DOI:
10.1039/A904591K
(Review Article)
Chem. Soc. Rev., 2001,
30, 16-25
Radical carboxylations of iodoalkanes and saturated
alcohols using carbon monoxide
Received 21st July 2000
First published on 15th December 2000
Abstract
This review covers two radical carboxylation methods using
carbon monoxide, both of which were developed by our group. The first
method, atom transfer carbonylation, converts alkyl iodides into
carboxylic acid esters or amides and the second method, remote
carboxylation, converts saturated alcohols into δ-lactones. Both
methods rely upon radical carbonylation chemistry to introduce carbon
monoxide, but the key steps are conceptually different. The first method
utilizes an atom transfer reaction from an alkyl iodide to an acyl radical
leading to an acyl iodide and the latter employs a one-electron oxidation
reaction to convert an acyl radical into an acyl cation. The iodine atom
transfer carbonylation process is reversible and therefore highly
inefficient unless it is performed in concert with an ionic system to shift
the equilibrium in the direction of an acyl iodide. In the latter process,
a 1,5-translocation scheme to shift the radical from oxygen to the
δ-carbon is successfully coupled with the
carbonylation–oxidation sequence. Carboxylations of alkyl halides by
transition metal catalyzed methods are often problematic because of the
inherent weakness of alkyl–metal bonds. Existing methods for
carbonylative δ-lactone synthesis using transition metal catalysts
are limited to unsaturated alcohols. Thus, these two radical
carboxylation methods nicely complement existing transition metal catalyzed
carboxylations.
 Ilhyong Ryu Ilhyong Ryu |
Ilhyong Ryu is a Professor of Chemistry at Osaka Prefecture University. He
was born in the suburbs of Nagoya in 1951. He received his BS (1973) from
Nagoya University, and his MS (1975) and PhD (1978) from Osaka University
working with Professor Noboru Sonoda. After spending some years as a JSPS
postdoctoral fellow and a research associate at Osaka University, he became
Assistant Professor at Osaka University in 1988 and was promoted to
Associate Professor of the Graduate School of Engineering, Osaka University
in 1995. He moved to Osaka Prefecture University as Full Professor in 2000.
He was a visiting scientist at the University of Ottawa with Professor
Howard Alper (1991–1992). He has been the recipient of the Progress
Award in Synthetic Organic Chem- istry, Japan (1990) and the Daicel
Chemical Award in Synthetic Organic Chemistry (1989). He has been a Fellow
of the Sumitomo Foundation (1996) and the Fujisawa Foundation (2000). His
research interests include the development of new synthetic methodologies
based on free radical species, catalytic synthetic transformations,
chemistry of dianions, concise synthetic reactions, and automated
synthesis. |
1 Introduction
Carboxylation with carbon monoxide is intrinsically important for the
synthesis of carboxylic acids and their derivatives. Much effort has been
devoted to transition metal catalyzed carboxylation chemistry to
date.1 Monsanto’s acetic acid
synthesis, which uses CO and methanol as the substrates, is a key example
of the industrial importance of the metal catalyzed carboxylation processes
(Scheme 1).2 This famous reaction is based on the rhodium
catalyzed carbonylation of methyl iodide, which is formed in situ
from methanol and HI. However, many hurdles still have to be surmounted
before the basic reaction can be generally applicable. The formation of
regioisomeric products is a serious problem for C3 and higher
alcohols, due to the tendency of the intermediate metal–alkyl species
to undergo isomerization. Research into methods to circumvent the
undesirable β-elimination is ongoing in the field of transition metal
chemistry.3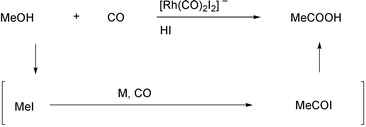 |
| | Scheme 1 | |
The controlled use of carbon dioxide as a feedstock for the
carboxylation of organic substrates represents a holy grail irrespective of
whether transition metal species or radical species are the intermediates.
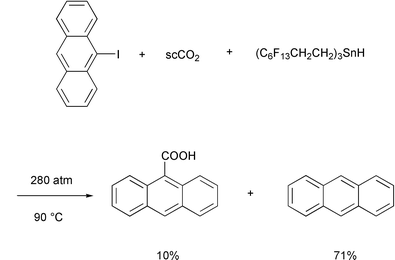
With respect to radical processes, to the best of my knowledge, there has
been only one recent report from the Curran group documenting the formation
of a carboxylic acid by radical carboxylation with carbon dioxide. In this
case, the formation of 9-anthracenecarboxylic acid was obtained in 10%
yield together with anthracene (71%), when the radical reduction of
9-iodoanthracene with the ethylene-spaced fluorous tin hydride reagent was
run at 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C using 280atm of supercritical CO2 as the
reaction medium (Scheme 2).4 As demonstrated in this rare example, the trapping
of CO2 by radicals is not an efficient process and therefore is
of limited synthetic utility.5
°C using 280atm of supercritical CO2 as the
reaction medium (Scheme 2).4 As demonstrated in this rare example, the trapping
of CO2 by radicals is not an efficient process and therefore is
of limited synthetic utility.5
 |
| | Scheme 2 | |
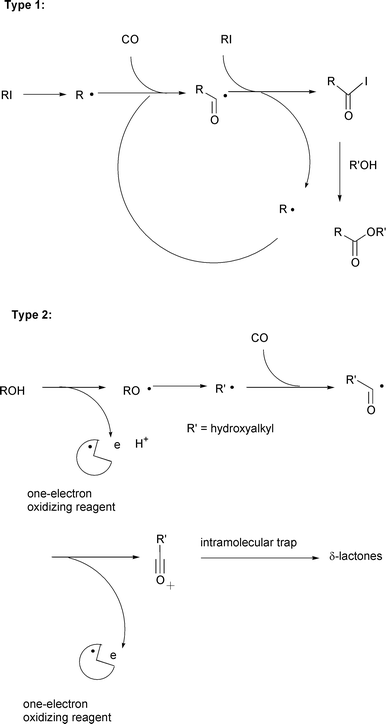
A surrogate method that relies upon the reaction between radicals and
carbon monoxide, in other words, radical carbonylations,6–8 can be used. Two methodologies
seem to have great potential for radical carboxylation with CO. One
promising approach is a radical–ionic hybrid method based on an
iodine atom transfer carbonylation that has been invented by us just
recently. In this approach, acyl iodides are the precursors for carboxylic
acid derivatives via ionic nucleophilic substitution reactions
(type 1 in Scheme 3).
