FTIR study of isopropanol reactivity on calcined layered double hydroxides
M. del Arco, S. Gutiérrez, C. Martín and V. Rives*
Departamento de Química Inorgánica, Uni![[italic v]](https://www.rsc.org/images/entities/char_e0f5.gif) ersidad de Salamanca, 37008-, Salamanca, Spain. E-mail: vrives@gugu.usal.es
ersidad de Salamanca, 37008-, Salamanca, Spain. E-mail: vrives@gugu.usal.es
First published on 8th December 2000
Abstract
A FTIR spectroscopy study on the surface reactivity of calcined layered double hydroxides (LDH), with different cations in the interlayer space (namely, carbonate, nitrate, silicate, and borate), for isopropanol oxidation is reported. The LDHs were previously calcined at 600°C and then their surface acidity was
quantitatively determined ![[italic v]](https://www.rsc.org/images/entities/i_char_e0f5.gif) ia volumetric determination of ammonia adsorption. Surface Lewis and/or Brönsted acid and basic sites were qualitatively studied by FTIR monitoring of the adsorption of pyridine and boric acid trimethyl ester. Carbonate and nitrate samples show surface acidity and basicity and hence the maximum activity for acetone formation through dehydrogenation of isopropanol. In contrast, although the silicate sample
displays maximum surface acidity, the lack of an appreciable amount of surface basic sites gives rise to a lower activity
in the named reaction, of the same order as that shown by the borate sample.
ia volumetric determination of ammonia adsorption. Surface Lewis and/or Brönsted acid and basic sites were qualitatively studied by FTIR monitoring of the adsorption of pyridine and boric acid trimethyl ester. Carbonate and nitrate samples show surface acidity and basicity and hence the maximum activity for acetone formation through dehydrogenation of isopropanol. In contrast, although the silicate sample
displays maximum surface acidity, the lack of an appreciable amount of surface basic sites gives rise to a lower activity
in the named reaction, of the same order as that shown by the borate sample.
Introduction
Layered double hydroxides (LDH), also known as anionic clays, consist of brucite-like layers, with partial Mg2+/Al3+ substitution; the positive charge of the layers thus introduced is balanced by anions located between the layers, coexisting with water molecules. They are also known as hydrotalcite-like or hydrotalcite-type materials, after the natural mineral hydrotalcite, [Mg6Al2(OH)16](CO3)·4 H2O usually written as [Mg0.75Al0.25(OH)2] (CO3)0.125 ·0.5 H2O to emphasize its relationship to brucite, Mg(OH)2. Their properties have been recently reviewed.1–4 These materials have received increasing attention in recent years because of their many applications as adsorbents, antacids, anion scavengers, and, mainly, as catalysts or catalyst supports. Upon calcination they form mixed, homogeneously dispersed, oxides; the nature of the cations in the layers, and of the anions in the interlayer can be changed widely, and so the opportunities for fine tuning of the catalytic properties are extremely high. Their catalytic activity, as that of the mixed oxides obtained after calcination, has been reviewed by several authors.5–7 Most of the studies carried out have dealt with the effect of substitution of the cations in the brucite-like layers, introducing several transition metal cations; however, the solids containing Mg or Al have also been used for catalytic processes, such as aldol condensation.In this paper we report on LDHs containing Mg and Al in the layers, and containing non-metal anions in the interlayer, i.e., carbonate, nitrate, borate and silicate. We have selected the carbonate and nitrate samples, in addition to the silicate and borate ones, for several reasons: the carbonate LDH because Mg,Al LDH with interlayer carbonate corresponds to the mineral hydrotalcite, and is the most widely studied member of this family; the nitrate LDH because this was used as the precursor to intercalate silicate and borate, as removal of interlayer carbonate is rather difficult. The solids obtained after calcination in air at 600°C (where the layered structure is completely lost and the interlayer anions, if volatile, are removed) have been used to study the adsorption and decomposition of isopropanol (ISP) in the vapour phase, in order to establish a correlation between the reactivity in this reaction and the surface acid–base properties of the solids, modified by the different anions originally existing in the interlayer. The study has been carried out by FTIR spectroscopy monitoring of ISP adsorption and decomposition; the technique was also applied to determine the surface acidity/basicity through adsorption of different probe molecules, pyridine (py) for acid sites, and boric acid trimethyl ester (bate) for basic sites.
