Microspectroscopic studies of template interactions in AlPO4-5 and SAPO-5 crystals
Sorina C. Popescu, Stuart Thomson† and Russell F. Howe*
School of Chemistry, Uni![[italic v]](https://www.rsc.org/images/entities/char_e0f5.gif) ersity of New South Wales, Sydney, NSW 2052, Australia
ersity of New South Wales, Sydney, NSW 2052, Australia
First published on 7th December 2000
Abstract
Individual
crystals of the aluminophosphate AlPO4-5 and the silicon-substituted aluminophosphate SAPO-5 synthesized
with a triethylamine template have been studied by FTIR and Raman microspectroscopy. Both materials
as synthesized contain the template in the protonated form. In AlPO4-5, the protonated template is charge balanced
by hydroxide ions. Dehydration causes elimination of water leaving the template in the free amine form,
but this process is reversible. Measurements with polarized infrared radiation reveal that both the protonated
and free template molecules are oriented with the C3 axes perpendicular to the channel direction. In SAPO-5,
deprotonation of the template also occurs on dehydration but to a lesser extent, and protonated template
is still present after heating in ![[italic v]](https://www.rsc.org/images/entities/i_char_e0f5.gif) acuo to 500 K. The balance between protonated and free template depends
on the extent of hydration of the sample and the presence or absence of negative charge on the framework.
After thermal calcination to remove the template completely, the single crystals of AlPO4-5 are devoid of
hydroxy groups, whereas
SAPO-5 shows two
types of hydroxy groups
oriented perpendicular to the channel axes.
acuo to 500 K. The balance between protonated and free template depends
on the extent of hydration of the sample and the presence or absence of negative charge on the framework.
After thermal calcination to remove the template completely, the single crystals of AlPO4-5 are devoid of
hydroxy groups, whereas
SAPO-5 shows two
types of hydroxy groups
oriented perpendicular to the channel axes.
Introduction
Many novel microporous materials have been synthesized using organic molecules or cations as templates around which the microporous structures are assembled. Understanding the template–microporous framework interactions is an important objective if the synthesis chemistry and the stability of the resulting structures are to be controlled. AlPO4-5 (AFI structure) is a microporous aluminophosphate structure consisting of alternating AlO4− and PO4+ units constituting an electrostatically neutral framework. AlPO4-5 was one of the first of many aluminophosphate zeolite analogs reported by Flanigen et al.1 Incorporation of a silicon source in the synthesis produces SAPO-5, in which Si may substitute for framework phosphorus in the AFI structure, producing a net negative charge on the framework which may be balanced by protons or other cations.2IR spectroscopy is widely used to study adsorbed species in zeolites and other microporous solids. Conventionally, either transmission spectroscopy on pressed disks of polycrystalline samples or diffuse reflectance measurements on powders are employed. A number of authors have shown recently however that IR microspectroscopy can be applied to individual single crystals of zeolites.3–8 Such studies offer two particular advantages: single crystal samples are free of amorphous impurities and other phases which may complicate the analysis, and orientation information about intra-zeolite species can be obtained by using polarized radiation.
Triethylamine (TEA) is the template most widely used for synthesis of AlPO4-5. Schnabel et al. recently described a study by FTIR and Raman spectroscopy of the state of the template in AlPO4-5.9 They report that both AlPO4-5 and SAPO-5 as synthesized contain protonated TEA, which on heating first eliminates water to form the neutral amine, then progressively loses ethylene to form lower amines and finally ammonia. In AlPO4-5 the protonated TEA was considered to be charge balanced by hydroxide ions, which is not the case in the negatively charged SAPO-5.
Schnabel et al. used macroscopic samples of AlPO4-5 and SAPO-5, and collected IR spectra by the technique of diffuse reflectance.9 In this study we have used IR and Raman microspectroscopy to examine individual crystals of AlPO4-5 and SAPO-5 which can be grown up to 300 microns or more in length,10 and to study the state of protonation of the TEA template in AlPO4-5 and SAPO-5 as a function of the extent of hydration of the materials. Polarized FTIR measurements were made of template orientation, and solid state 13C NMR studies were also undertaken on macroscopic samples of the same crystals.
Experimental
AlPO4-5 and SAPO-5 were synthesized hydrothermally using TEA as the structure directing agent. The reagents used were: pseudoboehmite (Catapal B), 85% orthophosphoric acid (Aldrich), triethylamine (Aldrich), and distilled water. For SAPO-5, the silicon source used was Ludox-40S (DuPont).Conditions used to synthesize large crystals were similar to those reported by Finger et al.10 Reagents in the ratios indicated below were mixed by slowly adding phosphoric acid with constant stirring to a saturated slurry of pseudoboehmite and water, then stirring the resultant gel for 30 min. In the case of SAPO-5, Ludox-40S was added to the slurry prior to the addition of acid. After the initial mixing the TEA template was introduced slowly with constant stirring, and the remaining water was added while stirring for a further 30 min, ensuring that the gel retained a uniform consistency. The gel was then placed in a Teflon-lined autoclave and heated at 190°C for 72 h. The gel compositions corresponded to P2O5:Al2O3:1.55 TEA:600 H2O for AlPO4-5, and 0.8 P2O5 :Al2O3: 0.2 SiO2:1.55 TEA: 500 H2O for SAPO-5. Upon completion of the synthesis the autoclaves were quenched and the products washed, sonicated, decanted to separate large single crystals from other material, filtered and dried.