 |
| | Scheme 3 | |
The other approach is more traditional, in which the radical
carbonylation sequence is combined with a subsequent one electron oxidation
process. Acyl cations, thus generated, serve as key precursors for the
carboxylated products. The original demonstration of this type of
carboxylation reaction dates from reports published in the 1950s, where the
Fenton reagent (FeII plus H2O2) was used
to create oxidative carbonylation systems.9
Among the recent studies aiming at carboxylating alkanes by an oxidative
carbonylation,10 Ishii’s method using
a mixed CO–O2 gas is quite remarkable, since the key
reagent NHPI (N-hydroxyphthalimide) is used in a catalytic
quantity.11 Nevertheless, the general
problem belying in this alkane functionalization is the marginal
regiocontrol, which limits the reaction to symmetrical substrates such as
methane, ethane, and cyclohexane. Furthermore, the reaction should be
stopped at low conversion to avoid complications associated with the
sequential carbonylations. The rare case of regiocontrolled C–H
functionalization with CO, which converts saturated alcohols and CO into
δ-lactones (type 2 in Scheme 3), also deserves mentioning in the
context of radical carboxylation strategies. Thus, two types of recently
invented carboxylation methodologies will be the focus of this review.
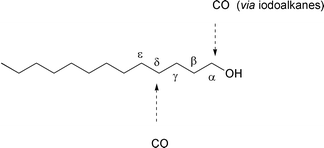
Since iodoalkanes can be synthesized from saturated alcohols, these
carboxylation reactions can be regarded as carboxylations of saturated
alcohols, which occur with a high level of regiocontrol at the α- and
δ-positions respectively.
 |
| | Scheme 4 | |
2 Atom transfer carbonylations of iodoalkanes with
CO
2.1 Irradiation system
Our initial idea of atom transfer carbonylation is the simple insertion
of carbon monoxide into an alkyl–halogen bond via homolysis.
To obtain this radical–radical pair, we chose the well known
photolytic homolysis of an alkyl iodide,12
carried out in the presence of pressurized carbon monoxide using a
stainless steel autoclave having two quartz windows. Using a 500 W xenon
lamp as the light source, a hexane solution of 2-iodooctane was irradiated
under 50atm of CO pressure for 15 h. We were unable to detect the desired
acyl iodide via this experiment. Since acyl iodides can decompose
by photo-irradiation, we decided to add an alcohol and potassium carbonate
in the hope that the acyl iodide, if formed, would immediately undergo
alcoholysis to give an ester that would be stable toward irradiation. This
idea is shown in Scheme 5. As we had
envisioned, a good yield of the desired carboxylic acid ester was obtained
using this reaction system.13 All of these
reactions were carried out using a pyrex glass liner, which appears to be
crucial for this catalyst free type of carboxylation reaction. When we
examined the reaction of 2-iodooctane using a quartz liner, formation of
the ester was completely suppressed. The major products obtained were an
isomeric mixture of 1- and 2-octenes. This is a known dehydroiodination
pathway iodoalkanes undergo when they are UV-irradiated.12 Remarkably, inefficient irradiation proved to be
crucial for efficient reaction. This strongly suggested to us that
irradation may simply initiate the process by generating a small amount of
an alkyl radical and a radical chain reaction may be involved in the
transformation (vide infra). Both organic and inorganic bases,
such as triethylamine, potassium hydroxide, sodium hydroxide, etc.
are all effective but in the absence of a base, no carbonylation took
place.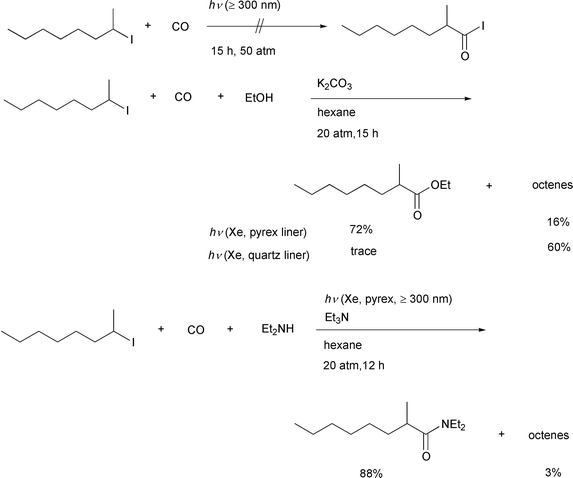 |
| | Scheme 5 | |
Scheme 6 illustrates some examples of
ester synthesis by this photo-irradiation reaction. This carboxylation
method which is based on atom transfer chemistry can be applied to a wide
range of alkyl iodides including tertiary alkyl iodides. Carbonylation of
secondary alkyl iodides by transition metals often results in the formation
of positional isomers associated with β-elimination of the key metal
alkyl species and, for similar reasons, the carbonylation of tertiary
iodides is thought to be attainable only with great difficulty. The present
carbonylation, however, takes place selectively at the carbon to which the
iodine had been attached and, thus, a wide range of aliphatic iodides
including tertiary substrates can be used. The reaction of primary alkyl
iodides was not fast due presumably to a slow iodine atom transfer. The
reaction can be extended to include the amide synthesis by simply changing
the trapping nucleophile from alcohols to amines.
