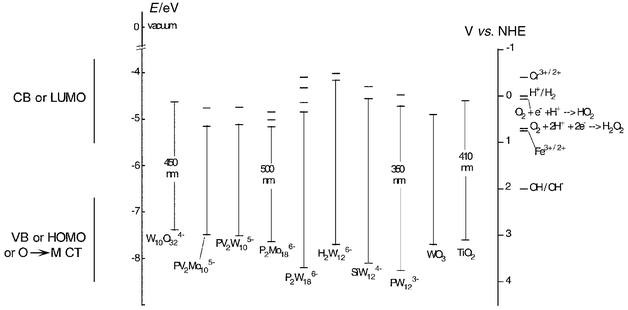 | ||
| Fig. 7 Ground state and excited state redox potentials and corresponding energy
levels, relative to vacuum and to NHE (normal hydrogen electrode), of some
POM together with those of characteristic SC, WO3 and
TiO2, for comparison. Only the first few energy levels of POM
are indicated on the diagram. For instance, for
PW12O403−
(PW123−) the first two energy levels are shown,
corresponding to the following reactions: Thus, the oxidizing ability of the ground state of PW12 O403− is 0.221 V vs. NHE (first reduction step), whereas the excited state potential is more positive by 350 nm (or 1240/350≅3.5 eV) more positive, i.e., ≅3.7 V vs. NHE, as shown in the diagram and explained in the text. | ||
To complete the argument in favour of OH radical formation, it has also been observed that the photodecomposition of trichloroacetic acid, which lacks α-hydrogen atoms, is much slower than that of chloroacetic acid (Fig. 8).29 As is already known from radiation chemistry studies in aqueous solutions OH radicals attack preferentially α-hydrogen atoms.
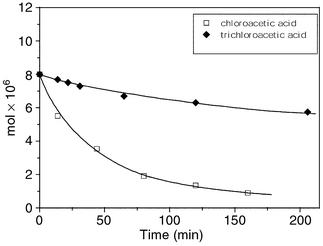 | ||
| Fig. 8 Comparison of the photodegradation of ClCH2COOH and CCl3COOH upon photolysis of an oxygenated aqueous solutions of PW12O403−. Solution volume 4 mL; catalyst, 0.7 mM; substrate, 2 mM; pH 1.0 (HClO4); λ > 320 nm. (Adapted from ref. 26, with permission). | ||
Most of the metal oxides are large band gap semiconductors, as so are the polyoxotungstates. Thus, only light below 400 nm is capable of producing electron hole pairs and so less than 5% of solar light can be utilized. One exception is iron(III) oxide with a band gap of 2.2 eV (for hematite α-Fe2O3).23 Hence, α-Fe2O3 absorbs in the visible below 560 nm. However, its photocatalytic action is limited to a few cases. Other SC particles, namely, CdS and GaP, also absorb larger fractions of the solar spectrum, but these catalysts are degraded during repeated photocatalytic cycles.23 Curiously enough, POM that absorb in the visible region, up to ca. 600 nm, such as molybdates, mixed tungstomolybdates and molybdovanadates, have limited photocatalytic activity and are unstable.12
From a philosophical point of view it seems that Nature preserved this world by not activating various metal oxides with visible light that would have destroyed organic matter. It is this goal, i.e., destruction of organic pollutants using solar light, that the scientific world is trying to achieve in a controlled manner.