Experimental
Preparation of the samples
All chemicals were from Fluka (Switzerland) and were used as purchased, without further purification. The LDH containing carbonate in the interlayer (MgAlC) was prepared with a molar Mg/Al ratio of 2, following the method described by Reichle.8 The nitrate-containing sample (MgAlN) was obtained similarly, by coprecipitation from Mg and Al nitrates in NaOH–NaNO3 aqueous solution at pH 10 using freshly decarbonated water; the method has been described previously.9 Both samples were submitted to hydrothermal treatment at 120°C for 15 days at autogenous pressure in a Teflon-lined stainless steel bomb, in order to improve the crystallinity and to increase the particle size. The borate (MgAlB) and silicate (MgAlSi) LDHs were obtained by anionic exchange of the nitrate precursor, using (NH4)2B4O7·4 H2O at pH 9.5, or (SiO2–NaOH–H2O), respectively.9 After preparation, the samples were washed with distilled water, filtered and dried at room temperature. The solids were characterized by powder X-ray diffraction (PXRD), thermogravimetric and differential thermal analysis, MAS-NMR for B and Si and FTIR spectroscopies, etc. and the results have been published elsewhere.9 The layered nature of all samples, containing, respectively, carbonate, nitrate, B4O5(OH)42− and (probably) [HSi2O5]nn− in the interlayer, was concluded from that study.The LDH precursors were calcined in air at 600°C for 3 h. This temperature was chosen after analyzing the results of the thermal study, and because: (i) these systems are usually more effective as catalysts after calcination; (ii) the FTIR spectra recorded for samples calcined at lower temperature were ill-defined, and (iii) the layered structure was lost in all four cases, according to the PXRD results, developing large specific surface areas. The calcined samples will be named as MgAlX/ 600, where X is the non-metal constituting the anion in the original LDH.
Characterization and reactivity
PXRD diagrams were collected on a Siemens D-500 instrument, using Cu-Kα radiation (λ = 1.54050 Å). The BET specific surface areas and pore size distributions were determined from the adsorption–desorption isotherms (−196°C) of nitrogen recorded in a Micromeritics Gemini apparatus, after degassing in N2 at 150°C for 2 h. The FTIR spectra were recorded in a Perkin-Elmer 1730 spectrometer in the wavenumber range 4000–300 cm−1; the KBr disc technique was used and 100 scans were averaged, to improve the signal-to-noise ratio, with a nominal resolution of 4 cm−1.Surface acidity/basicity properties were determined by FTIR monitoring of adsorption of pyridine (py) and boric acid trimethyl ester (bate) in a Perkin-Elmer 16PC spectrometer using self-supported discs; the samples were degassed in situ in a special cell with CaF2 windows, at 400°C for 2 h at 10−4 N m−2, prior to the adsorption experiments. After degassing, the probe vapour was adsorbed at room temperature and the spectrum recorded at room temperature after degassing at increasing temperatures, from room temperature to 400°C. The same method was used to study ISP reactivity on these solids; in this case, ISP was adsorbed at room temperature (after degassing the sample as described above) and the sample disc, without removing the gas phase, was heated at increasing temperatures; the gas phase spectrum was removed using the software facilities, and the liquids were previously degassed to remove dissolved air.
A quantitative determination of surface acidity was also carried out by adsorption of ammonia (from L'Air Liquide, Spain). This study was carried out in a home-built high vacuum system built in Pyrex glass; pressure changes were measured with a Baratron MKS model 220B instrument, coupled to a MKS model PDR-5B amplifier connected to a KNK-801-201 graphic recorded (pressure changes ± 1 N m−2); in order to obtain comparable results, the sample was outgassed in situ at 400°C for 2 h prior to the adsorption experiments.
Results and discussion
Characterization of the LDH precursors and the calcined solids
The PXRD diagrams for the original LDHs samples, and after calcination at selected temperatures, are included in Fig. 1. The patterns for the original samples evidence their layered structure, with intense (00l) diffraction maxima whose precise position is related to the interlayer spacing, depending on the size and orientation of the interlayer anion,9 the carbonate being flat, with its molecular plane parallel to the brucite-like layers, but the nitrate anion being tilted in an upwards position. Upon calcination, the behaviour shown by these samples is different: the PXRD patterns for samples MgAlC/600 and MgAlN/600 show only broad, medium-intense maxima close to 2.1 and 1.84 Å, ascribed to planes (200) and (220) of MgO,10 the Al3+ ions being probably dispersed in the MgO rock-salt structure, forming a solid solution. Crystallization of an MgAl2O4 spinel is observed after calcination at higher temperatures.11 | ||
| Fig. 1 PXRD diagrams of: (top) original samples MgAlC and MgAlN, and after calcination in air at 600°C, MgAlC/600 and MgAlN/600; (middle) original sample MgAlSi and after calcination at 600 and 800°C in air, and (bottom) original sample MgAlB and after calcination at 600 and 800°C in air. Peaks (*) correspond to the Al sample holder. The patterns have been displaced vertically for clarity. | ||
Samples with borate or silicate, calcined at 600°C, show PXRD diagrams with no defined maximum, characteristic of mostly amorphous materials. Only after calcination at 800°C do sharp peaks develop, corresponding to crystallization of Mg2B2O5 or MgSiO3, respectively. The stoichiometry in the initial solids and the fact that the anions are acidic and not volatile gives rise to formation of Mg salts, instead of MgO, as observed for the carbonate and nitrate precursors. However, as for these precursors, no defined phase containing Al was detected by PXRD, indicating that it forms very well dispersed, amorphous phases, or is dissolved in the Mg-compounds matrix. Also, we can assume that the same species detected in a crystallized state in the samples calcined at 800°C, is in a more or less amorphous state in the samples calcined at 600°C, where the layered structure of the precursor LDH has been completely destroyed, although experimental evidence is lacking at present.