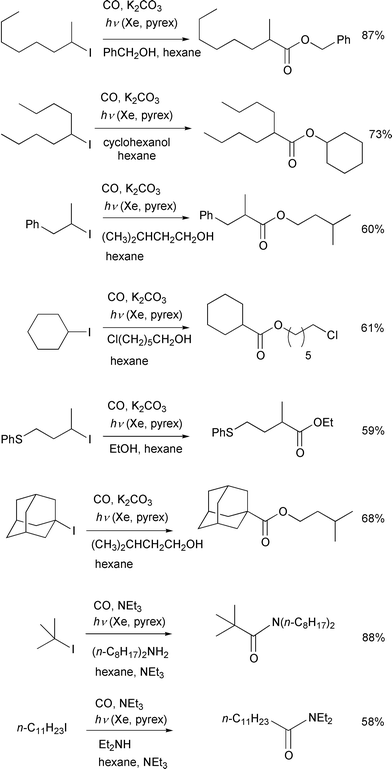 |
| | Scheme 6 Conditions: 20–50 atm, Xe irradiation through a pyrex
tube, 12–14 h. | |
It should be noted that the general idea of atom transfer carbonylation
originated from mechanistic considerations surrounding the unexpected
observation of lactone formation in the double carbonylation of 4-pentenyl
iodide using a slow radical mediator such as tributylgermane or
(TMS)3SiH (Scheme 7).14 Bicyclic lactone formation did not occur with
4-pentenyl bromide as the substrate and this clearly speaks against a rare
5-endo reaction mechanism.
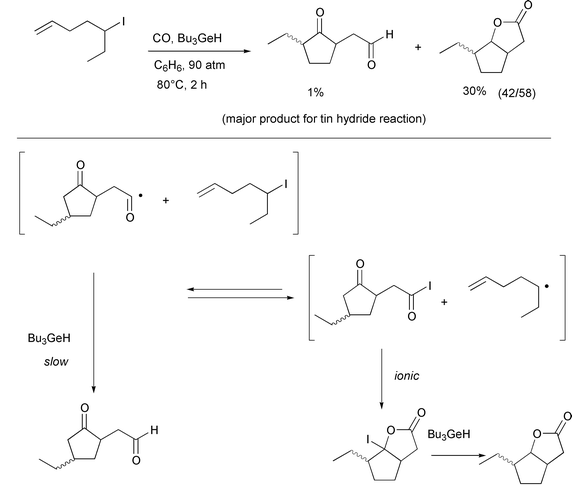 |
| | Scheme 7 | |
Group transfer ability of a phenylseleno functionality15,16 to an acyl radical was tested in the
photolysis of α-phenylselenoacetate and related compounds in the
presence of an alkene and CO. This three-component coupling reaction
involves the addition of an α-(alkoxycarbonyl)methyl radical to an
alkene, the trapping of the produced alkyl free radical by CO, and
termination of the reaction by a phenylseleno group transfer from the
starting material which leads to acyl selenides via group transfer
carbonylation. (Scheme 8).17 Unlike the case of the iodine atom transfer
system discussed above, the system shown in Scheme 8 does not need the
following ionic process, since the product acyl selenide is reasonably
stable under the irradiation conditions and group transfer is designed to
take place easily by employment of α-phenylseleno carbonyl compounds.
However, the fact that the reverse reaction was involved was determined by
photolyzing the product, which yielded the parent olefin (Scheme 8). This
competing process obviously lowers the yields of the product.
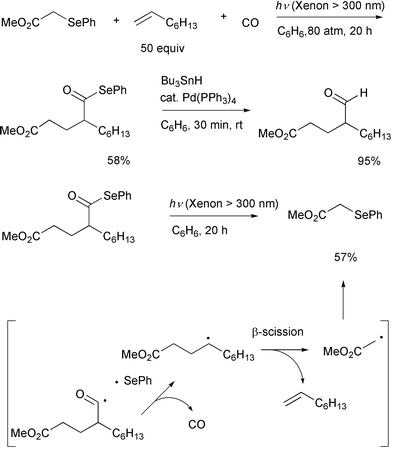 |
| | Scheme 8 | |
2.2 Thermal initiation system
What is the role of photo-irradiation? We suspected that the role is
merely to initiate the hybrid radical–ionic reaction by effecting the
homolysis of an R–I bond in a small quantity. If this was the case,
the thermal initiation process would allow the same reaction to take place.
We tested a variety of thermal initiation systems (Scheme 9),18 and
found that the AIBN–allyltributyltin system was quite promising. If
the avoidance of toxic tin compounds is preferred,
tris(trimethylsilyl)silane is equally effective.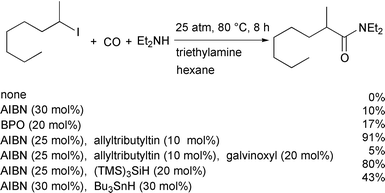 |
| | Scheme 9 | |
In these two systems, the abstraction of iodine by a tin or silyl
radical serves as the source of small amounts of an alkyl radical, which
participates in the subsequent chain reaction system comprised of (i)
radical carbonylation and (ii) iodine atom transfer reactions from an
iodoalkane to give an acyl radical. The latter does not necessarily appear
to be thermodynamically favorable. For example, a low level ab
initio MO calculation using UHF/3-21G* as a basis set suggests that
the conversion of isopropyl iodide and CO into the corresponding acyl
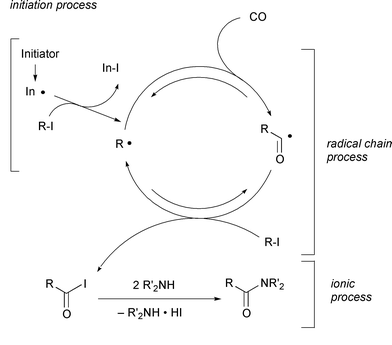
iodide is 1.92kcal mol−1 endothermic. The most likely
mechanism involves (i) radical initiation via either irradiation
or thermal initiation, (ii) radical chain propagation, composed of two
reversible radical reactions (carbonylation and iodine atom transfer) and
(iii) ionic quenching to shift the equilibria of the reversible reactions
(Scheme 10). In the hands of a tin
mediated radical carbonylation process, tertiary substrates require high CO
pressure to compete with the very rapid back decarbonylation reaction,
compared with secondary and primary alkyl radicals.19 However, the unique radical–ionic
cooperation to drive the carbonylation–decarbonylation equilibrium in
the forward direction could give an advantage for the carbonylation of
tertiary radicals.
