Nucleic acid conformation diversity: from structure to function and regulation
Philippe Belmont, Jean-François Constant and Martine Demeunynck
LEDSS, Laboratoire de Chimie Bioorganique, UMR CNRS 5616, Université Joseph Fourier, BP 53, 38041, Grenoble, cedex 9, France.. E-mail: Martine.Demeunynck@ujf-grenoble.fr;; Fax: +33 476 51 43 82
First published on 21st December 2000
Abstract
Nucleic acids are the structural supports of genetic material and therefore the key factors in many vital cellular processes. The double-stranded right-handed helix is a regular conformation adopted by both DNA and RNA in cells, but an increasing number of results point to the biological importance of alternative structures such as bulges, hairpins, branched junctions or quadruplexes. Progress in the chemical synthesis of oligonucleotides and in the knowledge of the factors that favour a particular conformation has opened new fields of research in molecular recognition and drug design.
 Philippe Belmont Philippe Belmont | Philippe Belmont was born in Paris in 1970. He grew up in the French Caribbean where he started a BS degree in Biology and Biochemistry. In 1990 he moved to the University Joseph Fourier (Grenoble, France), where in 1996 he obtained a PhD in Organic Chemistry (Advisor: M. Demeunynck) focused on the synthesis and biological properties of heterocyclic heterodimers that recognise specific lesions in DNA. Then, he moved successively as a post-doctoral fellow at Case Western Reserve University (Professor A. J. Pearson, Cleveland, USA) working on the total synthesis of cyclopeptide antibiotics and to the Collège de France (Professor J.-M. Lehn, Paris, France) designing new synthetic vectors for use in gene therapy. In 2000 he joined the group of Professor M. Ciufolini (Lyon, France) as a Researcher for the Centre National de la Recherche Scientifique. |
 Jean-François Constant Jean-François Constant | Jean-François Constant was born in 1956 in Annappes, France. After having completed a BS degree in Biochemistry, he received a PhD in 1986 from the University of Lille working in the group of Professor J. Lhomme. He moved then successively to Paris (Professor C. Helene’s group at the Muséum d’Histoire Naturelle) and Los Angeles (Professor D. Sigman’s group at UCLA) to work as a post-doctoral fellow on the regulation of gene expression by the antisense strategy and on the nuclease activity of derivatised copper phenanthroline complexes. In 1988, he took up his position as a Researcher for the Centre National de la Recherche Scientifique in the LEDSS at the Université Joseph Fourier (Grenoble, France). His research field is the study of DNA recognition and chemical modifications by specific binders. |
 Martine Demeunynck Martine Demeunynck | Martine Demeunynck is Researcher for the Centre National de la Recherche Scientifique. She was born in 1955 in Lille, France. After a BS degree in Biochemistry, she joined the group of Professor J. Lhomme to work on the reactivity of nitrenium ion precursors for a PhD in Chemistry. She spent one post-doctoral year working in the group of Professor J. W. Lown at the University of Alberta (Edmonton, Canada) studying the reactivity of α-acetoxynitrosamines. In 1987, she moved to the Université Joseph Fourier of Grenoble (France) to join the LEDSS. Her major field of interest is the chemistry of nitrogen heterocycles with the design, synthesis and chemical reactivity of nucleic acid binding molecules. |
1 Introduction
DNA and RNA encode biological information through their linear sequence of nucleotides to specify the composition of proteins, and through their shapes to control their assembly with other cellular macromolecules. Depending on their sequences and environmental conditions, DNA and RNA may adopt a variety of secondary structures such as double-stranded helices, bulges, hairpins, and three-way and four-way branches, which have been described in various reviews.1–4 The determination of the biological role of non-B DNA structures and of complex RNA edifices is an area of intense study. Important questions have to be answered, especially concerning their existence and role in vivo, their mode of formation and regulation, and the information they carry. The development of the chemical synthesis of oligonucleotides, now accessible in large quantities, allows the structural analysis of these conformations, their recognition by other macromolecules and the design of specific ligands.After a general introduction on DNA and RNA conformations and structures, emphasising their possible biological roles, we will show how the synthesis of oligonucleotides of definite sequence allows the detailed study of particular structures or conformations using a large variety of physical, biochemical and physicochemical methods. In the second part, the interaction with small molecules and macromolecules will be discussed in terms of biological significance and drug design.
2 Nucleic acid structures
The most usual structure of DNA is a double-stranded right-handed helix with a negatively charged backbone (deoxyribose phosphate) on the outside and stacked base pairs (adenine–thymine, guanine–cytosine) on the inside (Fig. 1). The primary structure of RNA differs only by the presence of 2′-OH group on the sugar unit and the replacement of thymine by uracil (which differs by the absence of the methyl group at C5). Yet RNAs display greater structural diversity and chemical reactivity; they can adopt double-stranded structures through base pairing, and also form folded structures composed of duplex domains connected by single-stranded regions.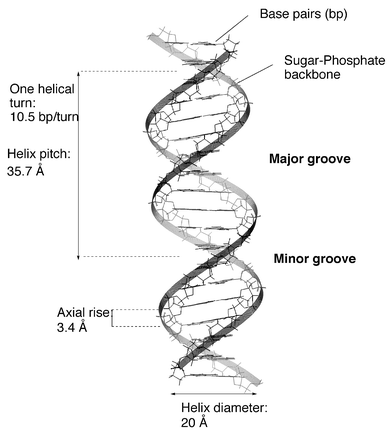 | ||
| Fig. 1 Structural parameters of B-form DNA. | ||
Different types of interaction control the structural changes observed in DNA and RNA: the charged phosphate groups of the backbone are mutually repulsive, and the formation of hydrogen bonds between bases and strong stacking interaction between the flat surfaces of the base pairs are implicated in the formation of multistranded structures (double- or triple-stranded helices, quadruplexes...) (Fig. 2).
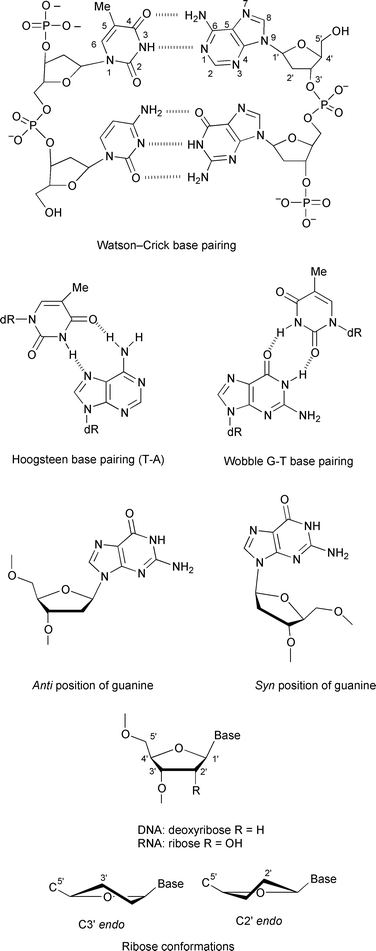 | ||
| Fig. 2 Secondary structure of DNA, base pairing and numbering, sugar and nucleoside conformations. | ||
The different conformations are not static entities. Each nucleotide has its own dynamics, thus conferring local motion on the helix in addition to the global motion of the macromolecule.