Where necessary, template removal was achieved by thermally calcining the as-synthesized materials in flowing oxygen. Samples were placed in a quartz tube containing a 10 μm frit in flowing oxygen and the temperature raised at 2 K min−1 to 423 K held at this temperature for 3 h, then heated at the same rate to 823 K, held for 30 h and cooled to room temperature at 2 K min−1.
Phase purity of the crystalline products was checked by powder diffraction using a Siemens D500 diffractometer; the only diffraction peaks detected were those of the AFI structure. Scanning electron microscopic examination of individual crystals was performed with an Hitachi 4500 SEM. Focussed ion beam miller (FIB) and EDAX (Oxford Instruments) were used for analysis of the crystals. Crystals were also checked by optical microscopy (Olympus).
Raman microspectroscopy was performed on single crystals under ambient conditions using a Renishaw 2000 Raman microprobe. All measurements used a 50 mW argon ion laser source (λ = 514 nm) and a 50 × objective, which allowed spectra to be collected from one crystal at a time. IR microspectroscopy used a Spectratech IRPlan microscope with 32 × objective coupled to a Nicolet Magna 860 spectrometer. A ZnSe polarizer attachment was used in polarization studies. Single crystals were mounted in an in situ vacuum cell similar to that described by Schuth et al.5 which allowed transmission spectra to be recorded while heating samples in a vacuum of better than 10−3 Torr. Spectra were in all cases collected by focussing the IR beam onto a single crystal and aperturing the beam above and below the sample to obtain a spectrum from part only of the crystal (typically 75 μm by 25 μm).
MAS NMR measurements were performed on a Bruker MSL300 spectrometer, using a 4 mm MAS probe. 27Al spectra were recorded at 78.2 MHz using single pulse Bloch decays (π/14 pulse width) and a spin rate of 10 kHz. 31P spectra were recorded at 121.4 MHz and 10 kHz spin rate using either proton cross-polarization or high power proton decoupling. 13C spectra were measured at 75.5 MHz and 4 kHz spin rate using proton cross polarization. Where necessary, dehydrated samples were transferred to the MAS rotor and sealed in a nitrogen atmosphere to prevent adsorption of water. Chemical shift references employed were kaolin (δ = 2.5 ppm for 27Al), ammonium dihydrogen phosphate (δ = 1 ppm for 31P) and adamantane (δ = 38.7 ppm for 13C).
Results and discussion
Synthesis and characterization of large crystals
The syntheses of SAPO-5 and AlPO4-5 resulted in the formation of hexagonal crystals with averge dimensions of 150 μm × 40 μm (SEM). Examination by optical microscopy and SEM showed no evidence of imperfections in the crystals chosen for study. EDAX measurements on the external regions and on ion beam milled cross sections of AlPO4-5 crystals showed a constant Al:P ratio of 1.00. As synthesized SAPO-5 crystals were also found to be homogeneous, with a silicon content Si/(Si + Al + P) = 0.035. The uniformity of the crystals was also confirmed by measuring identical infrared spectra from more than 20 different crystals and from different regions of the same crystal.27Al
NMR spectra of the as synthesized AlPO4-5 showed a dominant
signal at 42 ppm previously assigned to tetrahedral Al
in the AlPO4 framework.11 A second much weaker signal at
8 ppm, in the chemical shift range expected for 5 coordinate Al,12
was still present after dehydration by heating in acuo (see
below). After calcination to remove the TEA template, a single
27Al signal at 34 ppm was observed from dehydrated samples.
31P NMR spectra of as-synthesized AlPO4-5 showed a single broad signal at − 30 ppm; on dehydration this shifted to − 28.5 ppm and narrowed.