 |
| | Scheme 10 | |
Scheme 11 illustrates examples of
atom transfer carbonylation using allyltin–AIBN as the radical
initiator system. Generally, the amide synthesis proceeds more rapidly than
the corresponding ester synthesis. Indeed, even in the case where methanol
was used as the solvent, the amide was the only product derived from the
carbonylation reaction. Both primary amines and secondary amines were
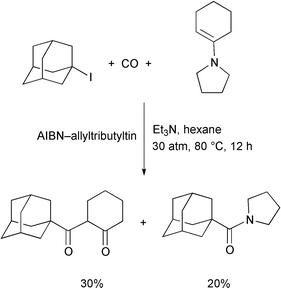
useful. We also tested trapping of an acyl iodide by an enamine, which gave
a mixture of a 1,3-diketone and an amide (Scheme
12).20
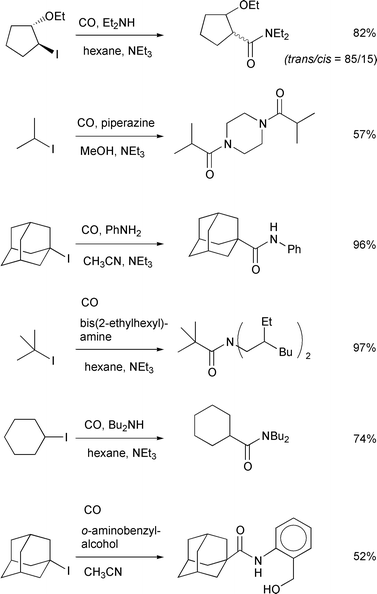 |
| | Scheme 11 Conditions: 20–25 atm, allyltributyltin (10
mol%)–AIBN (30 mol%), 80°C, 8 h. | |
 |
| | Scheme 12 | |
An intramolecular variant of the atom transfer based radical
carboxylation was examined using ω-iodo alcohols as the substrate
(Scheme 13). Thus, five- to
seven-membered lactones were prepared successfully in good yields by this
method.21 One easy way to prepare the
starting ω-iodo alcohols is carboiodination of unsaturated
alcohols.22 Scheme 13 also illustrates an
example where the three carbon components were combined via
successive carboiodination, atom transfer carbonylation, and intramolecular
ionic trap.
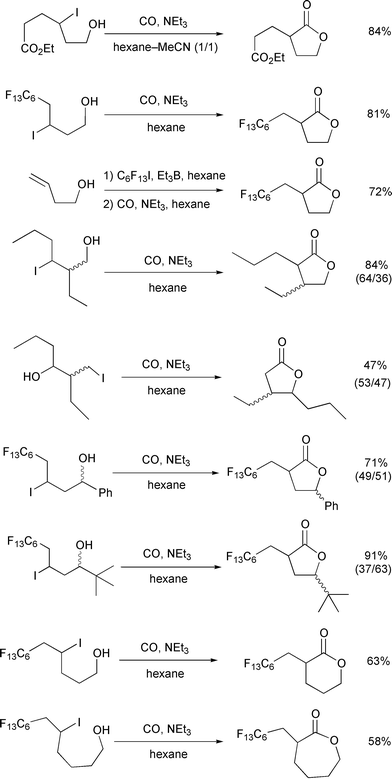 |
| | Scheme 13 Conditions: 45–90 atm, allyltributyltin (10
mol%)–AIBN (20–30 mol%), 80°C, 12 h. | |
Unfortunately the system cannot be applied to tandem double
carbonylation reactions even when 5-pentenyl iodide was used as the
starting substrate. It is probable that the iodine atom
transfer–ionic quenching protocol is not efficient enough to capture
the product over a multi-step equilibrium. However, we have recently found
that a Pd–light system enables us to execute such a reaction.23 Judging from the stereochemical outcome of the
cyclized product, it is concluded that the reaction includes an acyl
radical cyclization step.
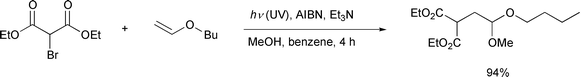
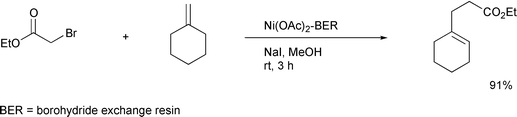
It is interesting to note that recently the Yoon24 and Curran25
groups independently have also reported that the use of a hybrid
radical–ionic system effected successful atom transfer reactions
which had been thought to be difficult (Schemes
14, 15). All these new
achievements based on a hybrid system may be a stimulus to reconsider some
other reactions whose potential has yet to be developed.
 |
| | Scheme 14 | |
 |
| | Scheme 15 | |
3 Remote carboxylations of saturated
alcohols
3.1 One-electron oxidation system
Alkoxy radical mediated δ-functionalization reactions of saturated
alcohols based on the 1,5-H shift have a long history.26,27 However, the examples of δ-functionalization
reactions have been largely restricted to cases of heteroatom introducing
reactions that occur at δ-carbons, represented by the Barton
reaction26d and a select few
intramolecular cyclization reactions. We became interested in the
carboxylative version of the Barton reaction, a regioselective
δ-carboxylation of saturated alcohols, which includes carbonylation
plus one-electron transfer (Scheme 16).