2.1 Double-stranded helices
Some structural features, such as the conformation of the sugar unit and the syn or anti conformation of the nucleic base relative to the sugar, are crucial for nucleic acid conformations (Fig. 2). DNA can adopt three major conformations, A-, B- and Z-forms (Fig. 3). The main parameters describing the helices are collected in Table 1. All three forms have antiparallel strands. A- and B-DNA are right-handed helices and Z-DNA is a left-handed helix. Double-stranded segments of RNA adopt the A-form structure that is very similar to the A-DNA conformation.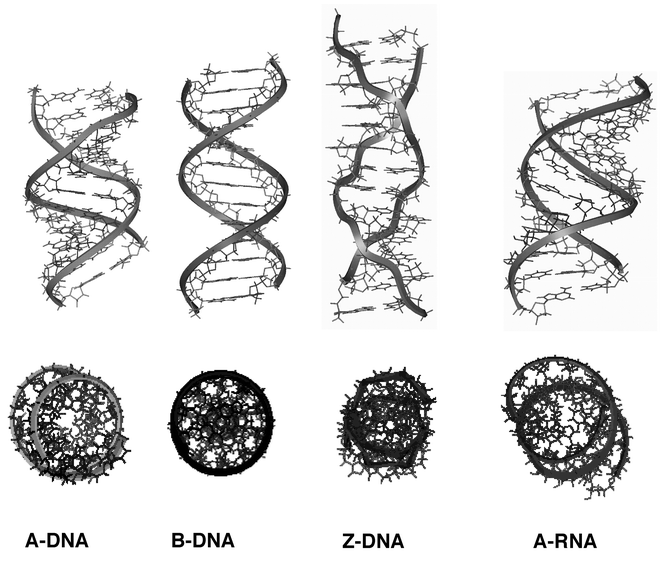 | ||
| Fig. 3 Molecular modelling representations of the major nucleic acid duplex conformations, the sugar–phosphate backbone is indicated with a ribbon. Bottom views: orthogonal representations. (Insight II (98) from MSI). | ||
| Parameter | A-DNA | B-DNA | Z-DNA | A-RNA |
|---|---|---|---|---|
| Helix sense | Right | Right | Left | Right |
| Residue per turn | 11 | 10.5 | 11.6 | 11 |
| Axial rise | 2.55Å | 3.4Å | 3.7Å | 2.8Å |
| Helix pitch | 28Å | 36Å | 45Å | |
| Rotation per residue | 32.7° | 36° | –9°, – 51° | 32.7° |
| Diameter of helix | 23Å | 20Å | 18Å | |
| Sugar pucker: | ||||
| dA, dT, dC | C3′ endo, | C2′ endo, | C2′ endo, | C3′ endo, |
| anti | anti | anti | anti | |
|---|---|---|---|---|
| dG | C3′ endo, | C2′ endo, | C3′ endo, | C3′ endo, |
| anti | anti | syn | anti |
|---|---|---|---|
| Major groove | Narrow, deep | Wide, deep | Flattened |
| Minor groove | Wide, shallow | Narrow, deep | Narrow, deep |
As shown in Fig. 3, A-DNA and A-RNA are wide and compact helices, with a cavernous major groove and a minor shallow groove. The base pairs are displaced off-axis. B-DNA is slimmer, more elongated with base planes essentially perpendicular to the helix axis, with a narrow minor groove and a wide major groove. The base pairs sit directly on the axis so that the major and minor grooves are of equal depth. For A- and B-conformations, the glycosylic bond is in the anti conformation. Z-DNA is thinner (diameter of approximately 18Å compared to 20Å for B-form) and more elongated (11.6 base pairs per helical turn compared to 10.5 base pairs for B-DNA). The major groove is completely flattened out on the surface of the molecule and the minor groove is very deep. The base pairs are displaced off-axis. The characteristic zigzag chain path in Z-DNA arises from the alternation of syn and anti conformations at guanine and cytosine respectively, which causes a local chain reversal and produces a helical repeat consisting of two successive bases, purine plus pyrimidine. Despite the fact that the two helices are ‘inverted’, B-DNA cannot be converted into Z-DNA by simply twisting the two ends of the helix. When converting a section of B-DNA into Z-DNA, the base pairs must flip to become upside down relative to the orientation they had in B-DNA. To do so, the purine residues rotate around their glycosylic bonds, from anti to syn positions and for the pyrimidine nucleosides, both the bases and the sugars rotate to produce the characteristic zigzag backbone conformation. The mechanism of this conversion is far from obvious and has received little attention compared to the physical and chemical studies of the Z-conformation itself. It should be noted that the position of the imidazole ring of guanine is profoundly affected by the conformation. In Z-DNA it is located on the outer side of the DNA molecule with easy access to the N7 and C8 atoms, whereas these two atoms are more shielded in B-DNA. This difference in accessibility accounts for the difference in reactivity of the two forms of the macromolecule.
In aqueous solution and in cells, DNA is essentially in B-form. Nevertheless, the existence of the Z-conformation in vivo and its biological role is still an intensive area of research. Most of the work dealing with in vivo studies of Z-DNA has been reviewed by Herbert and Rich;5 the major findings are the formation and stabilisation of Z-DNA by negative supercoiling. Formation of the Z-conformation unwinds DNA, thus relaxing the supercoils, which is a thermodynamically favoured process. It has been proposed that the energy necessary to form and stabilise Z-DNA in vivo can be generated during the transcription process. The difference in shape between B- and Z-DNA may also modulate the interaction with DNA-binding proteins, and Z-DNA-binding proteins have been isolated in different organisms.
Curvature is another significant feature of double-stranded DNA. The importance of curvature in DNA condensation and in the expression of genetic information justifies wide interest in this structural feature. It has been suggested that short runs of adenines, or A-tracts, separated by mixed sequences, produce substantial curvature of DNA. This hypothesis is the basis of sequence-directed curvature of DNA. Different models have been proposed to explain curvature, mostly based on interruption of the B-form DNA by an A-tract that adopts a non-B helix structure. Indeed, A tracts adopt a unique structure called B′ characterised by a reduced helical repeat (10 base pairs per turn instead of 10.5 base pairs per turn for B-DNA) in which the minor groove is narrower than in B-DNA.
2.2 Other secondary structures
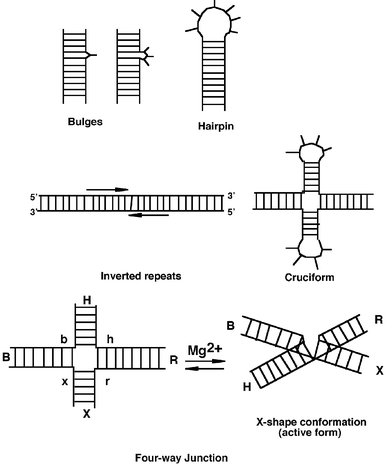 | ||
| Fig. 4 Simplified representation of RNA and DNA main secondary structures. | ||
In DNA, bulge formation may provoke mutations due to replication or transcription errors. In RNA, bulges are found in regions important for protein–RNA recognition or for the catalytic activity of RNA structures such as ribozymes.