The template in as-synthesized AlPO4-5 and SAPO-5
Fig. 1 shows Raman microspectra of as-synthesized AlPO4-5 and SAPO-5 individual crystals in the C–H stretching region, measured in air at room temperature. Also shown is the spectrum of liquid TEA in the same region. Using 514 nm excitation, the ν(C–H) bands were the most intense in the spectra. Other much weaker bands were observed in the C–H deformation region, and at lower frequencies. Table 1 summarizes the observed Raman frequencies of as-synthesized AlPO4-5 and SAPO-5, together with those of liquid TEA and solid triethylammonium chloride. | ||
| Fig. 1 Raman spectra in the ν(CH) region of (a) liquid triethylamine, (b) AlPO4-5 single crystal as synthesized, (c) SAPO-5 single crystal as-synthesized. | ||
| AlPO4-5 (as synthesized) | SAPO-5 (as synthesized) | TEA (l) | Et3NHCl (s) | Assignment |
|---|---|---|---|---|
| a Frequencies accurate to ± 2 cm−1. | ||||
| 2997 | ν(CH3) | |||
| symmetric | ||||
| 2986 | 2986 | 2966 | 2980 | ν(CH3) |
| symmetric | ||||
| 2950 | 2950 | 2934 | 2944 | ν(CH2) |
| asymmetric | ||||
| 2889 | 2889 | 2873 | 2916, 2897, | ν(CH3) |
| 2885 | asymmetric | |||
| 2800 | ν(CH2) | |||
| symmetric | ||||
| 1456 | 1458 | 1453 | 1480, 1464, | δ(CH3) |
| 1444 | ||||
| 1399, 1365 | δ(CH2) | |||
The Raman spectra show that the C–H stretching frequencies of the template are shifted significantly to higher frequency relative to liquid TEA. There are corresponding differences also in the other regions of the spectrum (Table 1). These differences are fully consistent with the conclusion of Schnabel et al.,9 reached from macroscopic Raman measurements, that the template is protonated in the as-synthesized aluminophosphates. The Raman-active vibrational frequencies of the template in AlPO4-5 and SAPO-5 are closer to (but not identical with) those of the triethylammonium chloride salt than to liquid TEA.
The 13C NMR spectra of the as-synthesized materials support this conclusion. Fig. 2 shows 13C CPMAS spectra of as-synthesized AlPO4-5 and SAPO-5 polycrystalline powders after dehydration at different temperatures and following exposure to air at room temperature. The initial spectrum of AlPO4-5 (Fig. 2(a)) shows two signals at 46.0 and 6.5 ppm respectively. The effects of dehydration at 450 K are loss in intensity of the 6.5 ppm signal and appearance of a new signal at 12.0 ppm (marked with an arrow in Fig. 2(b)). Higher dehydration temperatures reduced further the relative intensity of the 6.5 ppm signal (not shown). Subsequent exposure to air restored the original spectrum (Fig. 2(c)), with a slight shift of the low field component to higher field.
 | ||
| Fig. 2 13C CPMAS NMR spectra of (a) AlPO5-5 evacuated at 373 K, (b) at 450 K, (c) after subsequent exposure to air, (d) SAPO-5 evacuated at 373 K, (e) at 500 K, (f) after subsequent exposure to air. | ||
Similar experiments were undertaken with SAPO-5 crystals. The as-synthesized crystals in air gave a 13C spectrum showing signals at 46.5 and 8.5 ppm respectively (Fig. 2(d)). Dehydration at 500 K caused the appearance of weak additional peaks at 52.5 and 12.0 ppm, and a shift of the 8.5 ppm signal to 7.0 ppm with an increase in relative intensity (Fig. 2(e)). Subsequent exposure to air (Fig. 2(f )) caused little change. In particular, the new signals appearing after dehydration were not removed on exposure to air. Table 2 summarizes the 13C chemical shifts observed here and reported in the literature for TEA and triethylammonium chloride.
| Species | –CH2– | CH3 |
|---|---|---|
| a This work ± 0.5 ppm.b Ref. 13. | ||
| Triethylamine (l)b | 46 | 11 |
| Triethylammonium | 46 | 8.5 |
| chloride (aq)b | ||
| AlPO4-5 dehydrated 373 K | 46.0 | 6.5 |
| AlPO4-5 dehydrated 450 K | 46.0 | 12.0, 6.5 |
| AlPO4-5 dehydrated 500 K | 46.0 | 12.0, 7.0 |
| AlPO4-5 exposed to air | 46.8 | 7.8 |
| SAPO-5 hydrated | 46.5 | 8.5 |
| SAPO-5 dehydrated 500 K | 46.0, 52.5 | 12.0, 7.0 |
| SAPO-5 exposed to air | 46.5, 52.5 | 12.0, 7.0 |
The methylene 13C signal in TEA is insensitive to protonation, but in the liquid phase the methyl signal in triethylammonium chloride is shifted to lower field by about 3 ppm relative to the free amine. The 6.5–8.5 ppm signals in AlPO4-5 and SAPO-5 are thus assigned to methyl groups in the protonated template. The relative intensities of the methyl and methylene signals from the template in AlPO4-5 and SAPO-5 will depend on the cross-polarization efficiency in the CPMAS experiment; the fact that both signals have approximately equal intensity in the as-synthesized samples indicates that the template is rigidly bound, allowing effective dipolar coupling between 13C and protons.