The initial discovery in collaboration with Howard Alper that radical
carboxylation with CO can be carried out in a MnIII induced
one-electron oxidation system,28 encouraged
us to embark on this challenging C–H functionalization project. After
screening a variety of one-electron oxidizing systems, we determined that a
lead tetraacetate (Pb(OAc)4, LTA) promoted oxidative cyclization
system29 can be successfully applied to the
envisioned process. Thus, δ-hydroxyalkyl radicals can be generated in
high selectivity from the corresponding methanols via alkoxy
radicals. Subsequent trapping of the resulting radicals with CO results in
a δ-selective carbonylation. One-electron oxidation of the resulting
acyl radical generates an acyl cation which cyclizes to form the
δ-lactone ring.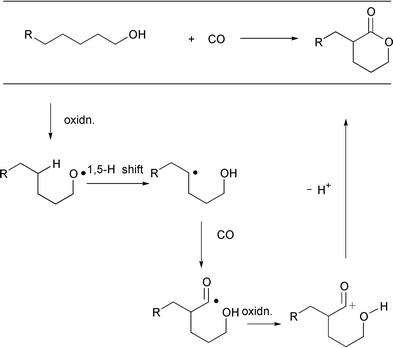 |
| | Scheme 16 | |
The results of control experiments using 1-octanol showed that lower
concentrations of 1-octanol, smaller amounts of LTA (1.5 mol equiv.),
higher pressures of CO (80–100atm), and lower reaction temperatures
(40°C) all favored carbonylation of the δ-hydroxyalkyl
radical over the undesirable direct oxidation leading to THF ring
formation.30
In Scheme 17, examples of
carboxylation of primary and secondary alcohols having 2° δ-carbons are provided. In all cases, the major competing reaction is
oxidation to give THF derivatives. On the other hand, small quantities
(<5%) of ε-lactones (via 1,6-H shifts) were occasionally
observed. These by-products can be eliminated by flash chromatography on
silica gel to afford pure δ-lactones. In general, the yields of
δ-lactones from secondary alcohols are higher than those from primary
alcohols. This may be due to a decrease in the rate of reaction leading to
THF derivatives from these secondary alcohols due to additional steric
congestion at the cyclization step.
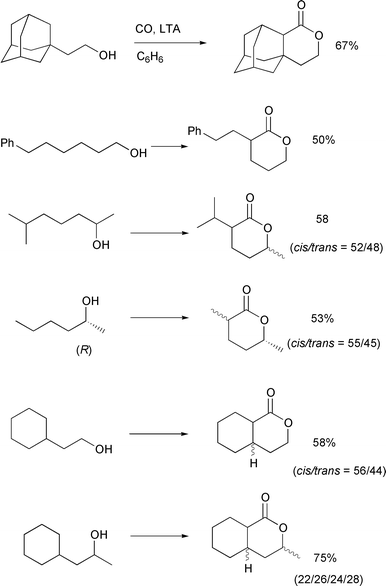 |
| | Scheme 17 Conditions: ROH (0.4–0.8 mmol), LTA (1.5–2.0
equiv.), C6H6 (20–40 mL), CO (80 atm),
40°C, 3–7 days. | |
This δ-carboxylation method was successfully applied to a concise
synthesis of carpenter bee pheromone from optically pure
(R)-(–)-2-hexanol (Scheme 17). Preparative HPLC separation
of the product mixture composed of cis- and
trans-2-methyl-5-hexanolide afforded pure
cis-(2S,5R)-2-methyl-5-hexanolide (ee∼100%),
a sex pheromone of the carpenter bee Xylocopa hirutissima. The
observed quantitative optical yield confirmed that the absolute
configuration at the hydroxy-bearing carbon was completely retained.
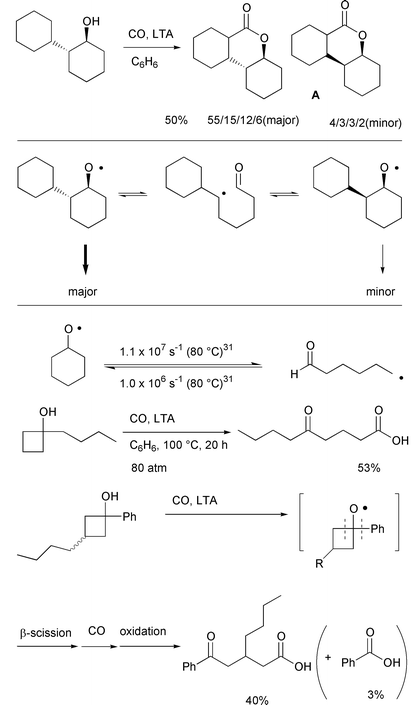
However, in some cases, for example
trans-2-cyclohexyl-1-cyclohexanol, the stereochemistry at the
hydroxy-bearing carbon was not retained (Scheme 18). Tricyclic
δ-lactone A was obtained as a mixture of eight
diastereomers, despite the fact that only four diastereomers were
considered to be possible if the configuration of hydroxy-bearing carbon
was retained. The four major diastereomers were assigned as trans
isomers with retention of ordinal stereochemistry, in a ratio of 55:15:12:6
and four minor diastereomers assigned as cis isomers with
inversion of ordinal stereochemistry, in a 4:3:3:2 ratio. The initially
generated alkoxyl radical having a trans configuration may undergo
β-scission, and cyclization of the resulting radical back onto the
internal aldehyde would afford an isomerized radical with cis
configuration. The isomerized radical undergoes 1,5-hydrogen transfer, CO
trapping, and oxidation to give the four minor products. This result is
supported by kinetic data which show that the
cyclization–fragmentation process is in equilibrium.31 The complete retention of stereochemistry
observed in the case of (R)-(–)-2-hexanol can be explained
in this context since β-scission yields two fragments, whose
recombination is entropically unlikely. In strained cyclic alcohols like
cyclobutanols, β-scission is irreversible and
carbonylation–oxidation of the resulting ring opened radicals leads
to carboxylic acids (Scheme 18).32
 |
| | Scheme 18 | |
The carboxylation of alcohols having 1° δ-carbons was also
successful. In this case, the reaction was not accompanied by the two types
of by-products that were observed with alcohols having 2° δ-carbons. ε-Lactones were not formed, since 1,6-H shifts are
no longer possible and very little competitive oxidation leading to the
tetrahydrofuran derivative was observed, which is ascribed to the slower
oxidation of primary radicals compared to secondary radicals. The slow side
reaction may enable us to carbonylate alcohols which have 1° δ-carbons at relatively low CO pressures (20–80atm) and at high
temperatures (60–80°C). Indeed in the first example shown
in Scheme 19, when
2,6-dimethyl-4-heptanol was treated with 20atm of CO, the δ-lactone
was obtained in 62% yield along with only 2% of the corresponding THF
derivative. The relatively low reactivity of the 1° δ-carbon
systems with carbon monoxide can, however, often lead to slightly reduced
conversions and recovery of the starting alcohols.