Cruciform structures6 are formed in defined ordered sequences (inverted repeats or palindromes) that read the same in either direction, from the 5′ to 3′ in either strand. Cruciforms contain three components: the loops, the stems and the four-way junction (Fig. 4). The stems are normally fully base paired in the B-conformation and are not subjected to supercoiling. The loops are single-stranded structures positioned at the end of each stem that contain two or more unpaired bases.
Cruciform extrusion and stabilisation require DNA supercoiling, and result in the relaxation of one negative supercoil per 10.5 base pairs of DNA. Two mechanisms (S- and C-pathways) account for the cruciform extrusion.1,2 In the S-pathway, 10 base pairs must unwind at the centre of the inverted repeat allowing intrastrand hydrogen bond formation. A mechanism of branch migration then forms the cruciform structure. The C-pathway is independent of the base composition of the inverted repeats but requires A + T rich regions flanking the inverted repeats. Denaturation of A + T rich sequences at low salt concentrations forms a denaturation bubble that may enlarge to the repeat allowing the formation of cruciforms.
Holliday junctions occur between two DNA regions of homologous sequences. This sequence identity allows one strand to base pair either with its original complementary strand or with a complementary segment of the second duplex. To be formed, Holliday junctions require the close proximity of the two helices that lead to groove–backbone interactions (DNA self-fitting).7 Once they are formed, Holliday junctions may undergo two types of processes: 1) branch migration, which is a progressive exchange of base pairing between homologous duplexes and which allows the junction to move along the DNA helices, and 2) resolution, where two of the four strands are enzymatically cleaved (by resolvases) to give two symmetrical helices.
The four-way junctions are involved in major biological events both in prokaryotes and in eukaryotes as reviewed in detail by Sinden2 and Pearson.6 It has been suggested that transient formation of cruciforms is a prerequisite for the initiation of DNA replication. Holliday junctions are formed during chromosome recombination, a major genetic process involved in gene expression and repair, which allows DNA to exchange sequences.
Three- and four-way junctions are also common in RNA and constitute the key elements for the folding into the three-dimensional structures responsible for the specific biological and catalytic activities of RNAs.4
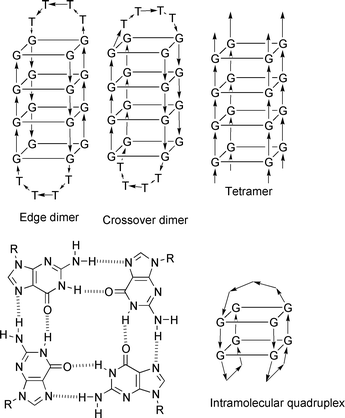 | ||
| Fig. 5 Quadruplex structure and major conformations. | ||
3 Oligonucleotides as tools for nucleic acid conformation studies
Most structural changes such as left-handed Z-DNA, quadruplex or cruciform formation require nucleotide sequences that are prone to adopt those conformations. The design and synthesis of specific oligonucleotides give access to the structural analysis of different conformations and to the study of the factors that control their formation. Z-DNA conformation has now been studied for years and the different factors that modify B–Z equilibrium have been analysed in detail using a wide range of techniques and are now well understood and controllable. Other structures have also been studied including bulges, hairpins, four-way junctions and (the most recently reported) quadruplexes. We will focus here on the structural description of short oligonucleotides that have been used to study different conformations and through this description we will emphasise the structural, chemical and environmental factors important for their formation and stabilisation.3.1 Z-Conformations
Many reviews have described in detail the Z-conformation and the factors that influence its formation.2,9 Rich published a major review in 1984.10In natural DNA, supercoiling is the major factor influencing Z-formation. At the oligonucleotide level, interconversion between right-handed and left-handed helices is dependent on environmental factors such as the presence of metal ions, polyamines or organic solvents and upon the presence of modifications on the bases. These factors have been extensively discussed in recent reviews2,9 and the main points are outlined here. Introduction of a bulky substituent, bromine or methyl group, at position 8 of guanines stabilises the Z-conformation since steric hindrance forces the deoxyribose to the syn position. Introduction of a methyl group or bromine at C5 of cytosine also favours the Z-conformation. The presence of a methyl group at position 5 of cytosine has been extensively studied as it stabilises the Z-conformation under physiological conditions, suggesting the biological interest of these structures. Indeed, poly(dG–dm5C).poly(dG–dm5C) sequences frequently occur in eukaryotic cells and it has been proposed that methylated sequences can play a role in the control of the gene expression.
Another feature of Z-DNA is that phosphate groups on opposite strands approach much closer than in B-DNA and therefore any factor that will reduce the phosphate–phosphate repulsion (shielding effect) will favour and stabilise the Z-form. Indeed, the formation of Z-DNA was first observed in a solution containing a high concentration of salt (4M NaCl). If high concentrations of monovalent cations (Na+, K+, Cs+, Li+) are required to achieve B to Z transitions, the shielding effect is much more effective with divalent cations (Mg2+, Mn2+). Typical salt conditions necessary to induce the Z-conformation in poly(dG–dC).poly(dG–dC) are 2.5 MNaCl or 0.7M MgCl2 or 0.02mM Co(NH3)63+. The effect is more pronounced with poly(dG–dm5C).poly(dG–dm5C) for which 0.6mM MgCl2 is sufficient to induce the transition. Polyamines have also been shown to stabilise the Z-conformation. The effect is strongly dependent on the sequence of the oligonucleotide and the nature of the polyamine. Poly(dG–dm5C).poly(dG–dm5C) undergoes B to Z transition in the presence of spermine4+ and spermidine3+, in concentrations where poly(dG–dC).poly(dG–dC) remains in the B-form and eventually aggregates.11 The effect of polyamines on DNA conformation is of major interest as polyamines are widely distributed in biological systems, and furthermore, increased concentrations of polyamines have been detected in some pathologies such as cancer, psoriasis and cystic fibrosis.