3.5 Direct reaction of the excited state of the catalyst with substrate
The organic substrates, for both systems, act as adsorbed traps for the photogenerated holes either directly or through the intermediacy of ‘surface’ hydroxyl radicals as mentioned earlier.Diffuse reflectance flash photolysis has been employed to distinguish the products of direct electron transfer to TiO2 in aqueous suspensions from those resulting from reaction with OH radicals. This, however, has not produced indisputable results.22,30
3.6 Mineralization of organic pollutants. Photodecomposition kinetics
The high oxidizing ability of the excited states and the formation of OH radicals by both polyoxotungstates and SC (TiO2), as mentioned above, are the causes of photodegradation (to CO2, H2O and inorganic anions) of a great variety of organic compounds.23,24 Thus mineralization has been reported in both systems for alcohols, aldehydes, ketones, acids, aromatic compounds substituted with Cl, Br, OH and for pesticides. Mineralization of carbon can be monitored through either the evolution of CO2 or the disappearance of all the organic carbon. Over 1500 papers have been published on the ability of TiO2 to cause mineralization, whereas the contribution of POM to the subject has been recognized only recently.21,24Fig. 9 shows a typical photodegradation process of chlorophenol, in which the disappearance of substrate and the formation of CO2 and Cl− are illustrated. It can be seen that CO2 is detected after an induction period, as is observed for all aromatic substrates used so far, indicating that the process of mineralization goes through several intermediates before the opening of the aromatic ring. For both systems, the disappearance of organic compounds as a function of irradiation time obeys first-order kinetics.22,24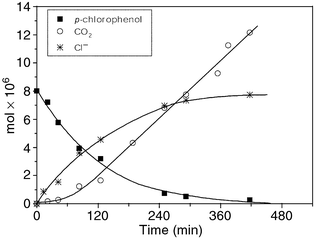 | ||
| Fig. 9 Photocatalytic degradation and formation of Cl− and CO2 of an oxygenated aqueous solution of 4-chlorophenol in presence of W10O324−, 0.7 mM; pH, 2.5; λ > 320 nm. Photolyzed solution 4 mL. | ||
3.7 Formation and decay of similar intermediates
The process of mineralization, for both categories of catalysts, passes through the formation and decay of several similar intermediates. This is to be expected, since their oxidizing ability is exercised mainly through OH radicals, as mentioned earlier. Thus, the characteristic reactions of OH radicals have been encountered: (a) hydroxylation of aromatic compounds (i.e. OH adducts) as mentioned earlier; (b) H-abstraction; (c) dehalogenations; (d) decarboxylations; (e) breaking of the aromatic ring and formation of short-chain aliphatic acids. Also addition or transfer of hydrogen atoms is observed.We present below examples of common intermediates encountered in the photodecomposition of, for example, phenol,23,31 (Scheme 1) and chlorophenols.24,32 (Scheme 2).
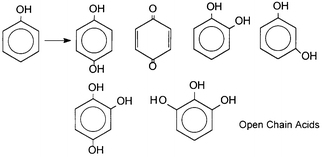 | ||
| Scheme 1 Common intermediates detected during photodegradation of phenol by polyoxotungstates and TiO2. (Reprinted from ref. 31, with permission from Elsevier Science). | ||
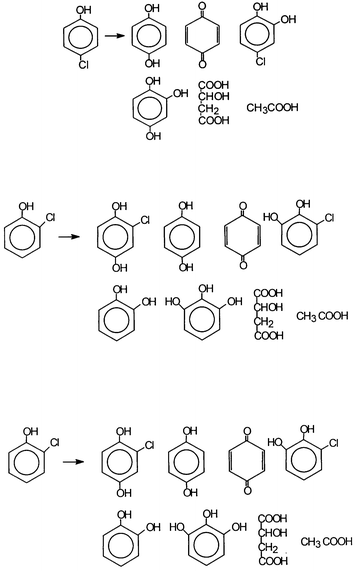 | ||
| Scheme 2 Intermediates detected during the course of mineralization of chlorophenols by both systems. (Reprinted from ref. 24, with permission from Elsevier Science). | ||
The formation of CH2 and CH3 groups from aromatic carbon (i.e. CH groups) implies that in the photocatalytic processes promoted by POM and TiO2 there is a reductive (hydrogenation) pathway, in addition to the oxidative process. On the other hand, the formation of aliphatic acids and the ultimate formation of CO2 by both systems implies addition of OH radicals to the organic ring, leading to the formation of CO bonds or hydration of the oxidized organic radical cation.23,24
An interesting case that also demonstrates the similarity of these processes is the photocomposition of nitrogen containing aromatic rings. In particular, the initial photodecomposition of atrazine, by both methods, takes place within a few minutes, leading, within a few hours, to cyanuric acid (CA). However, the latter resists photodegradation for tens of hours in the presence of either TiO2 or polyoxotungstates.33,34Scheme 3 shows the main common intermediates identified in the photodegradation processes of atrazine by the two systems and the final products. Notice that in both cases, the chlorine groups end up as chloride ions and amine nitrogen atoms as nitrate ions.