Fig. 3 and 4 show FTIR spectra of an individual crystal of as-synthesized AlPO4-5 measured in the vacuum cell after evacuation at room temperature, 373 and 500 K respectively. The spectra of such single crystals measured in air were dominated by bands due to adsorbed water, making identification of template bands impossible. After evacuation at room temperature, the spectrum show an intense band in the ν(OH) region at 3650 cm−1, a complex series of intense bands between 3200 and 2500 cm−1, a series of weaker bands between 2400 and 1500 cm−1 which are present under all circumstances and which are due to overtones and combinations of AlPO4 framework vibrations, some residual adsorbed water at 1645 cm−1, and several intense bands in the C–H deformation region below 1500 cm−1.
 | ||
| Fig. 3 FTIR spectra of an individual crystal of AlPO4-5 evacuated at (a) room temperature, (b) 373 K, (c) 573 K. | ||
 | ||
| Fig. 4 FTIR spectra of an individual crystal of AlPO4-5 evacuated at (a) room temperature, (b) 373 K, (c) 573 K. | ||
Evacuation at 373 K causes the 3650 cm−1 band to split into a doublet at 3662 and 3644 cm−1, removal of the adsorbed water band at 1645 cm−1, and changes in the relative intensities of the 3200–2500 cm−1 bands and the 1500–1300 cm−1 bands. Further changes in the intensities of these bands occurred on evacuation at 500 K; this process also completely removed the 3662, 3644 cm−1 doublet. Subsequent exposure to air completely reversed all of these changes. Closer examination of the C–H stretching and C–H bending regions reveals that the spectra obtained after outgassing at 373 K contain components of both the room temperature and 500 K spectra.
To further clarify the nature of the molecular vibrations responsible for the observed bands, an experiment was carried out in which an as-synthesized AlPO4-5 crystal was dehydrated by evacuation at 500 K, then exposed to D2O vapour at room temperature, followed by outgassing at 373 K to remove adsorbed D2O. Fig. 5(a) shows the spectrum obtained after adsorption of D2O into the fully dehydrated crystal and outgassing at 373 K. This shows that the 3662, 3644 cm−1 doublet originally present (Fig. 3(b)) is shifted to 2700, 2688 cm−1, and that several of the intense bands between 3200 and 2600 cm−1 are shifted to 2400–2100 cm−1, where they overlap with the AlPO4 framework bands in this region. The spectrum in the C–H deformation region after adsorption of D2O was identical to that obtained from the as-synthesized crystal (not shown).
 | ||
| Fig. 5 FTIR spectra of an individual crystal of AlPO4-5: (a), evacuated at 500 K, exposed to D2O vapour at room temperature, then evacuated at 373 K; (b), exposed briefly to air then evacuated at 373 K. | ||
The spectrum shown in Fig. 5(b) was measured after brief exposure of the crystal to air, followed by evacuation at 373 K. This shows partial reversal of the changes caused by adsorption of D2O.
Similar IR experiments have been undertaken with individual as-synthesized SAPO-5 crystals. Fig. 6 and 7 show spectra of such a crystal after evacuation at respectively room temperature, 373 and 500 K. Tables 3 and 4 summarize the vibrational frequencies of the major IR bands observed and the assignments of these to particular species, as discussed below.
 | ||
| Fig. 6 FTIR spectra of SAPO-5 crystal evacuated at (a), room temperature, (b) 373 K, (c), 500 K. | ||
 | ||
| Fig. 7 FTIR spectra of SAPO-5 crystal evacuated at (a), room temperature, (b) 373 K, (c), 500 K. | ||
| Chloride salt (s) | In AlPO4-5 | In SAPO-5 | Assignment |
|---|---|---|---|
| a Frequencies accurate to ± 2 cm−1. | |||
| 3150, 3050 | 3064 | ν(N–H) | |
| 3000 | 2999 | 3000 | ν(CH3) symmetric |
| 2976 | 2985 | 2986 | ν(CH3) symmetric |
| 2939 | 2952 | 2953 | ν(CH2) asymmetric |
| 2879 | 2891 | 2888 | ν(CH3) asymmetric |
| 2803 | 2808 | 2815 | ν(N–H···X) |
| 2757, 2739 | 2750 | 2740 | ν(N–H···X) |
| 2677, 2601, 2529, | ν(N–H···X) | ||
| 2491 | |||
| 1476, 1434 | 1474, 1460 | 1476, 1460 | δ(CH3) symmetric |
| 1398, 1384 | 1395 | 1395 | δ(CH3) |
| asymmetric | |||
| 1365, 1331 | 1360 | 1360 | δ(CH2) |
A full normal coordinate vibrational analysis of the triethylammonium cation has not been reported, to our knowledge. Information is available however about the vibrational modes in the corresponding trimethylammonium cation.15 In C3v symmetry the CH3 stretching vibrations divide into symmetric (A1 and E) and asymmetric (A1, A2, E) modes respectively (the A2 modes are IR-inactive), and the symmetric modes occur at higher frequencies than the asymmetric modes. Following Howe and Taylor,13 we assign the two highest frequency bands in the Raman spectrum of triethylammonium chloride (2997 and 2980 cm−1) to the symmetric C–H stretching vibrations of the methyl groups (Table 1). The remaining 4 bands in the C–H stretching region in the Raman spectrum of triethylammonium chloride cannot be assigned with such certainty, but we follow the usual convention of assuming that the asymmetric stretching modes of CH2 groups lie higher in frequency than the corresponding symmetric modes, and that the asymmetric CH3 modes occur at similar frequencies to those in the trimethylammonium cation. In the C–H deformation region, it is impossible to clearly separate CH3 and CH2 modes, although the higher frequency bands in this region (1444–1480) are most likely due to A1 and E modes of CH3 groups, given their similarity to those in the spectrum of the trimethylammonium cation.15Table 1 summarizes the assignments made in the Raman spectrum of triethylammonium chloride, and of the corresponding bands in the Raman spectra of the as-synthesized AlPO4-5 and SAPO-5.