The influence of drugs on the B–Z equilibrium has also been studied. Most of the drugs that intercalate between DNA base pairs (for example, proflavine, ethidium bromide, and the anticancer agent daunomycin shown in Fig. 6) have been found to induce the Z to B transition. This transition can be related to a higher affinity of the drugs for the B-form that progressively shifts the equilibrium in favour of B-DNA. Netropsin, which binds to the minor groove of DNA, also promotes the Z to B transition. An interesting behaviour was observed for poly(dG–dC).poly(dG–dC) in the presence of dinuclear platinum complex [(trans-PtCl(NH3)2)2(H2 N(CH2)nNH2)]2+ (Fig. 6), in which the polyamine chain induces the Z-conformation as previously described for polyamines, but due to the presence of the platinum complex, an additional step of interstrand covalent binding locks the Z-DNA and prevents the reversal to the B-conformation by ethidium bromide.12
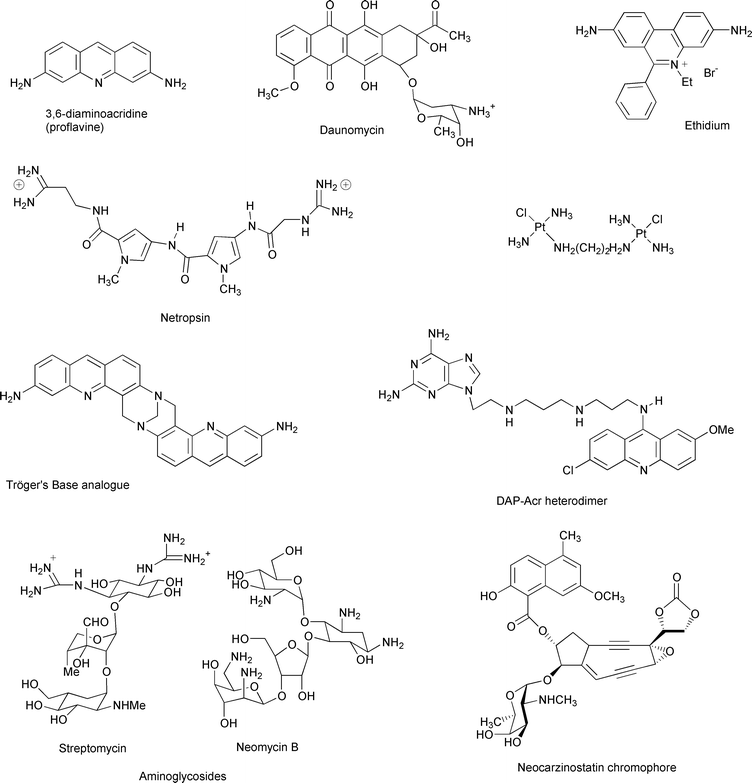 | ||
| Fig. 6 Representative binders of DNA and RNA secondary conformations | ||
3.2 Four-way junctions14,15
Gel electrophoresis is a very sensitive and non-perturbing technique that appeared to be a method of choice to study structural changes of pseudo-cruciforms. A stable junction is characterised by a much slower electrophoretic migration compared to linear sequences and this migration is dependent on the position of the junction relative to the ends of the arms. The conformations of the four-way junctions are highly influenced by the presence of cations. In the presence of Mg2+ the four-way junction folds into an X-shaped structure while in the absence of cations the unfolded extended conformation has a square geometry (Fig. 4). From these experiments, a structure was proposed for the Holliday junction, with an X-like conformation resulting from collinear association of two helices.14 The main characteristics of this conformation are: (1) the arms of the junction associate in pairs by helix–helix stacking and two conformers are possible, differing in their stacking partners; (2) there is a two-fold symmetry in the structure generating two sets of strands—two strands are continuous through the point of strand exchange and the two other strands interchange between the two helices.
This X-shaped conformation was confirmed by high-field NMR and fluorescence resonance energy transfer (FRET).4 This technique requires the synthesis of oligonucleotides in which two chromophores, a donor (fluorescein) and an acceptor (rhodamine), are covalently attached to two different 5′ ends. Upon excitation of the donor, energy is transferred to the acceptor through dipolar interaction between the two transition moments; the efficiency of the transfer is dependent on the distance between donor and acceptor fluorophores. Different donor–acceptor molecules can be used. Comparison of the efficiencies of FRET allows the determination of the preferred conformations. The data thus obtained fully confirmed the conformations proposed earlier.
The oligonucleotides used for the early electrophoresis measurements contained about 80 nucleotides and were too long for NMR methodology. Therefore for the NMR studies, shorter oligonucleotides were synthesised, with 16 residues per strand (64 for the full junction) and then later a stable four-way junction with 38 nucleotides was prepared and studied.16 To stabilise the junction, three of the four arms were ‘capped’ by small loops. Proton NMR data confirmed the stacked X-conformation and indicated that one single isomer was preferred. Another interesting finding was that full Watson–Crick base pairing was preserved at the junction site.
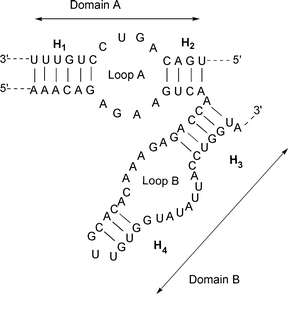 | ||
| Fig. 7 Secondary structure of the hairpin ribozyme showing the two domains | ||
3.3 Bulges
The structure determination and dynamics of bulges have been reviewed by Turner.17 The first technique used to compare different bulged duplexes was based on their mobility in gel electrophoresis. Different observations were made: duplexes containing bulges migrate more slowly than fully paired duplexes, and single-nucleotide bulges are faster than larger bulges. Electrophoretic migration is also dependent on both the nature and orientation of the base pairs flanking the bulges and this sequence effect is much more important for purine bulges than for pyrimidine ones. Similar results were also obtained with bulged RNA. More information on the conformation of bulges was gained from NMR experiments. NOE experiments providing quantitative and qualitative information on the distances between protons (distances < 5Å) were combined with molecular dynamics to generate pictures of possible conformations. Short oligonucleotides containing single- nucleotide bulges, such as d(CCGTGAATTCCGG)2 or d(CCGGAATTCTCGG)2 (where superscript bases designate the bulges), were studied by NMR spectroscopy. Factors such as the nature of the flanking base pairs or temperature were examined. Two positions for the unpaired bases related to the helix axis were observed, either looped out or stacked inside the helix, and this position was both sequence and temperature dependent. The major points that emerged from these experiments were that the structure of the bulge was very dependent on temperature for GTG and GCG but not for CTC, GAG or CAC, and that bulged purines kinked the helix axis more than did bulged pyrimidines.3.4 G-Quadruplexes
The first structural data of quadruplexes were obtained in 1992 from X-ray crystallography and NMR studies of guanine rich sequences. Since then a growing number of papers have reported on their structure determination and biological properties and have been recently reviewed.8,18It has been shown that guanine rich sequences form quadruplexes with different topologies and strand orientation depending on the sequence. Different types of quadruplexes, such as dimeric, tetrameric or intramolecular structures, have been reported (Fig. 5). One of the most extensively studied DNA sequences, d(G4T4G4), derived from the Oxytricha nova telomere, forms dimeric quadruplex structures [d(G4T4G4)]2 with four guanine-quartets. However, the structures obtained by X-ray crystallography and NMR spectroscopy display different conformations and folding topologies. One explanation for these differences might be the presence of different counterions (Na+versus K+). Other nucleic bases might also form quadruplexes as A- or T-quadruplexes have been recently reported.