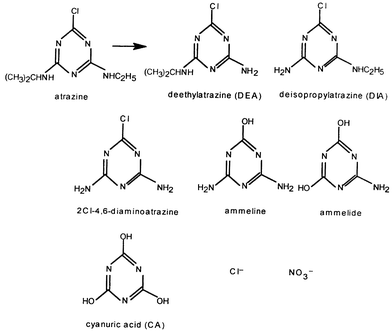 | ||
| Scheme 3 The main common intermediates and final products detected in the photodecomposition of atrazine by TiO2 and polyoxotungstates. | ||
We mentioned earlier that, getting down to specifics, the two processes might present differences. In this respect, we note that Texier et al.34 working with TiO2 and W10O324−, in the photodecomposition of atrazine, have identified with TiO2 two transient amides, which they did not observe with W10O324−. Even in this case, the authors point out that these amides might be too reactive toward W10O324− and so their steady-state concentration could be too low to be detected. A more serious argument on their part is that they doubt the formation of OH radicals with W10O324−. They argue that in absence of organic substrate, H2O2 should have been detected. We think that even in the case of W10O324− whose excited state life time is longer than those of most POM16 the indirect rate of electron hole recombination,
| h+ + OH− → OH | (5) |
| OH + e− → OH− | (6) |
3.8 Degradation pathways
As has been mentioned earlier, hydroxylated products may, in principle, arise from direct reaction of the aromatic ring with OH radicals and/or from the hydrolysis of the oxidized cation radical generated by direct hole transfer to the associated organic substrate, or to put it in more familiar chemical terminology, by direct electron transfer from the associated organic substrate to the photoexcited catalyst. Scheme 4 shows the common pathway of photodecomposition of an L-substituted aromatic compound as used by several workers working with OH radicals produced in aqueous solutions from 60Co-γ-radiolysis, TiO2 photolysis or POM.24Scheme 4 provides details of the fate of the organic substrate, accounting for the formation of hydroxy aromatic derivatives and the overall mineralization process.24 For an ortho OH addition in the L-substituted benzene ring above, the reactions depicted in Scheme 5 explains, according to Matthews,26 the mineralization process. Dioxygen is also involved, the role of which is considered below. Finally, a more specific example is the photodecomposition of 2,4-dichlorophenol.35 Several common intermediates, identified in the decomposition by both systems, necessitate the degradation pathway shown in Scheme 6.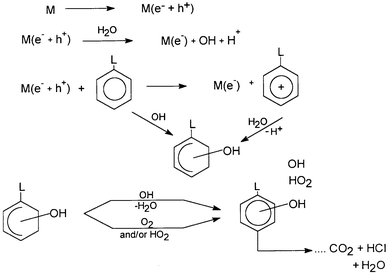 | ||
| Scheme 4 The common pathway of photodecomposition of an L-substituted aromatic compound, as used by several workers working with OH radicals. (Reprinted from ref. 24, with permission from Elsevier Science). | ||
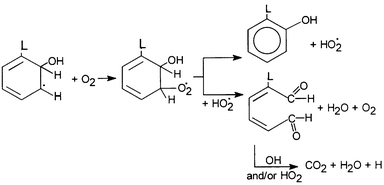 | ||
| Scheme 5 The mechanism of mineralization after an ortho OH addition in the L-substituted benzene ring, showing, also, the participation of dioxygen, according to Matthews.26 (Reprinted from ref. 24, with permission from Elsevier Science). | ||
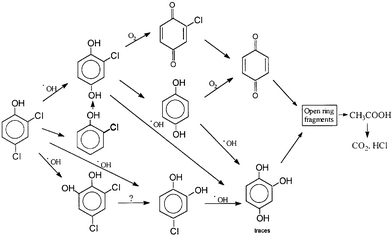 | ||
| Scheme 6 Photodegradation pathway according to the common intermediates found in the photocatalytic decomposition of 2,4-dichlorophenol by both systems. (Reprinted from ref. 35, with permission from Elsevier Science). | ||
3.9 Regeneration of catalysts
Both systems can serve as electron relays, i.e. they are able to transfer the accumulated electrons to a variety of compounds with a less negative reduction potential than the conduction band of SC or the highest occupied molecular orbital (HOMO) of POM and thus close the photocatalytic cycle, which is represented for both systems in Fig. 10. For both categories of catalysts, the rate-determining step in the photocatalytic cycle has been reported to be the reoxidation (regeneration) of catalyst.12,13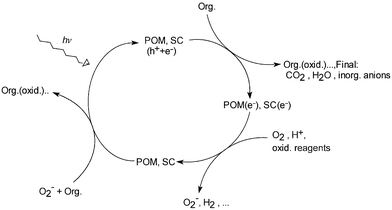 | ||
| Fig. 10 The photocatalytic cycle representing the reactions involved for both POM and SC. | ||
Generally, photocatalytic reductions with SC (TiO2) are less frequently encountered than are oxidations, probably because, as has been stated, the reducing ability of the conduction band electrons is significantly lower than the oxidizing ability of a valence band hole.30 This also holds true for POM as one can see from the redox potential of Fig. 7 and for one further reason: POM are in the d0 electronic state (i.e. the highest, W6+, oxidation state); therefore there are no electrons available for reduction.