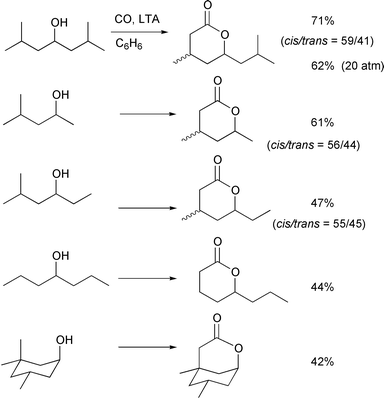 |
| | Scheme 19 Conditions: ROH (0.4–0.8 mmol), LTA (1.5–2 equiv.),
C6H6 (20 mL), CO (80 atm), 60°C,
2–5 days. | |
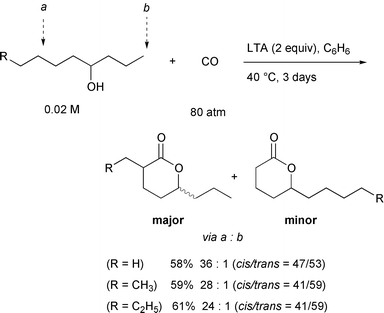
This lower reactivity of the primary carbon can be used to our advantage
in the selective carbonylation of an alcohol which has both 2° and
1° δ-carbons, a and b. In this case, carbon
monoxide could be incorporated at the 2° δ-carbon a,
with high regiochemical selectivity (Scheme
20). For example, 2-propyl-5-octanolide was formed predominantly
over 5-undecanolide in a 24∶1 ratio. Purification to eliminate the
minor product was easily accomplished by flash chromatography on silica
gel. The preferential formation of the secondary carbonylation product is
reasonable in light of the weaker C–H bond strength of methylene
relative to that of methyl (ΔE = ca.
3kcal mol–1).33 The
efficiency of the 1,5-hydrogen transfer correlates well with the C–H
bond dissociation energies.
 |
| | Scheme 20 | |
3.2 Irradiation system through alkyl
benzenesulfenates
In contrast to primary and secondary alcohols, tertiary alcohols were
virtually inert under the reaction conditions employed. This is presumably
because of the difficulty of the formation of alkoxylead species due to
steric congestion at the initial step under the standard conditions.In order to circumvent this limitation, we considered generating the
alkoxyl radical without LTA and we were particularly interested in the
methods involving photolytic cleavage of O–X bonds.34 It is well known that O–S bonds undergo
homolytic cleavage to give alkoxyl radicals,35 as does O–O homolysis. It has recently
been reported that photolytic cleavage of the O–S bond in alkyl
4-nitrobenzenesulfenates,36 which are
readily prepared by the reaction of the alcohols with
4-nitrobenzenesulfenyl chloride, provides a convenient method for the
generation of alkoxyl radicals. Thus, the reaction of an alkyl
4-nitrobenzenesulfenate derived from a tertiary alcohol and
4-nitrobenzenesulfenyl chloride with CO was conducted under sunlamp
irradiation in the hope of effecting δ-carboxylation to give a
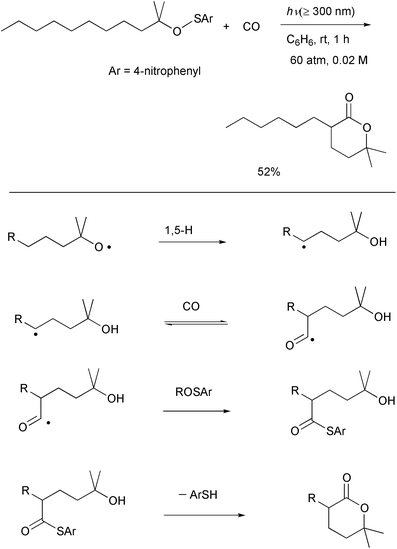
thioester. The requisite lactone was formed in 52% yield (Scheme 21).30b The formation of the lactone can be
rationalized as being derived from an initially produced thioester which is
formed via an SH2 reaction at sulfur. Therefore, group
transfer from a benzenesulfenate to an acyl radical is a key step for this
reaction. Although the system requires optimization in terms of the yields
of the δ-lactones, we believe that the photolytic
δ-carbonylation of alkyl 4-nitrobenzenesulfenates is complementary to
our LTA-induced oxidation system.
 |
| | Scheme 21 | |
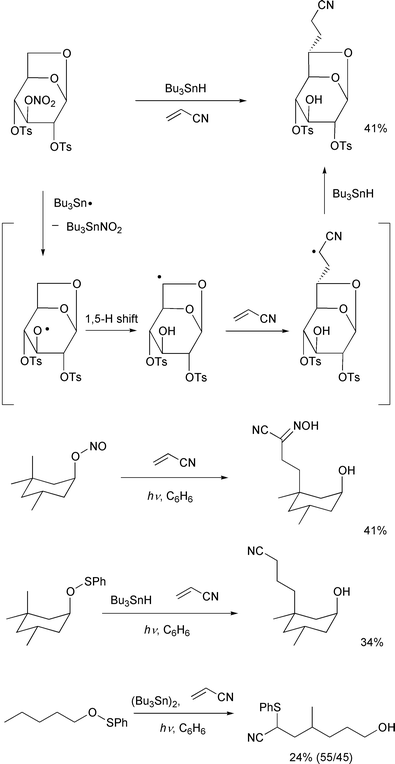
Finally, though quite restricted, some interesting examples of
intermolecular C–C bond forming reactions using an alkene are listed
in Scheme 22. The first example was
reported by Fraser-Reid and co-workers in 1989,37 and the latter three examples were recently
reported by Cekovic and co-workers.38,39 These results are a proof of the continuing vitality
of remote-carbon-functionalization.
 |
| | Scheme 22 | |
4 Conclusion
Two types of radical carboxylation methods are described in this
account. Radical carboxylations based on iodine atom transfer chemistry are
found to be particularly useful for the conversion of a wide variety of
alkyl iodides into the one carbon homologated carboxylic acid derivatives.