4 DNA and RNA molecular recognition
4.1 Recognition by small molecules
The interaction of neocarzinostatin chromophore with bulged DNA was studied in detail by Goldberg and co-workers23 Neocarzinostatin (Fig. 6), the active chromophore of a macromolecular protein, contains the ene-diyne functionality. As with other members of the ene-diyne family, the neocarzinostatin chromophore is activated by the formation of a biradical species that induces DNA cleavage by hydrogen abstraction. The specific interaction of the active form of the drug with a bulge appeared to modify its chemical behaviour. The cleavage efficiency was enhanced and the reaction led to a new drug product. A detailed NMR study of the complex formed between the drug and a bulge revealed a perfect fit between the active form of the drug and the bulge. Surprisingly, while a number of DNA bulges have been shown to interact with neocarzinostatin, the drug cleaves RNA bulges very poorly. Recent results24 suggest the existence of a steric clash between 2′-OH of the ribose and OMe moiety of the drug that might account for the lower binding to RNA. These experiments also point out the necessity of a bulge-specific drug binding for efficient cleavage.
Different classes of compounds have been shown to bind specific sites (such as bulges and hairpins) of structured RNAs. These include, for example, small organic and inorganic ligands, short peptides. This area has been reviewed recently.25 We designed a series of heterodimers containing an intercalator (aminoacridine) and a 2,6-diaminopurine linked together by a polyaminolinker (Fig. 6). These molecules were shown to interact with a bulge containing RNA.26 Our hypothesis is that the aminoacridine moiety intercalates in the RNA duplex and that the 2,6-diaminopurine residue may base pair with the unpaired uracil of the bulge. Although most of the small RNA ligands appeared to bind DNA as well, aminoglycoside antibiotics (i.e. streptomycin, neomycin shown in Fig. 6) revealed a promising preference for RNA motifs such as hairpins and bulges.
A promising application involves RNA aptamers.27 RNA aptamers are short RNA sequences that are selected in vitro from randomised pools of RNA to bind small ligands with high affinity and selectivity. This field has been reviewed recently by Tor27 and a recent work of Werstuck and Green28 reported on the use of small organic molecules to control gene expression in living cells. To do so, they incorporated a small aptamer into a specific region (5′-untranslated region or UTR) of a messenger RNA (mRNA). The presence of the aptamer alone did not alter the translation process of the modified mRNA, but the binding of the aptamer specific ligand, an aminoglycoside antibiotic, induced a conformational change that blocked the mRNA translation in vitro as well as in cultured mammalian cells.
 | ||
| Fig. 8 Possible sites for intramolecular quadruplex specific binders. | ||
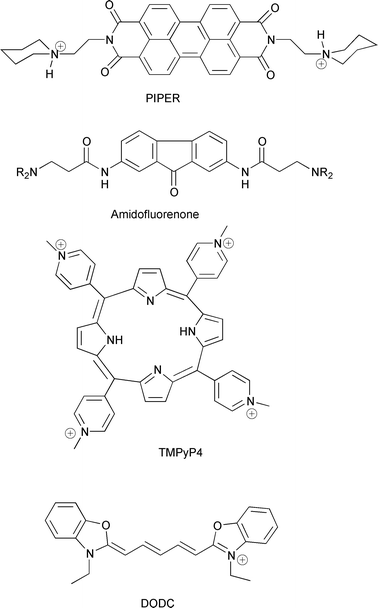 | ||
| Fig. 9 Chemical structures of quadruplex binders. | ||
4.2 Protein–nucleic acid interactions
Z-RNA is also immunogenic.36 Anti-Z-RNA antibodies recognise Z-RNA but not natural or synthetic Z-DNA conformations. The major chemical difference between Z-RNA and Z-DNA being the presence of 2′-hydroxy groups exposed on the surface of the Z-RNA helix, one explanation for the observed specificity is that these hydroxy groups might be strongly involved in the binding of the antibody to Z-RNA sequences. Anti-Z-antibodies have been used to probe the presence of Z-RNA in cytoplasm and later, to show that Z-RNA was synthesised in the nucleolus. Similarities in the behaviour of Z-RNA and ribosomal RNA suggested that some detected cellular Z-RNA might in fact be double-stranded sequences of ribosomal RNA of nucleolar origin. Furthermore, it was also proposed that A to Z transitions in ribosomal RNA might act as molecular switches for controlling protein synthesis. This hypothesis was supported by the inhibition of protein synthesis by anti-Z-RNA antibodies bound to RNA duplexes.36
Branched structures are also targets for specific antibodies. Anti-cruciform antibodies recognise some structural features of the cruciforms but not the corresponding linear inverted repeats. Fluorescence labelling of eukaryotic cell nuclei with anti-cruciform antibodies has been used to localise and quantify cruciform structures. The presence of cruciforms was found to be intimately linked to DNA replication.6
Recently, a new class of antibodies was isolated and found to have a preference for quadruplex DNA37 and more specifically for parallel four-stranded structures. However, the detection of quadruplex structures in vivo remains to be achieved.
Specific recognition of Z-DNA sequences. The specific recognition of Z-DNA by enzymes has been the subject of intense study in order to grasp the biological role of Z-conformation. Different enzymes were found to recognise Z-DNA and among them, double-stranded RNA deaminase (ADAR1) displayed the highest affinity for Z-DNA. Interaction of this enzyme with Z-DNA was studied in detail by Rich and co-workers.38 The enzyme includes a DNA binding domain, Zα, which is specific for left-handed DNA. Very recently, a crystal structure of Zα complexed to Z-DNA (d(TCGCGCG)2 hexamer) was obtained and showed that interactions occur between the sugar–phosphate backbone of the oligonucleotide and Zα domain of the enzyme and that both the ‘zigzag’ shape of the backbone and the anti–syn alternation of the bases are recognised by the Zα domain.