The IR spectra of the as-synthesized AlPO4-5 and SAPO-5 crystals outgassed at room temperature show IR bands in the ν(C–H) and δ(C–H) regions at frequencies closely similar to those in the Raman spectra (Table 3). The IR spectra show however a number of additional features which are not present in the Raman spectra. In AlPO4-5, there is an intense band at about 3650 cm−1 which splits into a doublet at 3662 and 3644 cm−1 on outgassing at 373 K (Fig. 3). The polarized spectra in Fig. 8 (discussed further below) show that the 3650 cm−1 band in fact comprises the 3662 and 3644 cm−1 bands which are not however resolved in the unpolarized spectrum after outgassing at room temperature. In addition, intense broad bands are observed at 3150, 3052, 2810 and 2748 cm−1, which are reduced in intensity on outgassing at 373 K.
 | ||
| Fig. 8 Polarized FTIR spectra of AlPO4-5 crystal. (a), evacuated at room temperature, polarized along crystal c-axis; (b) polarized perpendicular to crystal c-axis; (c), evacuated at 500 K, polarized along crystal c-axis; (d), polarized perpendicular to crystal c-axis. | ||
The origin of these additional features becomes clear from the experiments with samples exposed to D2O (Fig. 5). After exchange with D2O, the (3662, 3644 cm−1) doublet shifts to (2700, 2688 cm−1), a frequency shift close to that calculated for free hydroxy groups. The bands at 3150, 3052, 2810 and 2748 cm−1 are completely removed from the spectrum after exchange with D2O, indicating that they are also associated with exchangeable protons, but only the two highest frequency bands can be identified in Fig. 5(a) at about 2320 and 2230 cm−1, frequency shifts expected for replacement of NH by ND. Corresponding frequency shifts would place the 2810 and 2748 cm−1 bands between 2000 and 2100 cm−1, but no such bands are seen after D2O exchange.
The explanation for these anomalies can be found in hydrogen bonding. The N–H bonds in alkylammonium salts are known to be extensively perturbed by hydrogen bonding. For example, Odinokov et al.16–18 have described an extensive study of hydrogen bonding of the triethylammonium cation in salts with a range of different anions. Hydrogen bonding causes resonance perturbation of the ν(NH) bands, resulting in frequency shifts, splitting into three or more components, and dramatic intensity enhancements (see, for example, the data for triethylammonium chloride in Table 3). These effects are attributed to Fermi resonance between the totally symmetric ν(NH) mode and the first overtone of the δ(NH) deformation mode (1420 cm−1) or the combination mode δ(NH) + ν(CN). In the case of the triethylammonium template in AlPO4-5, the ν(NH) multiplet bands occur between 3150 and 2750 cm−1, so that Fermi resonance with the overtone of the deformation mode is the most likely source of the observed multiplet structure. Odinokov et al. established correlations between the mean ν(NH) frequency and the hydrogen bond energy for triethylammonium salts in solution; the observed mean frequency for the triethylammonium cation in AlPO4-5 of ca. 2940 cm−1 is similar to that observed for the perchlorate salt in solution, corresponding to a hydrogen bond energy of about 22 kJ mol−1. On deuteration, the first overtone of the δ(NH) deformation mode will shift to below 2100 cm−1, thus reducing Fermi resonance with the ν(ND) mode, and hence removing the multiplet structure in the ν(ND) band.
The IR spectra of as-synthesized SAPO-5 are similar (although not identical to) those of AlPO4-5. The ν(CH) and δ(CH) bands in particular occur at the same frequencies, and there is a similar ν(NH) multiplet structure, although the highest frequency ν(NH) band is now only a single component. The triethylammonium cation in SAPO-5 is thus in a similar hydrogen bonding environment to that in AlPO4-5.
The most striking difference between the IR spectra of as-synthesized AlPO4-5 and SAPO-5 is the presence of an intense ν(OH) band or bands in the spectra of AlPO4-5 which are not seen in SAPO-5. Schnabel et al.9 attributed such bands to hydroxide ions present in the pores of AlPO4-5 to balance the positive charge of the triethylammonium cations. Charge compensating anions are not, in principle, required in SAPO-5, since the framework carries a negative charge due to the silicon substitution. The frequency shifts observed here on deuteration support the assignment of Schnabel et al. The close similarity in the ν(NH) spectra for AlPO4-5 and SAPO-5 indicates however that the triethylammonium cation is in both cases hydrogen bonded to oxide ions of the aluminophosphate framework. The formation of hydrogen bonded ion pairs between the cation and hydroxide anions in AlPO4-5 would be expected to perturb the ν(NH) bands considerably more than is observed, and would also have a marked influence on the ν(OH) band of the hydroxide anion. The observation of narrow ν(OH) bands at relatively high frequencies suggests that the hydroxide ions are not interacting directly with the template.