This work demonstrated that thermodynamically unfavorable radical chain
reactions can be carried out if they are performed in conjunction with
ionic reaction systems. Remote carboxylation provides a convenient one-step
synthesis of δ-lactones from a cheap and readily available feedstock,
saturated alcohols. Carboxylation of tertiary alcohols, although
unsuccessful with the LTA-induced system, can be achieved via
photolysis of alkyl 4-nitrobenzenesulfenates, which may prove to be a
highly efficient alternative to the LTA system. We believe that these two
topics will stimulate further developments in carboxylation chemistry.5 Acknowledgements
I would like to thank the students whose names are listed in the
published papers for their sincere dedication to the chemistry through
their hard work. I also thank my colleagues for many useful discussions.
Thanks are especially due to Dr Kiyoto Nagahara and Dr Shinji Tsunoi who
did the majority of the work in the atom transfer carbonylation and remote
carboxylation reactions, respectively. I also thank Professors Noboru
Sonoda and Mistuo Komatsu for their warm-hearted encouragement throughout
the course of this work. Professor Cathleen M. Crudden is thanked for
useful comments on this manuscript. Financial support from the Monbu-sho is
gratefully acknowledged.6 References
- H. M. Colquhoun,
D. J. Thompson and
M. V. Twigg,
Carbonylation: Direct Synthesis of Carbonyl Compounds,
Plenum, New York,
1991. Search PubMed.
- T. W. Dekleva and D. Forster, J. Am. Chem. Soc., 1985, 107, 3565 CrossRef CAS.
-
(a) R. Takeuchi, Y. Tsuji and Y. Watanabe, J. Chem. Soc., Chem. Commun., 1986, 351 RSC; R. Takeuchi, Y. Tsuji, M. Fujita, T. Kondo and Y. Watanabe, J. Org. Chem., 1989, 54, 1831 CrossRef CAS; T. Kondo, Y. Sone, Y. Tsuji and Y. Watanabe, J. Organomet. Chem., 1994, 473, 163 CrossRef CAS; H. Urata, H. Maekawa, S. Takahashi and T. Fuchigami, J. Org. Chem., 1991, 56, 4320 CrossRef CAS.
- S. Hadida, M. S. Super, E. J. Beckman and D. P. Curran, J. Am. Chem. Soc., 1997, 119, 7406 CrossRef CAS.
-
(a) For decarboxylation kinetics, see: J. W. Hilborn and J. A. Pincock, J. Am. Chem. Soc., 1991, 113, 2683 CrossRef CAS; D. E. Falvey and G. B. Schuster, J. Am. Chem. Soc., 1986, 108, 7419 CrossRef CAS.
- For a review, see: I. Ryu and N. Sonoda, Angew. Chem., Int. Ed. Engl., 1996, 35, 1050 CrossRef.
- For a review on radical C1 synthons, see: I. Ryu, N. Sonoda and D. P. Curran, Chem. Rev., 1996, 96, 177 CrossRef CAS.
- For a comprehensive review on acyl radical chemistry: C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chem. Rev., 1999, 99, 1991 CrossRef CAS.
-
(a) D. D. Coffman, R. Cramer and W. E. Mochel, J. Am. Chem. Soc., 1958, 80, 2882 CrossRef CAS; G. P. Chiusoli and F. Minisci, Gazz. Chim. Ital., 1958, 88, 43 Search PubMed; C. Walling and E. S. Savas, J. Am. Chem. Soc., 1960, 82, 1738 CrossRef CAS.
-
(a) M. Lin and A. Sen, J. Chem. Soc., Chem. Commun., 1992, 892 RSC; D. H. R. Barton, E. Csuhai and D. Doller, Tetrahedron Lett., 1992, 33, 4389 CrossRef CAS; Y. Fujiwara, K. Takaki and Y. Taniguchi, Synlett, 1996, 591 CrossRef CAS; M. Asadullah, Y. Taniguchi, T. Kitamura and Y. Fujiwara, Tetrahedron Lett., 1999, 40, 8867 CrossRef CAS.
- S. Kato, T. Iwahama, S. Sakaguchi and Y. Ishii, J. Org. Chem., 1998, 63, 222 CrossRef CAS.
-
(a) P. J. Kropp, Acc. Chem. Res., 1984, 17, 131 CrossRef CAS; P. J. Kropp, G. S. Poindexter, N. J. Pienta and D. C. Hamilton, J. Am. Chem. Soc., 1976, 98, 8135 CrossRef CAS.
- K. Nagahara, I. Ryu, M. Komatsu and N. Sonoda, J. Am. Chem. Soc., 1997, 119, 5465 CrossRef CAS.
- S. Tsunoi, I. Ryu, S. Yamasaki, H. Fukushima, M. Tanaka, M. Komatsu and N. Sonoda, J. Am. Chem. Soc., 1996, 118, 10670 CrossRef CAS.
- J. H. Byers and G. C. Lane, J. Org. Chem., 1993, 58, 3355 CrossRef CAS.
- D. P. Curran, E. Eichenberger, M. Collis, M. G. Roepel and G. Thoma, J. Am. Chem. Soc., 1994, 116, 4279 CrossRef CAS.
- I. Ryu, H. Muraoka, N. Kambe, M. Komatsu and N. Sonoda, J. Org. Chem., 1996, 61, 6396 CrossRef CAS.
- I. Ryu, K. Nagahara, N. Kambe, N. Sonoda, S. Kreimerman and M. Komatsu, Chem. Commun., 1998, 1954 Search PubMed.
-
(a) Y. P. Tsentalovich and H. Fischer, J. Chem. Soc., Perkin Trans. 2, 1994, 729 RSC; C. E. Brown, A. G. Neville, D. M. Rayner, K. U. Ingold and L. Lusztyk, Aust. J. Chem., 1995, 48, 363 CAS; C. Chatgilialoglu, C. Ferreri, M. Lucarini, P. Pedrielli and G. F. Pedulli, Organometallics, 1995, 14, 2672 CrossRef CAS.