The FTIR spectra of the solids calcined at 600°C, recorded using the KBr pellet technique, are shown in Fig. 2. These are rather similar for the calcined carbonate and nitrate precursors. A broad band around 3442 cm−1 is recorded in the high wavenumber region, which corresponds to the ν(OH) stretching mode of adsorbed water molecules, whose δ(H2O) deformation mode is responsible for the band recorded at 1630 cm−1. It should be taken into account that despite the fact that these samples have been calcined at 600°C and completely dehydrated and dehydroxylated, they adsorb water vapour from the atmosphere during handling to prepare the KBr disc. A broad band at ca. 1489 cm−1 is recorded in the lower wavenumber region corresponding to an overtone of lattice Mg–O vibrations,12 recorded between 800 and 400 cm−1, together with Al–O modes.13 The presence of weakly adsorbed carbonate or bicarbonate species, formed through interaction with atmospheric CO2, cannot be discounted, and would be responsible for the broad, weak bands around 1500–1400 cm−1. For the other two samples, in addition to the bands due to water and hydroxy groups, intense bands due to the anions existing in these samples are recorded. For sample MgAlB/600 a broad band is recorded between 1590 and 1050 cm−1, which contains several overlapped bands due to modes ν(BO3) and ν(BO4) of borate species.13,14 A broad, intense band is recorded between 1200 and 800 cm−1 for sample MgAlSi/600, which corresponds to Si–O vibrations.15 Also for these two samples, bands characteristic of ν(Mg–O) and ν(Al–O) modes are recorded below 800 cm−1.
 | ||
| Fig. 2 FTIR spectra (KBr discs) of samples calcined at 600°C in air, MgAlC/600, MgAlN/600, MgAlSi/600 and MgAlB/600. The spectra have been displaced vertically for clarity. | ||
The nitrogen adsorption–desorption isotherms, recorded at − 196°C for all four precursors calcined at 600°C, are shown in Fig. 3. All correspond to type II in the IUPAC classification,16 corresponding to mesoporous solids. A narrow type H216 hysteresis loop is recorded above P/P0 = 0.4 for sample MgAlN/600, while for the other samples the loops extend over a narrower relative pressure range. The specific surface areas (Table 1), calculated following the BET, Cranston and Inkley (for cumulative surface area)17 and t-plot method proposed by Lippens and de Boer18 are coincident in all cases, confirming the absence of micropores in these samples calcined at 600°C. However, if the surface area values are compared with those for the uncalcined, parent LDHs (Table 1), it can be observed that the values for samples originally containing borate and silicate are almost the same for the uncalcined and calcined samples (i.e., 72 and 75 m2 g−1 for MgAlSi and MgAlSi/600, respectively, and 56 and 55 m2 g−1, respectively, for MgAlB and MgAlB/600), while for the solids prepared from the carbonate and nitrate precursors the surface areas are appreciably larger for the calcined samples. These results are in agreement with previous19,20 results for LDHs containing carbonate and nitrate in the interlayers: upon calcination, the evolved gases (CO2 and NO/NO2, respectively) escape, forming craters through the brucite-like layers, giving rise to surface area enhancement. The pore size distribution curves, following the method of Cranston and Inkley,17, show that pores with a diameter in the 2–4 nm range are predominant in all calcined samples, except for MgAlC/600, for which an important contribution to the surface area by pores with a diameter close to 5 nm is also observed.
 | ||
| Fig. 3 Nitrogen adsorption desorption isotherms (−196°C) for the samples calcined at 600°C in air: (a) MgAlC/600, (b) MgAlN/600, (c) MgAlSi/600 and (d) MgAlB/600. The plots have been displaced vertically for clarity. | ||
Surface acid–base properties
Pyridine (py) was chosen as the probe molecule to study the surface acid sites in the calcined samples.21,22 The shift in the positions of the bands due to py is related to the deformation originated in its electron density through interaction with surface acid sites, while the persistence of these bands as the outgassing temperature is increased can be related to the strength of the surface acid sites.In all cases, a constant pressure of py vapour (400 N m−2) was left in contact with the in situ outgassed, self-supported disc of the sample, and the FTIR spectrum was recorded after outgassing at room temperature. For sample MgAlC/600, bands are recorded at 1612 (shoulder), 1602, 1592, 1574, 1488 and 1445 cm−1, Fig. 4 (left). The intensities of these bands decrease after outgassing at 100°C, and are almost completely removed after outgassing at 200°C. They correspond to modes 8a, 8b, 19a and 19b of py molecules coordinated to surface Lewis acid sites.23,24 The splitting observed for the band originated by mode 8a (1612, 1602 and 1592 cm−1) clearly shows the presence on the surface of different types of surface Lewis acid sites. Bands in positions close to these have been previously recorded upon adsorption of py on alumina (1622, 1614, 1596, 1579, 1493 and 1449 cm−1), and have been ascribed to py coordinated to tetrahedrally (1622 and 1614 cm−1) or octahedrally coordinated (1596 cm−1) Al3+ cations,25 although the band recorded at 1614 cm−1 has also been ascribed to octahedrally coordinated Al3+ ions, that at 1622 cm−1 to tetrahedrally coordinated Al3+ ions, and that at 1596 cm−1 to hydrogen-bonded pyridine molecules.25 So, we may conclude that although no crystalline phase containing Al3+ was detected by PXRD, these cations are located in an octahedral environment in the sample calcined at 600°C, similar to that existing in corundum. The surface Lewis acid sites, i.e., coordinatively unsaturated Al3+ ions, are not too strong, as coordinated pyridine is removed after outgassing at 200°C.