Hole oxidation of organic compounds accumulates electrons on the catalyst, which can be delivered to various reducible species in the solution. Thus, for instance, H+ has been reported to be reduced to H2,12,30 haloaromatics undergo reducible dehalogenation23,24 and nitrobenzene has been reported to be reduced to aniline.
Various oxidants have been used as photoelectron traps (or oxidizing reagents for reduced catalysts) such as S2O82−, IO4−, BrO3−, ClO3− and H2O2.23 Dioxygen is the most effective and the cleanest electron trapping reagent for both types of catalysts. In the process, the superoxy radical is formed,
| M(e−) + O2 → M + O2− | (7) |
4 Conclusions
(a) Metal oxide particulates and POM upon excitation with near visible and UV light serve as photocatalysts in a variety of similar processes. (b) For photoreaction to occur, before deactivation of the excited catalyst takes place, both systems require the formation of a preassociated complex or preassociation equilibrium or, for that matter, a supramolecular species between the catalyst and substrate. (c) For both systems, the rates of photooxidation of substrate are the result of two competitive reactions: (i) direct reaction of the excited catalyst with substrate and (ii) reaction through OH radicals. (d) Both systems cause mineralization (i.e. formation of CO2, H2O and inorganic anions) of a great variety of organic pollutants, via similar intermediates. (e) The photodecomposition rates obey first-order kinetics with respect to the organic species. (f) For both systems, the rate-determining step in the photocatalytic cycle has been reported to be the regeneration (reoxidation) of the catalyst with dioxygen being the most effective and the cleanest oxidizing reagent. (g) Using chlorophenol as an example, the overall photodecomposition reaction is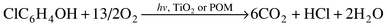 | (8) |
Acknowledgements
We thank The Ministry for Development, General Secretariat of Research and Technology for financing part of this work.References
- L. C. W. Baker and D. C. Glick, Chem. Rev., 1998, 98, 3 CrossRef.
- M. T. Pope, Heteropoly and Isopoly Oxometalates in Inorganic Chemistry Concepts 8, ed. C. K. Jorgensen, M. F. Lappert, S. J. Lippard, J. L. Margrave, K. Niedenzu, H. Noth, R. W. Parry and H. Yamatera, Springer Verlag, West Berlin, 1983. Search PubMed.
- F. A. Cotton and G. Wilkinson, Advanced Inorganic Chemistry, J. Wiley and Sons, New York, 5th edn, 1988, 776–819. Search PubMed.
- M. T. Pope and A. Muller, Angew. Chem., Int. Ed. Engl., 1991, 30, 34 CrossRef.
- A. Muller, F. Peters, M. T. Pope and D. Gatteschi, Chem. Rev., 1998, 98, 239 CrossRef.
- M. R. Hoffmann, T.S. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69 CrossRef CAS.
- M. Gratzel, Photocatalysis and Environment, ed. M. Schiavello, NATO ASI Series, Kluwer, Dordrecht, 1987, pp. 367–398. Search PubMed.