Recognition and manipulation of four-way junctions. Four-way junctions play a central role in genetic recombination and in the repair of double-strand breaks caused by radiation and chemical damage. Different proteins bind more or less specifically to four-way junctions and manipulate the junctions; the subject has been reviewed recently.39 Studies of genetic recombination pathways in Escherichia coli have helped to define each stage of the process and to elucidate the DNA–protein interactions that occur during DNA pairing, strand exchange (branch migration) and resolution of recombination intermediates. In Escherichia coli, three enzymes, RuvA, RuvB and RuvC, are implicated in recombination of Holliday junctions: the RuvAB complex catalyses branch migration and RuvC ultimately resolves recombination intermediates into two separate duplex DNAs. In the recent years, use of synthetic Holliday junctions has allowed a better understanding of the different processes and detailed characterisation of protein–Holliday junction complexes. RuvB, which may be considered as the motor of branch migration, is a ring-shaped hexameric protein that belongs to the helicase family. RuvA is a tetrameric protein that selectively recognises and binds Holliday junctions. Finally, RuvC is a member of the resolvases (or junction resolving enzymes) whose mechanisms of action have been extensively studied.39 A recent crystal structure of a RuvA–Holliday junction complex40 and detailed biochemical studies41,42 give a more accurate picture of the recombination process. RuvA binds with high affinity to Holliday junctions. The crystal structure of RuvA protein (isolated from Mycobacterium leprae) in complex with a synthetic four-way junction indicates that two RuvA tetramers are associated into an octameric shell, generating a cruciform cavern sandwiching the DNA junction. The complexation of RuvA to four-way junctions induces an important distortion of the junction structure from the X-shape conformation, characteristic of the free junctions, to a cruciform structure as recently shown by atomic force microscopy.43 Tethered to the RuvA–Holliday junction complex are two hexameric rings of RuvB protein encircling the DNA duplex arms in opposite directions.41 The two RuvB rings act as DNA motors driving branch migration through the RuvA-bound junction. Ultimately, RuvC achieves the junction resolution. From the biochemical studies of the mechanism of action of RuvC and other resolvases (such as T4 endonuclease VII from bacteriophage or CCE1 from yeast),44 the following points emerge: (1) the binding of the enzyme is a structure-selective process in which the base sequence does not play any role; (2) the resolving enzymes bind to the junction as dimers in which two sub-units cleave two opposite strands of the four-way junction; (3) the binding of a resolving enzyme to a four-way junction induces a change in the structure of the junction, CCE1 introducing the greatest distortion as it unfolds the X-structure into an extended square configuration of the arms; and (4) the enzymes catalyse the hydrolysis of the phosphodiester backbone in a sequence-specific process. It has been proposed that the binding of RuvC to a junction is stabilised by RuvB, which also stimulates its resolvase activity.42 In the hypothetical scheme, the three enzymes form a RuvABC complex with the four-way junction in which RuvC ‘scans’ the DNA sequences as they pass through the complex, resulting in efficient resolution at preferred sites.
5 Tertiary structures and the importance of folding in nucleic acid biology
5.1 Structures
The folding or the association of secondary structures of DNA or RNA creates a remarkable variety of different shapes. The three-dimensional architecture of DNA and RNA provides potentially catalytic centres and binding pockets for cations, proteins and other nucleic acids. Metal ions are known to play an important role in the folding process dominated by electrostatic interactions.Structural studies of RNAs taken from a wide variety of organisms have dramatically increased during the past few years. Despite the diversity of the RNA sources, only a limited number of structural motifs have been identified, suggesting that the complex folding responsible for the specific recognition in vitro is due to the way the simple elements are pieced together.45 Branch points formed by the global folding may create specific binding pockets for metal ions or specific target sites for proteins.4 Internal loops and bulges are among the simplest secondary structures that create branch points, which may act as specific recognition sites for protein binding. Interactions between secondary structures can be of different nature.46 Helical motifs may interact with each other or with unpaired motifs. In the first case, coaxial stacking of helical regions allows higher order organisation of RNA through stacking interactions between nucleic bases. This contribution in the folding process was demonstrated first in tRNA structures (Fig. 10). These RNAs are organised into three hairpin structures. About 60% of the bases are implicated in stacking interactions in double-stranded sequences, but actually, more than 90% of the bases are involved in stacking interactions due to coaxial stacking occurring between arms in the three-dimensional structure. Such an organisation is also observed in natural catalytic RNAs. For these structures, the coaxial stacking is often completed by the binding of divalent ions close to the stacking site. The interactions between helical and unpaired regions lead to the formation of new motifs such as the four nucleotide loop sequences or tetraloops. Furthermore, new helices can be created through base pairing interactions between the nucleotides of two hairpin loops (‘kiss complex’) or between hairpin loops and single-stranded sequences (pseudo-knots).46
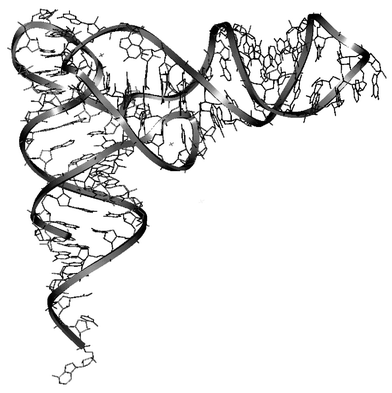 | ||
| Fig. 10 Three-dimensional structure of tRNAs (Insight II (98) from MSI). | ||
An important aspect of the biological activities of RNAs resides in their ability to catalyse chemical processes such as formation and hydrolysis of phosphodiester bonds or formation of peptide bonds. These natural catalysts are named ribozymes. We will comment only on small catalytic motifs such as the hammerhead and hairpin ribozymes expressed during the replication cycle of Tobacco ringspot virus satellite. We will not discuss the more complex structures found in Group I introns, hepatitis delta virus or ribosomes. It is interesting to note that recent technical progress in the field of combinatorial chemistry of nucleic acids and their in vitro selection has led to a broadening of the field of the chemical transformations catalysed by nucleic acids:47 cleavage of amide or carboxylic ester bonds, C–C bond formation, isomerisation reactions, etc.
The hammerhead and hairpin ribozymes catalyse the site specific cleavage and ligation reactions that transform the oligomeric forms of the circular RNAs into monomeric circular forms. The hydrolysis of the phosphodiester bond generates 2′,3′-cyclic phosphate and 5′-OH extremities. The two ribozymes differ in their structural organisation and their cleavage mechanism. The hammerhead ribozyme (Fig. 11) is composed of three double-stranded domains connected by single-stranded regions containing conserved nucleic bases that are necessary for the cleavage activity. The reaction requires the activation of a ribose 2′-hydroxy group, which then attacks its 3′-attached phosphate. The activation step involves a hydroxide ion bridged by two Mg2+ ions.48 The hairpin ribozyme (Fig. 7) bound to its substrate constitutes two domains termed A and B which contain an internal loop flanked by two helices (H1 and H2 in domain A and, H3 and H4 in domain B as shown in Fig. 7). The folding of the two domains is independent, but tertiary contacts between them are necessary to form a functional complex. Metal ions facilitate this global folding but appear not to be required for the catalytic reaction. It has been suggested that the role of the tertiary contacts might be to organise the active site in such a way that the reactions can be catalysed by RNA functional groups. This hypothesis is supported by a recent study on another self-cleaving ribozyme.49 In this example, it was convincingly suggested that a cytosine residue acts as the general base catalyst which activates a specific ribose 2′-hydroxy group for attacking the 3′ adjacent phosphodiester bond.