 | ||
| Fig. 4 FTIR spectra (self-supported discs) recorded after adsorption of pyridine at room temperature on MgAlC/600 (left) and MgAlN/600 (right) and degassed at (a) room temperature, (b) 100°C and (c) 200°C. | ||
Similar results were obtained upon adsorption of py on sample MgAlN/600, Fig. 4 (right). The bands, corresponding to adsorption of py on surface Lewis acid sites (coordinatively unsaturated Al3+ cations), are recorded at 1612 (shoulder), 1602, 1592, 1578, 1490 and 1445 cm−1. The bands are also removed after outgassing at 200°C, indicating that the strength of the surface Lewis acid sites is similar to those on the surface of sample MgAlC/600. Note that no band corresponding to adsorption on surface Brönsted acid sites was recorded for these two samples.
The spectra recorded after adsorption of py at room temperature on sample MgAlSi/600 are rather similar, Fig. 5. The bands are in the same positions, but their intensities are much larger than for the two samples discussed above (note the scale bars in both figures). In addition, an extremely weak band is recorded close to 1545 cm−1, which was absent in the spectra recorded after adsorption of py on samples MgAlC/600 and MgAlN/600. This band corresponds to mode 19b of the pyridinium species, i.e., py adsorbed on surface Brönsted acid sites. It is well known that py is only physisorbed on the surface of silica (silicates, clearly detected in a crystalline form after calcination of this sample at 800°C, should exist, although in an amorphous state, in this sample calcined at 600°C), but, when silica is mixed with alumina, both surface Lewis and Brönsted acid sites develop, giving rise to adsorption of coordinated py and formation of protonated py species, respectively.23
 | ||
| Fig. 5 FTIR spectra (self-supported discs) recorded after adsorption of pyridine at room temperature on MgAlSi/600 and degassed at (a) room temperature, (b) 100°C and (c) 200°C. | ||
The spectrum recorded after adsorption of py on sample MgAlB/600 is absolutely different from those described above. Bands are now recorded at 1590, 1597, 1490 and 1449 cm−1, but they disappear when the sample is outgassed, even at room temperature, and thus should correspond to physisorbed py; a careful examination of the spectrum permits detection of an extremely weak band at 1602 cm−1, which disappears after outgassing at 100°C, and, as in the cases previously discussed, corresponds to py weakly coordinated to dispersed coordinatively unsaturated Al3+ species.
A quantitative analysis of total surface acidity was carried out by volumetric measurement of ammonia adsorption. The values obtained (for the calcined solids) are summarized in Table 1. Although in this case the probe molecule was ammonia, and in the FTIR experiments it was py, the amount of adsorbed ammonia decreases as the intensities of the bands due to adsorbed py do in the FTIR study. Thus, we can conclude that the surface acid sites are exposed on “open ” portions of the surface of the solids, where access to small (ammonia) or slightly larger (py) molecules is not restricted.
In order to obtain a complete picture of the surface of these solids, the basicity was also studied by FTIR spectroscopy. The selected probe molecule was boric acid trimethyl ester (bate),26 which behaves as a Lewis acid, being able to accept electron density on the empty “ p” orbital of the boron atom, which is trigonally bonded to the methoxy groups. The free molecule shows IR bands at 1485, 1360 and 1036 cm−1, which correspond to the deformation mode of the methyl group, and to the B–O and C–O stretching modes, respectively. If electron density is accepted in the non-bonding p orbital of the boron atom, the bands closer to the p orbital than the methyl groups, mainly those corresponding to ν(B–O) and, to a lesser extent, ν(C–O), will be modified in position and may be split.
The spectra recorded upon adsorption of bate at room temperature on all four calcined samples are shown in Fig. 6. Those for samples MgAlC/600 and MgAlN/600 were very similar as were those recorded for MgAlSi/600 and MgAlB/600. For the first pair of samples (MgAlC/600 and MgAlN/600) bands in positions close to those of gaseous bate were recorded at 1491, 1361 and 1039 cm−1; in addition, other bands, more intense, were recorded at 1275, 1203 and 1108 cm−1, which remain after outgassing above room temperature. Thus, two types of surface basic sites exist on the surface of these two samples, one on which adsorption is very weak, and electron transfer from the solid to the probe molecule is very weak (thus accounting for the bands being recorded in almost the same positions as for gaseous bate), and one, on which adsorption is much stronger; probably, these two types of surface basic sites correspond to oxide anions in different geometric environments, or different electron density.
 | ||
| Fig. 6 FTIR spectra (self-supported discs) recorded after adsorption of bate at room temperature on (a) MgAlC/600 and MgAlN/600 and (b) MgAlSi/600 and MgAlB/600. | ||
Adsorption on the other two samples gives rise to completely different spectra. The bands recorded are in the same positions as for the gaseous probe molecule, at 1493–1483, 1391 (shoulder), 1363 and 1040 cm−1, with intensities lower than those of the bands recorded upon adsorption on samples MgAlC/600 and MgAlN/600; the bands are removed by simply outgassing at room temperature. The spectrum, also shown in Fig. 6, is very similar to that reported for adsorption of bate on SiO2.