- A. P. Alivisatos, Science, 1996, 271, 933 CrossRef CAS.
- I. Bedja, S. Hotchandani and P. V. Kamat, J. Phys. Chem., 1993, 97, 11064 CrossRef CAS.
- P. Gomez-Romero, Solid State Ionics, 1997, 101, 243 Search PubMed.
- G. Rothengberger, J. Moser, M. Gratzel, N. Serpone and D. K. Sharma, J. Am. Chem. Soc., 1985, 107, 8054 CrossRef CAS.
- E. Papaconstantinou, Chem. Soc. Rev., 1989, 16, 1 and references therein. Search PubMed.
- H. Gerischer and A. Heller, J. Phys. Chem., 1991, 95, 5261 CrossRef CAS.
- M. T. Nenadovic, T. Rajh, O. I. Micic and A. J. Nozik, J. Phys. Chem., 1984, 88, 5827 CrossRef CAS.
- M. T. Pope and G. M. Varga, Inorg. Chem., 1966, 5, 1249 CrossRef CAS.
- C. Tanielian, Coord. Chem. Rev., 1998, 178-180, 1165 CrossRef CAS and references therein.
- R. C. Chambers and C. L. Hill, Inorg. Chem., 1991, 30, 2766.
- A. Maldotti, R. Amadelli, G. Varani, S. Tollar and F. Porta, Inorg. Chem., 1994, 33, 2968 CrossRef CAS.
- M. A. Fox, R. Cardona and E. Gailard, J. Am. Chem. Soc., 1987, 109, 6347 CrossRef CAS.
- E. Coronado, J. R. Galan-Mascaros and C. Gimenez-Saiz, J. Am. Chem. Soc., 1998, 120, 4671 CrossRef CAS and references therein..
- H. Einaga and M. Misono, Bull. Chem. Soc. Jpn., 1996, 69, 3435 CAS.
- P. Pichat, Handbook of Heterogeneous Catalysis, eds. G. Ertl, H. Knozinger and J. Weitkamp, Wiley-VCH, New York, 1997, vol. 4, pp. 2111–2122. Search PubMed.
- D. Bahnemann, J. Cunningham, M. A. Fox, E. Pelizzetti, P. Pichat and N. Serpone, Aquatic and Surface Photochemistry, eds. G. R. Helz, R. G. Zepp and D. G. Grosby, Lewis Publ., Boca Raton, 1994, pp. 261-316. Search PubMed.
- A. Mylonas and E. Papaconstantinou, J. Photochem. Photobiol. A, 1996, 94, 77 Search PubMed and references therein..
- D. F. Ollis, C.-Y. Hsiao, L. Budiman and C.-L. Lee, J. Catal., 1984, 88, 89 CrossRef CAS.
- R. W. Matthews, J. Catal., 1988, 111, 264 CrossRef CAS.
- T. Yamase and T. Kurozumi, J. Chem. Soc., Dalton Trans., 1983, 2205 RSC.
- M. Anpo, T. Shima and Y. Kubakawa, Chem. Lett., 1995, 1799.
- A. Mylonas, A. Hiskia and E. Papaconstantinou, J. Mol. Catal., 1996, 114, 191 Search PubMed.
- M. A. Fox and M. T. Dulay, Chem. Rev., 1993, 93, 341 CrossRef CAS.
- A. Mylonas, V. Roussis and E. Papaconstantinou, Polyhedron, 1996, 15, 3201 CrossRef.
- G. Al-Sayed, J.-C. D’Oliveira and P. Pichat, J. Photochem. Photobiol. A, 1991, 58, 99 Search PubMed.
- C. Minero, V. Maurino and E. Pelizzetti, Res. Chem. Intermed., 1997, 23, 291 Search PubMed.
- I. Texier, J. Ouazzani, J. Delaire and C. Giannotti, Tetrahedron, 1999, 55, 3401 CrossRef CAS.
- A. Hiskia, E. Androulaki, A. Mylonas, S. Boyatzis, D. Dimotikali, C. Minero, E. Pelizzetti and E. Papaconstantinou, Res. Chem. Intermed., 2000, 26, 235 Search PubMed.
| This journal is © The Royal Society of Chemistry 2001 |