SAPO-5 dehydrated at room temperature still contains some adsorbed water, but evacuation at 373 K completely removes the ν(OH) (3600–3000 cm−1) and δ(OH) (1642 cm−1) bands of water.
The template in dehydrated AlPO4-5 and SAPO-5
Dehydration of AlPO4-5 by evacuation at 500 K causes almost complete removal of features in the IR spectrum which were assigned to protonated template. The intense bands between 3200 and 2500 cm−1 associated with hydrogen bonded NH stretching vibrations and the ν(OH) bands due to charge compensating hydroxide ions are no longer present. The ν(CH) and δ(CH) bands remaining are shifted to lower frequency relative to those of the protonated template; their frequencies are close to those of free triethylamine (Table 4). These changes suggest that dehydration at higher temperatures cause elimination of water through a reaction such as: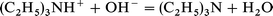
Evidence for triethylamine formation in dehydrated AlPO4-5 samples is also seen in the appearance of a signal at 12 ppm in the 13C NMR spectra, close to the chemical shift of methyl groups in liquid TEA. The relative intensity of this signal remains low compared with the methylene signal; this can be attributed to lower cross-polarization efficiency for the methyl groups in the free amine compared with the protonated template, due to motion on the NMR timescale. The presence of a residual signal due to protonated template in the NMR spectra of samples dehydrated at high temperature when no IR bands of this species remain is due to the difficulties of achieving and maintaining complete dehydration of the macroscopic sample required for the NMR experiment.
The IR spectra of individual crystals of SAPO-5 dehydrated at 500 K (Fig. 6 and 7) still show the presence of protonated template. The bands assigned to hydrogen bonded NH vibrations are still present, although reduced in relative intensity. In the δ(CH) region (Fig. 7) the 1476 and 1395 cm−1 bands due to protonated template are also still present, although the additional bands due to free triethylamine which dominate the spectrum of AlPO4-5 after high temperature dehydration (cf. Fig. 5) can also be seen in SAPO-5 to a lesser extent. In the ν(CH) region, bands of free triethylamine are clearly seen (Fig. 6(c)), although these overlap with their higher frequency counterparts due to the protonated template.
The 13C NMR spectra of SAPO-5 dehydrated at 500 K are still dominated by the signals of protonated triethylamine. The 12 ppm signal due to methyl groups in free triethylamine can be seen however as a weak shoulder in Fig. 2(e) for example (much less intense than in the corresponding spectrum of AlPO4-5). The other new (weak) signal at 52.5 ppm in dehydrated SAPO-5 is attributed to some partial decomposition product.
We conclude that in the case of SAPO-5 dehydration at 500 K produces a mixture of protonated template, some free triethylamine, and small amounts of decomposition products. The important differences between AlPO4-5 and SAPO-5 are that protonated template remains after dehydration of the SAPO-5, and that free triethylamine generated during high temperature dehydration of SAPO-5 is not re-protonated on exposure to air. The changes in the NMR spectrum of SAPO-5 are not reversed on subsequent exposure to air.
It may be asked why triethylamine (with a lower boiling point) remains in the pores of the AFI structures following elimination of water. This must be attributed to the steric restraints on desorption of the larger TEA molecule, and possibly to specific interactions between TEA and aluminium cations in the framework.
Template orientation
Polarization studies were undertaken to investigate the orientations of the template species within the AlPO4-5 channels. Fig. 8 shows spectra in the C–H stretching region of the AlPO4-5 crystal after evacuation at room temperature and at 500 K respectively with the polarization of the infrared radiation oriented either parallel or perpendicular to the crystal c-axis. The effects of polarization on the C–H deformation bands were much less pronounced, and are not shown. Corresponding spectra for SAPO-5 are shown in Fig. 9. | ||
| Fig. 9 Polarized FTIR spectra of SAPO-5 crystal. (a), evacuated at room temperature, polarized along crystal c-axis; (b) polarized perpendicular to crystal c-axis; (c), evacuated at 500 K, polarized along crystal c-axis; (d), polarized perpendicular to crystal c-axis. | ||
The most striking feature of the polarized infrared spectra measured from both AlPO4-5 and SAPO-5 crystals when fully hydrated is the strong polarization dependence of the ν(NH) bands of the protonated template. As shown in Fig. 8 and 9, these bands are most intense when the infrared polarization is oriented perpendicular to the crystal axis i.e. perpendicular to the channel direction in the AFI structure. This implies immediately that the protonated template molecules are oriented with their N–H bonds perpendicular to the channel walls. Given such a preferred orientation, we expect also significant polarization effects in the ν(CH) and δ(CH) vibrations of the protonated templates, and these are indeed evident in the spectra. Accounting for these effects is however difficult, in light of the complexity of the spectra and the extensive coupling between vibrational modes.