It can be concluded that samples MgAlC/600 and MgAlN/600 have two types of surface basic sites, while seem to be absent on the surfaces of the other two samples studied.
Adsorption of isopropanol
After outgassing the self-supported disc in situ as described above, ISP was admitted to the vacuum-IR cell and the spectrum recorded; the sample was then heated and the spectra were recorded at increasing temperatures.The results obtained for MgAlC/600 are shown in Fig. 7.
On the left-hand side, trace (a) corresponds to the solid calcined
at 600°C and outgassed in situ at 400°C. The broad,
weak band centered at ca. 3585 cm−1 corresponds to
hydrogen-bonded hydroxy groups on the surface of MgO,
while the weak, but distinguishable peak at 3726 cm−1
corresponds
to free, non-associated hydroxy groups27,28 on the
MgO surface; however, their positions are very close to those
of the bands recorded for alumina.28,29 Upon adsorption of
ISP these bands disappear, and a much broader, intense band
centered at ca. 3500–3000 cm−1 is recorded, which corresponds
to new surface hydroxy groups formed on the surface
of the solid by dissociative adsorption, although the simultaneous
presence of molecularly adsorbed, hydrogen-bonded
ISP cannot be ignored. The sharp bands at 2970, 2937 and
2895 cm−1 correspond to stretching modes of the methyl
group of ISP. In the low wavenumber region (Fig. 7B), bands
are recorded at 1468, 1382, 1371, 1259 (shoulder), 1168, 1133
and 1030 cm−1, ascribed, Table 2, to alcoholate species
formed ia dissociative adsorption of the ISP molecule, or to
molecularly adsorbed ISP; note that dissociative adsorption
could also account for development of new surface hydroxy
groups, through reaction between the dissociated proton
(from the ISP molecule) and surface oxide anions.
 | ||
| Fig. 7 FTIR spectra (self-supported discs) recorded after adsorption of isopropanol on MgAlC/600. (a) Original sample degassed at 400°C, (b) after adsorption of isopropanol at room temperature, (c) at 200°C and (d) at 300°C. Note different y-axis in part A (high wavenumber section, transmittance) and part B (medium wavenumber section, absorbance). | ||
| Mode | MgAlC/600 | MgAlN/600 | MgAlSi/600 | MgAlB/600 |
|---|---|---|---|---|
| a Undissociated isopropanol. | ||||
| ν(OH) | 3500 | 3659 | 3414 | 3500–3400 |
| ν(CH3)as | 2970 | 2970 | 2984 | 2979 |
| 2 × δ(CH3)as | 2937 | 2937 | 2939 | 2937 |
| ν(CH3) | 2895 | 2895 | 2897 | 2878 |
| δ(CH3)as | 1468 | 1477, 1468 | 1482, 1465 | 1462 |
| δ(CH3)s | 1382, 1371 | 1380, 1370 | 1381 | 1393 |
| δ(CH) | 1345 | |||
| δ(OH)a | 1259 | 1259 | 1259 | 1259 |
| ν(CO) | 1168 | 1179 | 1153 | 1163 |
| ν(CC) | 1133 | 1138, 1130 | 1080 | 1079 |
| ρ(CH3) | 1030 | 1038 | 1036 | |
When the solid is heated in situ at increasing temperatures,
the intensities of the bands originated by the isopropoxide
species decrease, while those due to molecularly adsorbed
(undissociated) ISP are completely removed; above 200°C
new bands develop, at 1717 and 1232 cm−1; these bands are
in positions very close to those previously reported after
adsorption of acetone on several metal oxides,30 and corresponding
to ν(C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) O) and
νas(C–C–C) modes of acetone weakly coordinated
through the oxygen atom of the carbonyl group to surface Lewis
acid sites; the corresponding bands for gaseous acetone
are recorded at 1734 and 1215 cm−1.31 The intensities
of the bands due to acetone increase with temperature, but
decrease sharply after outgassing, even at rather low temperatures.
Note, however, that no band due to the presence of
carboxylate species, is recorded.
O) and
νas(C–C–C) modes of acetone weakly coordinated
through the oxygen atom of the carbonyl group to surface Lewis
acid sites; the corresponding bands for gaseous acetone
are recorded at 1734 and 1215 cm−1.31 The intensities
of the bands due to acetone increase with temperature, but
decrease sharply after outgassing, even at rather low temperatures.
Note, however, that no band due to the presence of
carboxylate species, is recorded.