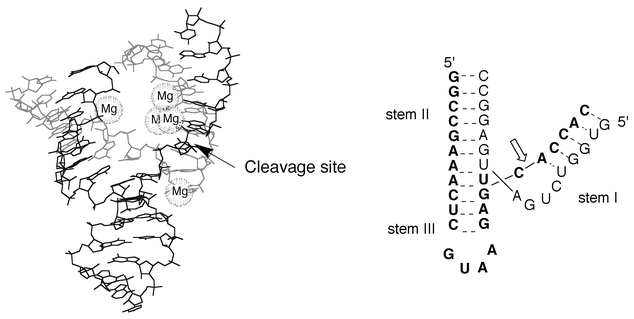 | ||
| Fig. 11 The active form of the hammerhead ribozyme (adapted from Scott et al.57). The cleavage site in the substrate strand (in bold character) is marked by an arrow. | ||
5.2 Folding interactions
Structural information on large and complex RNAs is not always fully addressed by structural techniques such as NMR or X-ray crystallography. The use of organic chemistry to vary individual functional groups in a nucleic acid is referred to as ‘atomic mutagenesis’. This concept has found many applications in the study of RNA, for which a great variety of chemical modifications has been envisaged. Photo-cross-linking agents or photoreactive nucleic bases (such as 4-thiouridine, 5-bromouridine.) are used to probe RNA–RNA and RNA–protein interactions. Modified ribose and phosphates are very useful to analyse RNA structure and function. Fluorescent labels can also be incorporated to investigate RNA folding pathways and kinetics by using the fluorescence resonance energy transfer (FRET) technique. Many different strategies have been devised to incorporate chemical modifications into RNA (for a recent review, see Zimmerman et al.50)Methods based on RNA–protein cross-linking have found application in the study of nucleoproteins and the recognition of folded RNAs by proteins or peptides. An example of secondary structure recognised by specific proteins is given by the HIV life cycle.51 The transcriptional regulation of this virus requires specific binding of Tat protein with the transcription responsive region (TAR) RNA, a stem-loop structure containing a bulge and present at the 5′-end of all messenger RNA (mRNA). This binding causes a substantial increase in mRNA levels. Structurally, the contact extends over five base pairs and includes a trinucleotide pyrimidine bulge and hexanucleotide loop. The role of the bulge is most likely to make an accessible Tat site in the otherwise deep and narrow major groove of the duplex RNA.
HIV-1 Tat comprises 86 amino-acid residues organised in six regions. TAR is a stem-loop structure of 59 residues found at the 5′-end of all HIV transcripts. The interaction of Tat with TAR is essential for the virus cell cycle. Atomic mutagenesis allowed the identification of the essential residues of TAR required for Tat interaction.50 Functional group mapping of TAR showed that in the TAR–Tat complex, the basic region lies in the major groove of the duplex segment of TAR. The phosphate groups appeared also to actively participate in Tat binding. Replacement of precise phosphate groups by methyl phosphonates revealed their indirect interaction with basic residues (arginine or lysine) in Tat.
Other good examples of the use of modified nucleotides are found in the ribozyme field.50 The hammerhead and hairpin ribozymes, which contain less than 50 nucleotides, can be prepared by total chemical synthesis and thus can easily be site specifically modified. The functional groups required for the cleavage activity have been studied by modifying the bases, sugar or phosphate at almost every position. These chemical modifications have given new insights into the mechanistic questions unanswered by the crystallographic approach.
6 Future prospects
As already mentioned above, the easy generation of large combinatorial libraries of RNA or DNA molecules has extended the fields of application of oligonucleotides from molecular biology to pharmacology and even organic chemistry.52 Selection experiments afford oligonucleotides, commonly named aptamers, which may selectively bind to bioactive macromolecules (proteins or nucleic acids) or small molecules (dyes, amino acids, antibiotics, etc.). The presence of secondary structures such as bulges, hairpins or junctions is frequently associated with the recognition, binding or catalytic processes. The structural studies of aptamer–small molecule complexes should give new insights into the understanding of molecular recognition of nucleic acids.DNA nanotechnology, i.e. the construction of specific geometrical or topological DNA objects, is a new field of investigation that has been developed by Seeman and recently reviewed.53 The goals include the use of DNA molecules to organise the assembly of other macromolecules for crystal facilitation or memory device construction. Topological objects such as knots and catenates have also been designed; their central features are their crossings or nodes. Right-handed B-DNA generates negative nodes and left-handed Z-DNA generates positive nodes, therefore controlling the sequence of a single-stranded DNA and the ion content allows the specific formation of various types of knot. RNA knots have also been prepared and used to demonstrate an RNA topoisomerase activity. Various 3-D DNA objects have been made using branched DNA components as building blocks, as for example a cube-like object corresponding to a hexacatenate of single-stranded DNA. Each of the six faces contains a cyclic strand doubly linked to its four neighbouring strands. Very recently,54 the control of the B–Z transition has been used to design a nanomechanical device allowing rotation between two objects.
The use of DNA in computing science is another exciting area of research. A self-assembly model of computing, based on different equilibria of B- and Z-DNA has been already proposed.55
RNA and DNA are no longer considered as only passive carriers of genetic information but have been shown to act as active biomolecules involved in many key cellular processes. This functional diversity has been attributed to the complexity of the edifices generated by secondary structures and tertiary interactions. Because DNA and RNA are polyelectrolytes, their tertiary structures may create binding pockets for ions, small ligands and proteins. These recognition sites can be promising targets for designing specific binders that could modulate the cellular processes involving these biologically active structures. Furthermore, the duality of ribozymes which cleave other RNA molecules with high sequence specificity are promising in therapeutical practices that require the control of gene expression. A recent review on the use of catalytic RNA ongene function inhibition has been published by Muotri et al.56
7 Acknowledgement
The authors thank M. L. Dheu-Andries for technical assistance with the Insight II generated figures.8 References
- G. M. Blackburn and M. J. Gait, ‘Nucleic Acids in Chemistry and Biology’, ed. G. M. Blackburn and M. J. Gait, Oxford 1996. Search PubMed.
- R. R. Sinden, C. E. Pearson, V. N. Potaman and W. D. Ussery, Adv. Genome Biol., 1998, 5A, 1–141 Search PubMed.
- D. M. J. Lilley, ‘The formation of alternative structures in DNA’, ed. S. M. Hecht, Oxford University Press, Oxford, 1996. Search PubMed.
- D. M. J. Lilley, Biopolymers, 1998, 48, 101 CrossRef CAS.
- A. Herbert and A. Rich, J. Biol. Chem,, 1996, 271, 11595 CrossRef CAS.
- C. E. Pearson, H. Zorbas, G. B. Price and M. Zannis-Hadjopoulos, J. Cellular Biol., 1996, 63, 1 Search PubMed.
- Y. Timsit and D. Moras, Quart. Rev. Biophys., 1996, 29, 279 CAS.
- D. E. Gilbert and J. Feigon, Curr. Opin. Struct. Biol., 1999, 9, 305 CrossRef CAS.
- B. H. Johnston, Methods Enzymol., 1992, 211, 127 Search PubMed.
- A. Rich, A. Nordheim and A. H.-J. Wang, Ann. Rev. Biochem., 1984, 53, 791 Search PubMed.
- L. van Dam and L. Nordenskiöld, Biopolymers, 1999, 49, 41 CrossRef CAS.
- P. K. Wu, M. Kharatishvili, Y. Qu and N. Farrell, J. Inorg. Biochem., 1996, 63, 9 CrossRef CAS.
- C. C. Hardin, G. T. Walker and I. Tinoco, Biochemistry, 1988, 27, 4178 CrossRef CAS.