Simple consideration of dipole moment changes in a protonated template oriented perpendicular to the channel walls suggests that totally symmetric in phase (A1) CH3 stretching vibrations should show a maximum intensity for polarization along the C3 axis of the molecule i.e. perpendicular to the channel wall, whereas out of phase (E) modes should be more evident for polarization along the channel direction. Such considerations would suggest, for example, that the 2999 cm−1 band for the protonated template in AlPO4-5 (Fig. 3) is due to out of phase CH3 stretching modes, whereas the 2985 cm−1 band may be assigned to an in-phase CH3 mode. The 2952 cm−1 band, assigned to the asymmetric CH2 stretching mode, should show maximum intensity for polarization along the channel direction. This description is oversimplified however, since there is evidence in the polarized spectra of partially resolved additional bands, and the intensity variations with polarization of the ν(CH) bands are distorted by the corresponding changes in the underlying ν(NH) bands. For these reasons, we have not attempted a more detailed analysis of the polarization dependence.
The two ν(OH) bands associated with charge compensating hydroxide ions in hydrated AlPO4-5 show different polarization behaviour. The 3662 cm−1 band is most intense when the polarization is along the channel direction, and the 3644 cm−1 band most intense when the polarization is perpendicular to the channel direction. Both bands can be observed however at all polarization angles, suggesting that there is a distribution of orientations for both. It is clear however that these two hydroxide ions occupy different sites; this is also evident from the changes in relative intensity of the two bands on partial dehydration (Fig. 3).
Polarization effects are also seen in the ν(CH) bands of triethylamine remaining in AlPO4-5 after dehydration at 500 K (Fig. 8(c) and (d)), indicating that the free template also has a preferred orientation within the channels. The highest frequency ν(CH) band shows little polarization dependence, which is expected if this band contains both in-phase and out-of-phase CH3 symmetric stretching modes, although the intensity is slightly higher for polarization perpendicular to the channel axis. The 2935 and 2875 cm−1 bands both show maximum intensity when the polarization direction is parallel to the channel axes, which is consistent with these bands being due to asymmetric stretching modes of CH2 and CH3 groups respectively of a triethylamine molecule aligned with its C3 axis perpendicular to the channel direction. The 2804 cm−1 band assigned to CH2 symmetric stretching modes would then be expected to show maximum intensity when the polarization direction is perpendicular to the channel axis, as is observed. The polarization data thus suggest that the template retains its preferred orientation following deprotonation, consistent with the presence of a specific interaction between the lone pair on the nitrogen and aluminium ions in the AFI framework. Examination of molecular models shows that such “sideways ” orientation of TEA in the AFI pores is also sterically favoured over an orientation in which the C3 axis is parallel to the channel direction.
The polarized spectra of hydrated SAPO-5 (Fig. 9) are broadly similar to those of AlPO4-5, with the exception of the ν(OH) bands (the ν(OH) bands of residual adsorbed water in SAPO-5 show no evidence of preferred orientation, as expected). In particular, the broad intense band structure associated with hydrogen bonded N–H groups of the triethylammonium cation is much more intense when the polarization direction is perpendicular to the channel axis, as for AlPO4-5, and the intensity variations with polarization of the ν(CH) bands indicate a similar orientation of the template in SAPO-5 to that in AlPO4-5.
As discussed above, dehydration of SAPO-5 at 500 K leaves a mixture of protonated template and some free triethylamine. This is evident in the lower resolution of the ν(CH) bands in the polarized spectra shown in Fig. 9(c) and (d). The polarization effects noted in the ν(CH) bands are still present however, indicating that template orientation is retained.
Calcined samples
Calcination of AlPO4-5 and SAPO-5 in flowing oxygen at 550°C caused complete removal of all template bands from the IR spectra. The microspectra of individual crystals showed no evidence of hydroxy bands remaining in calcined AlPO4-5. In contrast, the diffuse reflectance spectra reported by Schnabel et al.9 do show the presence of hydroxy groups in calcined polycrystalline samples, possibly due to the presence of defects or impurity phases in the macroscopic samples. Calcined crystals of SAPO-5 showed the two O–H stretching bands reported previously by Schuth et al.5 3630 and 3530 cm−1. Polarization measurements on these bands confirmed the earlier report that their intensity is a maximum when the incident radiation is polarized perpendicular to the crystal c-axis.In
the case of AlPO4-5, template decomposition occurs ia dehydration
of the protonated template to form triethylamine, which
subsequently decomposes ia elimination of ethylene and
finally ammonia.9 Hydroxy groups are generated on calcination
of SAPO-5. Decomposition of the triethylammonium cation
in SAPO-5 leaves protons to charge balance the negative
charge on the framework resulting from silicon substitution.
The ν(OH) bands at 3630 and 3530 cm−1 are similar to those reported
for polycrystalline SAPO-5 samples.9,19–24 Schuth et al.5
assigned the higher frequency band to hydroxy groups in the
main channel, and the lower frequency band to hydroxy groups
in a 6-membered ring, both oriented perpendicular to the
channel axis.