Similar results were obtained upon adsorption of ISP on
sample MgAlN/600, Fig. 8. In this case, the original outgassed
solid did not show any detectable absorption band in the high
wavenumber region, indicating the absence of detectable
amounts of free or hydrogen-bonded hydroxy groups. Upon
adsorption of ISP, bands are recorded at 3659 (OH groups),
2970, 2937 and 2895 cm−1 (methyl groups of isopropoxide
species), and 1477, 1468, 1380, 1370 (shoulder), 1259, 1179,
1138 (shoulder), 1130 and 1038 cm−1. Most of these band are
coincident with those ascribed above to isopropoxide species,
formed following dissociative adsorption of ISP, Table 2,
although in general, some bands are broader or even split.
Also, the band at 1259 cm−1, ascribed to mode δ(OH) of ISP undissociated
(physisorbed or hydrogen-bonded to the solid surface) is much stronger than for sample MgAlC/600; note also the development of the intense band at 3659 cm−1, which could be ascribed to rather free OH groups, formed on the surface of the catalyst. When the sample is heated at or above 200°C, new bands at 1745, 1719 and 1238 cm−1, ascribed to ν(CO) and ν(C–C–C) of acetone, well gaseous (1745 cm−1) or weakly coordinated to surface Lewis acid sites (1719 cm−1), develop, with a
concomitant decrease in the intensity of the band due to the
δ(OH) mode of undissociated ISP. As for the previous catalyst, acetone is easily desorbed, and is not further oxidized to carboxylate species.
 | ||
| Fig. 8 FTIR spectra (self-supported discs) recorded after adsorption of isopropanol on MgAlN/600. (a) Original sample degassed at 400°C, (b) after adsorption of isopropanol at room temperature and (c) at 300°C. Note different y-axis in part A (high wavenumber section, transmittance) and part B (medium wavenumber section, absorbance). | ||
The spectrum of sample MgAlSi/600 outgassed at 400°C shows a sharp, rather intense band at 3737 cm−1, together with a much broader absorption at 3581 cm−1; these positions are very close to those reported for free (3740 cm−1) and hydrogen-bonded (3650 cm−1) hydroxy groups on the surface of silica, Fig. 9; on this oxide, the bands persist even after outgassing at 800°C.32 Adsorption of ISP almost cancels the sharp absorption at 3737 cm−1, while the minimum of the band due to hydrogen-bonded hydroxy groups shifts towards lower wavenumber; in addition, bands due to the methyl groups are also recorded below 3000 cm−1. The spectrum in the low wavenumber region follows a similar behaviour to that described for the other two samples, but in this case bands due to acetone are recorded only after heating the sample at 300°C, although even at this temperature the intensities of the bands are rather low.
 | ||
| Fig. 9 FTIR spectra (self-supported discs) recorded after adsorption of isopropanol on MgAlSi/600. (a) Original sample degassed at 400°C, (b) after adsorption of isopropanol at room temperature and (c) at 300°C. Note different y-axis in part A (high wavenumber section, transmittance) and part B (medium wavenumber section, absorbance). | ||
Finally, the spectra recorded after adsorption of ISP on
sample MgAlB/600 are shown in Fig.
10. Weak bands at 3700
and 3587 cm−1 due to hydroxy groups, recorded in the spectrum of the original sample outgassed at 400°C, disappear
after adsorption of ISP at room temperature; the intensity of
the broad band due to hydrogen-bonded hydroxy groups is
also enhanced after adsorption of ISP, while below 3000 cm−1
the bands due to the methyl groups (2979, 2937 and 2878
cm−1) of the adsorbed molecule are recorded. The bands due
to adsorbed ISP are recorded at 1462, 1393, 1259, 1163 and
1079 cm−1, Table 2, and they seem to correspond mostly to
undissociated ISP, weakly (physically) adsorbed or hydrogen-bonded to the surface, although the presence of isopropoxide
species, formed ia dissociative adsorption, cannot be completely ruled out. The characteristic bands of acetone at 1740
and 1720 cm−1 are recorded above 300°C but are much
weaker than those recorded for the other samples; however
the interaction of acetone with the solid surface should again
be rather weak, as the bands disappear (acetone is removed)
after outgassing at low temperature.
 | ||
| Fig. 10 FTIR spectra (self-supported discs) recorded after adsorption of isopropanol on MgAlB/600. (a) Original sample degassed at 400°C, (b) after adsorption of isopropanol at room temperature and (c) at 300°C. Note different y-axis in part A (high wavenumber section, transmittance) and part B (medium wavenumber section, absorbance). | ||
The results obtained are somewhat similar to those previously reported for adsorption of alcohols on different metal oxides, such as TiO2, ZrO2, MgO, Al2O3, etc.;30,33–37 in fact, the samples studied here constitute a mixture of ill-defined oxides (Mg, Al, Si, B).
If we assume mainly ionic character for the oxides studied here, and heterolytic breaking of the bonds, the mechanism shown in Scheme 1 can be assumed to describe the oxidation of isopropanol to acetone on the surface of these samples. In a first step, the alcohol is dissociatively adsorbed, forming the corresponding alkoxide (isopropoxide in our case), detected by FTIR spectroscopy, and bonded to coordinatively unsaturated surface metal sites (surface Lewis acid sites). Then, the C–H bond is broken, this being the rate-determining step of the process. This bond breaking can be described as a consequence of the direct interaction of the hydrogen atom with the coordinatively unsaturated surface site (1), or with a polarized hydrogen atom originating from the previous breakage of the O–H bond in the alcohol molecule (2). For either step, molecular hydrogen and acetone would be formed in the final step. The facile desorption of acetone, weakly interacting with the surface, avoids its oxidation to carboxylate species, which were not detected in any of the four samples studied here.