- I. Ryu,
S. Kreimerman and
M. Komatsu,
unpublished results..
- S. Kreimerman, I. Ryu, S. Minakata and M. Komatsu, Org. Lett., 2000, 2, 389 CrossRef CAS.
- Y. Takeyama, Y. Ichinose, K. Oshima and K. Utimoto, Tetrahedron Lett., 1989, 30, 3159 CrossRef CAS.
- I. Ryu,
S. Kreimerman,
F. Araki and
M. Komatsu,
unpublished..
- M. J. Joung, J. H. Ahn, D. N. Lee and N. M. Yoon, J. Org. Chem., 1998, 63, 2755 CrossRef CAS.
- D. P. Curran and S.-B. Ko, Tetrahedron Lett., 1998, 39, 6629 CrossRef CAS.
-
(a) Reviews on remote functionalization: G. Majetich and K. Wheless, Tetrahedron, 1995, 51, 7095 CrossRef CAS; R. Bleslow,
Comprehensive Organic Synthesis, B. M. Trost and I. Fleming,
Eds, Pergamon, Oxford,
1991, vol. 7, chapt. 1.3; (c)
Search PubMed; P. Brun and
B. Waegell,
Reactive Intermediates; R. A. Abramovitch, Ed.,
Plenum Press, New York,
1983, vol. 3, chapt. 6; (d)
Search PubMed; D. H. R. Barton, Pure Appl. Chem., 1968, 16, 1 Search PubMed.
-
(a) For kinetic and theoretical aspects, see: B. C. Gilbert, R. G. G. Holmes, H. A. H. Laue and R. O. C. Norman, J. Chem. Soc., Perkin Trans. 2, 1976, 1047 RSC; K. N. Houk, J. A. Tucker and A. E. Dorigo, Acc. Chem. Res., 1990, 23, 107 CrossRef CAS; A. E. Dorigo and K. N. Houk, J. Org. Chem., 1988, 53, 1650 CrossRef CAS; A. E. Dorigo, M. A. McCarrick, R. J. Loncharich and K. N. Houk, J. Am. Chem. Soc., 1990, 112, 7508 CrossRef CAS.
- I. Ryu and H. Alper, J. Am. Chem. Soc., 1993, 115, 7543 CrossRef CAS also see: K. Okuro and H. Alper, J. Org. Chem., 1996, 61, 5312 CrossRef CAS.
-
(a) For reviews, see: M. L. Mihailovic,
Z. Cekovic and
L. Lorenc,
Organic Syntheses by Oxidation with Metal Compounds, W. J.
Mijs and C. R. H. I. de Jonge, Eds., Plenum Press,
New York, 1986, chapt.
14; (b) Search PubMed; M. L. Mihailovic and Z. Cekovic, Synthesis, 1970, 209 Search PubMed; G. M. Rubottom,
Oxidation in Organic Chemistry, Academic
Press, New York, 1982,
part D. chapt. I (d) CrossRef CAS; G. W. Rotermund,
Methoden der Organischen Chemie (Houben-Weyl), E.
Müller, Ed., Georg Thieme,
Stuttgart, 1975, Band 4, Teil
1b, Oxidation II, p. 204; (e) CrossRef CAS; K. Heusler and J. Kalvoda, Angew. Chem., Int. Ed. Engl., 1964, 3, 525 CrossRef CAS.
-
(a) S. Tsunoi, I. Ryu and N. Sonoda, J. Am. Chem. Soc., 1994, 116, 5473 CrossRef CAS; S. Tsunoi, I. Ryu, T. Okuda, M. Tanaka, M. Komatsu and N. Sonoda, J. Am. Chem. Soc., 1998, 120, 8692 CrossRef CAS.
-
(a) A. L. J. Beckwith and B. P. Hay, J. Am. Chem. Soc., 1989, 111, 2674 CrossRef CAS; A. L. J. Beckwith and B. P. Hay, J. Am. Chem. Soc., 1989, 111, 230 CrossRef CAS.
- S. Tsunoi, I. Ryu, Y. Tamura, S. Yamasaki and N. Sonoda, Synlett, 1994, 1009 CrossRef CAS.
- D. D. M. Wayner and
D. Griller,
Advances in Free Radical Chemistry, D. D. Tanner, Ed.,
JAI Press, Greenwich,
1990, Vol. 1, p. 159. Search PubMed.
-
(a) C. Walling, Pure Appl. Chem., 1967, 15, 69 CrossRef CAS; J. Kalvoda and K. Heusler, Synthesis, 1971, 501 CrossRef CAS; P. Brun and
B. Waegell,
Tetrahedron, 1976, 32,
517; also see ref. 26d. Search PubMed.
- A. L. J. Beckwith, B. P. Hay and G. M. Williams, J. Chem. Soc., Chem. Commun., 1989, 1202 RSC.
-
(a) D. J. Pasto and G. L’Hermine, J. Org. Chem., 1990, 55, 5815 CrossRef CAS; D. J. Pasto and G. L’Hermine, Tetrahedron, 1993, 49, 3259 CrossRef CAS; D. J. Pasto and F. Cottard, Tetrahedron Lett., 1994, 35, 4303 CrossRef CAS.
- J. C. Lopez, R. Alonso and B. Fraser-Reid, J. Am. Chem. Soc., 1989, 111, 6471 CrossRef CAS.
- G. Petrovic, R. N. Saicic and Z. Cekovic, Tetrahedron Lett., 1997, 38, 7107 CrossRef CAS.
-
(a) G. Petrovic and Z. Cekovic, Tetrahedron, 1999, 55, 1377 CrossRef CAS; G. Petrovic, R. N. Saicic and Z . Cekovic, Synlett, 1999, 635 CAS.
|
| This journal is © The Royal Society of Chemistry 2001 |
Click here to see how this site uses Cookies. View our privacy policy here.