Conclusions
We conclude from this study of individual crystals of AlPO4-5 and SAPO-5 that:The triethylamine template in as-synthesized AlPO4-5 is fully protonated, and hydrogen bonded to oxide ions of the framework. Charge balance is achieved through the inclusion of hydroxide ions in the channels. This finding fully supports the earlier conclusions reached from measurements on polycrystalline samples.9
Dehydration of as-synthesized AlPO4-5 by heating causes a reversible deprotonation of the template, forming triethylamine and eliminating hydroxide ions as water. The relative concentrations of protonated and free template depend on the extent of dehydration, and the process can be reversed by adding water.
Both the protonated and free triethylamine species are oriented in the channels, with the C3 axis approximately perpendicular to the channel axis.
In as-synthesized SAPO-5 the template is also fully protonated, and is charge balanced by the negative charge on the framework resulting from silicon substitution.
SAPO-5 largely retains the protonated template on heating and dehydration; some decomposition to triethylamine and lower amines cannot be reversed by adding water.
The protonated template in SAPO-5 is hydrogen bonded to the framework and oriented in a similar manner to that in AlPO4-5.
Acknowledgements
This work was supported by funding from the Australian Research Council. We thank Professor Michael Taylor, University of Auckland, for helpful discussions.References
- S. T. Wilson, B. M. Lok, C. A. Messina, T. R. Cannan and E. M. Flanigen, J. Am. Chem. Soc., 1982, 104, 1146 CrossRef CAS.
- B. M. Lok, C. A. Messina, R. L. Patton, T. R. Cannan and E. M. Flanigen, J. Am. Chem. Soc., 1984, 106, 6092 CrossRef CAS.
- M. Nowotny, J. A. Lercher and H. Kessler, Zeolites, 1991, 11, 454 CAS.
- F. Schuth, J. Phys. Chem., 1992, 96, 7493 CrossRef.
- F. Schuth, D. Demuth, B. Zibrowius, J. Kornatowski and G. Finger, J. Am. Chem. Soc., 1994, 116, 1090 CrossRef.
- F. Marlow, D. Demuth, G. Stucky and F. Schuth, J. Phys. Chem., 1995, 99, 1306 CrossRef CAS.
- K. Jackson and R. F. Howe, Stud. Surf. Sci. Catal., 1994, 83, 187 Search PubMed.
- K. Jackson and R. F. Howe, in Surface Science: Principles and Applications, ed. R. J. MacDonald, E. C. Taglauer and K. R. Wandelt, Springer, Heidelberg, 1996, p. 331. Search PubMed.
- K. H. Schnabel, G. Finger, J. Kornatowski, E. Loeffler, C. Peuker and W. Pilz, Microporous Mater., 1997, 11, 293 CrossRef CAS.
- G. Finger, J. Richter-Mendau, M. Bulow and J. Kornatowski, Zeolites, 1991, 11, 443 CAS.
- S. M. Bradley, R. F. Howe and R. A. Kydd, Magn. Reson. Chem., 1993, 31, 883 CAS.
- C. A. Fyfe, K. C. Wong-Moon and Y. Huang, Zeolites, 1996, 16, 50 CrossRef CAS.
- The Aldrich Library of 13C and 1H FTNMR Spectra, ed. C. J. Pouchert and J. Behnke, Aldrich, Milwaukee, 1980. Search PubMed.
- P. Sauvageau and C. Sandorfy, Can. J. Chem., 1960, 38, 1901 CAS.
- R. F. Howe and M. J. Taylor, Spectrochim. Acta, Part A, 1987, 43, 73 CrossRef.
- V. P. Glazunov, A. A. Mashovsky and S. E. Odinikov, J. Chem. Soc., Faraday Trans. 2, 1979, 75, 629 RSC.
- A. A. Mashkovsky, A. A. Nabiullin and S. E. Odinokov, J. Chem. Soc., Faraday Trans. 2, 1983, 79, 951 RSC.
- S. E. Odinokov, V. P. Glazunov and A. A. Nabiullin, J. Chem. Soc., Faraday Trans. 2, 1984, 80, 899 RSC.
- B. Zibrowius, E. Loeffler and M. Hunger, Zeolites, 1992, 12, 167 CrossRef CAS.
- J. A. Martens, M. Mertens, P. J. Grobet and P. A. Jacobs, Stud. Surf. Sci. Catal., 1988, 37, 97 Search PubMed.
- C. Halik, J. A. Lercher and H. Mayer, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 4457 RSC.
- U. Zscherpel, B. Staudte, E. Loeffler, C. Peuker and E. Z. Jahn, Z. Phys. Chem. (Leipzig), 1989, 270, 207 Search PubMed.
- S. G. Hegde, P. Ratnasamy, L. M. Kustov and V. B. Kazansky, Zeolites, 1988, 8, 137.
- J. A. Martens, P. J. Grobet and P. A. Jacobs, J. Catal., 1990, 126, 299 CrossRef CAS.
Footnote |
| † Present address: Max Planck Institute für Kohlenforschung, P.O. Box 10 13 53, D-45466 Mulheim, Germany. |
| This journal is © the Owner Societies 2001 |