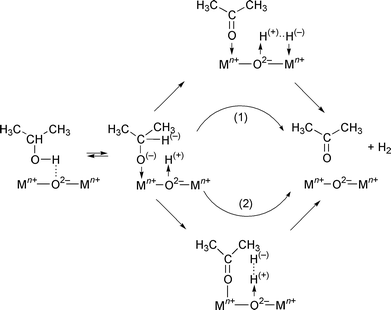 | ||
| Scheme 1 | ||
An alternative mechanism, consisting of the reaction of ISP with surface hydroxy groups:

should be discarded; although the broad band recorded around 3500 cm−1 is due to the stretching mode of OH groups, the absence of the δ(H2O) band around 1630 cm−1 indicates that water is not formed during the reaction.
On the other hand, the loss of bands due to surface free hydroxy groups upon adsorption of ISP, together with the development of the broad band due to hydrogen-bonded hydroxy groups, suggests the possible simultaneous molecular, non-dissociative adsorption of ISP according to:

Conclusion
The results described indicate that MgAlC/600 and MgAlN/600 consist of low crystalline MgO; Al3+ ions are dissolved in the MgO matrix, or form amorphous alumina (as they were not detected by PXRD). In the other two samples, amorphous Mg (and probably Al) borates and silicates are formed, which crystallize when the solids are calcined at 800°C.The spectra recorded after adsorption of pyridine indicate
the presence of surface Lewis acid sites on both MgAlC/600
and MgAlN/600. The highest surface acidity is observed for
sample MgAlSi/600 (both Lewis and Brönsted acidities), but
there is no indication of the presence of surface basic sites
(from bate adsorption). Finally, no detectable acidity nor
basicity was observed for sample MgAlB/600. The qualitative
results derived from the FTIR study are quantitatively confirmed by volumetric measurement of ammonia adsorption.
Altogether, the acidity of the samples decreases as: MgAlSi/600>MgAlC/600≈MgAlN/600![[double greater-than, compressed]](https://www.rsc.org/images/entities/char_2aa2.gif) MgAlB/600 while the basicity
decreases as: MgAlC/600≈MgAlN/600MgAlSi/600≈MgAlB/600.
MgAlB/600 while the basicity
decreases as: MgAlC/600≈MgAlN/600MgAlSi/600≈MgAlB/600.
However, the presence of Lewis acidity on the surface of sample MgAlB/600 would be expected: both the magnesium borate identified by PXRD in the sample calcined at 800°C (see Fig. 1), and alumina, show surface acidity (actually, surface acidity shown by the other samples was mostly related to the presence of amorphous alumina), but in our sample calcined at 600°C such acidity was not detected. This apparent disagreement could be explained if we assume that the well dispersed, amorphous phases existing after calcination at 600°C would show different surface acidity/basicity, and may interact strongly forming the amorphous precursor of the salt crystallizing at 800°C, where Al3+ cations could be embedded, thus remaining inaccessible to the probe basic molecule. Such a strong interaction would also account for the lack of surface basic sites in sample MgAlB/600, compared with those detected in samples MgAlC/600 and MgAlN/600.
The activity of the catalysts towards acetone formation decreases as MgAlN/600>MgAlC/600>MgAlSi/600≈MgAlB/600 and is in agreement with the mechanism proposed above. Both surface acidic and basic sites exist in samples MgAlC/600 and MgAlN/600, and such sites are necessary to dissociate ISP forming protons and the alkoxide species. The silicate sample, although displaying the maximum surface acidity and high surface hydroxylation, does not contain an appreciable amount of surface basic sites; both samples MgAlB/600 and MgAlSi/600 show similar activities for acetone formation. Surface acid/basic properties and activity for dehydration (alkene formation) or oxidation (acetone formation) are intimately related, and ISP has even been used in some cases to determine surface acid/basic properties of metal oxides.38–40
Our results on the catalytic conversion of ISP are in line with previous results reported by Reichle,9,19 who found a higher activity for oxides prepared from LDH precursors containing vaporizable anions. However, this fact seems to be more related to development of porosity (larger surface area, larger number of surface active sites) during thermal decomposition and evolution of gaseous compounds (CO2 and NO/NO2) than to the precise chemical composition of the sample.
Acknowledgements
Thanks are due to DGES (grant PB96-1307-C03-01) for financial support and to Junta de Castilla y León (Consejería de Educación y Cultura, grant SA45/98) for financial support and a grant to S.G.References
- A. de Roy, C. Forano, K. El Malki and J. P. Besse, in Expanded Clays and other Microporous Solids, Synthesis of Microsporous Materials, ed. M. L. Occelli and H. Robson, van Nostrand Reinhold, New York, 1992, ch. 7, p. 108. Search PubMed.