- D. R. Duckett, A. J. H. Murchie, R. M. Clegg, G. S. Bassi, M.-J. E. Giraud-Panis and D. M. J. Lilley, Biophys. Chem., 1997, 68, 53 Search PubMed.
- A. I. H. Murchie and D. M. Lilley, Methods Enzymol., 1992, 211, 158 Search PubMed.
- F. J. J. Overmars, V. Lanzotti, A. Galeone, A. Pepe, L. Mayol, J. A. Pikkemaat and C. Altona, Eur. J. Biochem., 1997, 249, 576 CrossRef CAS.
- D. H. Turner, Curr. Opin. Struct. Biol., 1992, 2, 334 CrossRef CAS.
- S. Borman, Chem. Eng. News, 1999, 77, 36 Search PubMed.
- T. D. Tullius, Curr. Opin. Struct. Biol., 1991, 1, 428 CrossRef CAS.
- A. Tatibouët, M. Demeunynck, C. Andraud, A. Collet and J. Lhomme, Chem. Commun., 1999, 161 RSC.
- K. E. Erkkila, D. T. Odom and J. K. Barton, Chem. Rev., 1999, 99, 2777 CrossRef CAS.
- C.-C. Cheng, Y.-N. Kuo, K.-S. Chuang, C.-F. Luo and W. J. Wang, Angew. Chem. Int. Ed., 1999, 38, 1255 CrossRef CAS.
- Z. Xi, Q. K. Mao and I. H. Goldberg, Biochemistry, 1999, 38, 4342 CrossRef CAS.
- L. S. Kappen, Z. Xi and I. H. Goldberg, Bioorg. Med. Chem., 1997, 5, 1221 CrossRef CAS.
- C. S. Chow and F. M. Bogdan, Chem. Rev., 1997, 97, 1489 CrossRef CAS.
- W. D. Wilson, L. Ratmayer, M. T. Cegla, J. Spychala, D. Boykin, M. Demeunynck, J. Lhomme, G. Krishnan, D. Kennedy, R. Vinayak and G. Zon, New J. Chem., 1994, 18, 419 Search PubMed.
- Y. Tor, Angew. Chem. Int. Ed., 1999, 38, 1579 CrossRef CAS.
- G. Werstuck and M. R. Green, Science, 1998, 282, 296 CrossRef CAS.
- P. J. Perry and T. C. Jenkins, Expert Opin. Invest. Drugs, 1999, 8, 1981 Search PubMed.
- J. L. Mergny, P. Mailliet, F. Lavelle, J. F. Riou, A. Laoui and C. Helene, Anti-Cancer Drug Des., 1999, 14, 327 Search PubMed.
- H. Han, C. L. Cliff and L. H. Hurley, Biochemistry, 1999, 38, 6981 CrossRef CAS.
- M. A. Read, A. A. Wood, J. R. Harrison, S. M. Gowan, L. R. Kelland, H. S. Dosanjh and S. Neidle, J. Med. Chem., 1999, 42, 4538 CrossRef CAS.
- S. Y. Rha, E. Izbicka, R. Lawrence, K. Davidson, D. Sun, M. P. Moyer, G. D. Roodman, L. Hurley and D. Von Hoff, Clin. Cancer Res., 2000, 6, 987 Search PubMed.
- H. Arthanari, S. Basu, T. L. Kawano and P. H. Bolton, Nucleic Acids Res., 1998, 26, 3724 CrossRef CAS.
- Q. Chen, I. D. Kuntz and R. H. Shafer, Proc. Natl. Acad. Sci. USA, 1996, 93, 2635 CrossRef CAS.
- D. A. Zarling, C. J. Calhoun, B. G. Feuerstein and E. P. Sena, J. Mol. Biol., 1990, 211, 147 CrossRef CAS.
- B. A. Brown, Y. Li, J. C. Brown, C. C. Hardin, J. F. Roberts, S. C. Pelsue and L. D. Shultz, Biochemistry, 1998, 37, 16325 CrossRef CAS.
- T. Schwartz, M. A. Rould, K. Lowenhaupt, A. Herbert and A. Rich, Science, 1999, 284, 1841 CrossRef CAS.
- M. F. White, M.-J. E. Giraud-Panis, J. R. G. Pöhler and D. M. J. Lilley, J. Mol. Biol., 1997, 269, 647 CrossRef CAS.
- S. M. Roe, T. Barlow, T. Brown, M. Oram, A. Keeley, I. R. Tsaneva and L. H. Pearl, Mol. Cell, 1998, 2, 361 CrossRef CAS.
- M. Grigoriev and P. Hsieh, Mol. Cell, 1998, 2, 373 CrossRef CAS.
- A. J. van Gool, R. Shah, C. Mézard and S. C. West, EMBO J., 1998, 17, 1838 CrossRef CAS.
- L. S. Shlyakhtenko, P. Hsieh, M. Grigoriev, V. N. Potaman, R. R. Sinden and Y. L. Lyubchenko, J. Mol. Biol., 2000, 296, 1169 CrossRef CAS.
- M. F. White and D. M. Lilley, Nucleic Acids Res., 1998, 26, 5609 CrossRef CAS.
- G. L. Conn and D. E. Draper, Curr. Opin. Struct. Biol., 1998, 8, 278 CrossRef CAS.
- R. T. Batey, R. P. Rambo and J. A. Doudna, Angew. Chem. Int. Ed., 1999, 38, 2326 CrossRef.
- M. Famulok and A. Jenne, Top. Curr. Chem., 1999, 202, 102 Search PubMed.
- T. Hermann, P. Auffinger, W. G. Scott and E. Westhof, Nucleic Acids Res., 1997, 25, 3421 CrossRef CAS.
- A. Perrotta, I.-H. Shih and M. D. Been, Science, 1999, 286, 123 CrossRef CAS.
- R. A. Zimmermann, M. J. Gait and M. J. Moore, ‘Incorporation of modified nucleotides into RNA for studies on RNA structure, function and intermolecular interactions’, ed. H. Grosjean and R. Benne, ASM Press, Washington, 1998, 59. Search PubMed.
- A. D. Frankel and J. A. Young, Ann. Rev. Biochem., 1998, 67, 1 Search PubMed.
- M. Famulok and A. Jenne, Curr. Opin. Chem. Biol., 1998, 2, 320 CrossRef CAS.
- N. C. Seeman, Angew. Chem. Int. Ed., 1998, 37, 3220 CrossRef CAS.
- C. Mao, W. Sun, Z. Shen and N. C. Seeman, Nature, 1999, 397, 144 CrossRef CAS.
- M. Conrad and K.-P. Zauner, BioSystems, 1998, 45, 59 CrossRef CAS.
- A. R. Muotri, L. D. Pereira, L. D. Vasques and C. F. M. Menck, Gene, 1999, 237, 303 CrossRef CAS.
- W. G. Scott, J. B. Murray, J. R. P. Arnold, B. L. Stoddard and A. Klug, Science, 1996, 274, 2065 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2001 |