- F. Trifirò
and A. Vaccari, in
Comprehensie Supramolecular Chemistry. Volume 7. Solid-State
Supramolecular Chemistry: Two- and Three-Dimensional Inorganic Networks, ed. J. L. Atwood, J. E. D. Davies, D. D. MacNicol, F. Vögtle, J.-M. Lehn, G. Alberti and T. Bein, Pergamon, Oxford, 1996, p. 251. Search PubMed.
- S. P. Newman and W. Jones, New J. Chem., 1998, 105 RSC.
- V. Rives and M. A. Ulibarri, Coord. Chem. Re., 1999, 181, 61 Search PubMed.
- F. Cavani, F. Trifirò and A. Vaccari, Catal. Today, 1991, 11, 173 CrossRef CAS.
- H. H. Kung and E. I. Ko, Chem. Eng. J., 1996, 64, 203 CrossRef CAS.
- A. Vaccari, Appl. Clay Sci., 1999, 14, 161 CrossRef CAS.
- W. T. Reichle, J. Catal., 1980, 63, 295 CrossRef CAS.
- M. del Arco, S. Gutiérrez, C. Martín, V. Rives and J. Rocha, J. Solid State Chem., 2000, 151, 272 CrossRef CAS.
- T. Sato, H. Fujita, T. Endo and M. Shimada, React. Solids, 1988, 5, 219 CrossRef CAS.
- F. M. Labajos, V. Rives and M. A. Ulibarri, J. Mater. Sci., 1992, 27, 1546 CrossRef CAS.
- R. A. Niquist and R. O. Kagel, Infrared Spectra of Inorganic Compounds, Academic Press, New York, 1971. Search PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Wiley, New York, 5th edn., 1997. Search PubMed.
- L. Li, S. Ma, X. Liu, Y. Yue, J. Hui, R. Xu, Y. Bao and J. Rocha, Chem. Mater., 1996, 8, 204 CrossRef CAS.
- A. N. Lazarev, in Vibrational Spectra and Structure of Silicates, C/B Consultants Bureau, New York, 1972. Search PubMed.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. Pierotti, J. Rouquerol and T. Sieminiewska, Pure Appl. Chem., 1985, 57, 603 CrossRef CAS.
- R. W. Cranston and F. A. Inkley, Ad. Catal., 1957, 9, 143 Search PubMed.
- B. C. Lippens and J. H. de Boer, J. Catal., 1965, 4, 319 CrossRef.
- W. T. Reichle, J. Catal., 1985, 94, 547 CrossRef CAS.
- W. Kagunya and W. Jones, Appl. Clay Sci., 1995, 10, 95 CrossRef CAS.
- H. Knözinger, Ad. Catal., 1976, 25, 184 Search PubMed.
- H. A. Benes and B. H. C. Winsquist, Ad. Catal., 1978, 27, 97 Search PubMed.
- E. P. Parry, J. Catal., 1963, 2, 371 CrossRef CAS.
- E. B. Wilson, Phys. Re., 1934, 45, 706 Search PubMed.
- C. Morterra, S. Coluccia, E. Garrone and G. Ghiotti, J. Chem. Soc., Faraday Trans. 1, 1979, 75, 289 RSC.
- C. Li, Sh. Fu and H. Zhang, J. Chem. Soc., Chem. Commun., 1994, 17 RSC.
- E. Echterhoff and E. Knözinger, J. Mol. Struct., 1988, 178, 343 CrossRef.
- A. Zecchina, S. Coluccia and C. Morterra, Appl. Spectrosc. Re., 1985, 21, 259 Search PubMed.
- J. B. Peri, J. Phys. Chem., 1965, 69, 220 CAS.
- C. Martín, I. Martín and V. Rives, J. Catal., 1994, 145, 239 CrossRef CAS.
- D. Delle Piane and J. Overend, Spectrochim. Acta, 1966, 22, 593 CrossRef.
- L. H. Little, Infrared Spectra of Adsorbed Species, Academic Press, New York, 1966. Search PubMed.
- G. A. G. Hussein, N. Sheppard, M. I. Zaki and R. B. Fahin, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 1723 RSC.
- H. Miyata, Y. Nakagawa, T. Ono and Y. Kubokawa, J. Chem. Soc., Faraday Trans. 1, 1983, 79, 2343 RSC.
- R. O. Kagel, J. Phys. Chem., 1967, 71, 844 CrossRef CAS.
- A. V. Deo, T. T. Chuang and I. G. Dalla-Lana, J. Phys. Chem., 1971, 75, 234 CrossRef CAS.
- H. Knözinger and A. Scheglila, J. Catal., 1970, 17, 252 CrossRef.
- A. Ouqour, G. Coudurier and J. C. Vedrine, J. Chem. Soc., Faraday Trans., 1993, 89, 3151 RSC.
- B. Grybowska-Swierkosz, Mater. Chem. Phys., 1987, 17, 121 CrossRef.
- M. Ai, J. Catal., 1977, 49, 305 CrossRef CAS.
| This journal is © the Owner Societies 2001 |