Antibody-catalyzed hydrolysis of oligomeric esters: a model for the degradation of polymeric materials†
Oliver
Brümmer
,
Timothy Z.
Hoffman
,
Da-Wei
Chen
and
Kim D.
Janda
*
Department of Chemistry, The Scripps Research Institute and The Skaggs Institute for
Chemical Biology, 10550 North Torrey Pines Road, La Jolla, CA 92037, USA.. E-mail: kdjanda@scripps.edu
First published on 11th December 2000
Abstract
A catalytic antibody has been discovered that degrades oligomeric ester substrates.
A topic of current interest to both the chemical and global community alike concerns the development of readily degradable polymers and benign agents necessary for polymer breakdown.1,2 From an environmental perspective, microbial/enzymatic approaches for polymer breakdown are very attractive, and while a number of commercially used polymers are sensitive to biodegradation, a large variety of commonly used materials are essentially resistant.3–5 Furthermore, our current understanding is not sufficient to fully predict a degradability profile or to specifically target an enzyme/microorganism that could degrade a man-made, synthetic polymer.6
Antibodies bind biological macromolecules or haptens with high affinities and specificities.7 We and others have reported antibodies that bind phosphorus transition-state analogues can catalyze the hydrolysis of carbonate, ester, carbamate and amide substrates.8 Yet, to date there have been no reports of antibody catalysts that accept oligomeric versions of these simple substrate classes. Herein we report the first example of a catalytic antibody capable of degrading synthetic oligomeric esters.
Several tactics could be envisioned in a hapten design for an antibody-catalyzed hydrolysis of an oligomer. Among the ones we considered include: (1) a transition-state analogue used as a repeating unit, and (2) a pseudo-symmetric transition-state analog. Both approaches have merit, but to date no methodology for antibody-catalyzed oligomer degradation has been disclosed.8 We recently synthesized the new transition-state analogue phosphorodithioate hapten 1 and used this molecule attached to keyhole limpet hemocyanin (KLH) to obtain antibodies with excellent catalytic activity for the hydrolysis of carbonate 2a (kcat 2.7 min−1) and good activity to ester 2b (kcat 0.27 min−1).9 Based on the carbonate and esterolytic activities seen from antibodies derived from 1 and its pseudo-symmetric structure we reexamined this panel of antibodies for possession of ‘deoligomerase’ activity. Our objective was thus to see if an antibody could perform either a stepwise degradation from an oligomer’s terminus or mediate nonselective endo-cleavage on a starting oligomer and resulting subunit structures.
Before antibody screening could be engaged a set of oligomers that could be recognized based on 1’s haptenic structure needed to be prepared. While aromatic oligocarbonate structures were deemed to be the most highly congruent substrates for antibody library recognition we found such molecules to be unstable for screening. As a second choice, consideration was given to oligoesters. Polyesters are notoriously insoluble in both aqueous and organic solvents.2 However, we found that smaller molecular weight oligoesters could be synthesized in a controlled stepwise and highly pure manner.10 Furthermore, these oligomers displayed good stability and workable solubility parameters for the kinetic analysis of the appearance and disappearance of distinct oligomeric subunits. Based on these considerations (vide supra), and the need for maximal antibody substrate recognition, 4-hydroxy- phenylacetic acid 3 was chosen as our monomer for oligomeric synthesis.
Twenty-five monoclonal antibodies (mAbs) were screened against tetramer 4, where the term ‘tetramer’ refers to the total number of aromatic residues, that contained distinct terminal units (Fig. 1). Though 4 was only soluble up to 250 μM in aqueous buffer (50 mM PIPES, 50 mM NaCl, pH 6.7, 5% DMSO cosolvent), we were still able to conduct the assay at 500 μM of 4 under heterogeneous conditions using an orbital shaker. Three mAbs were found that cleaved the tetramer by cutting at all three ester linkages. Interestingly, none of these antibodies were noted previously to be catalysts for the hydrolysis of the simple carbonate 2a, hence they had not been previously examined as catalytic antibodies for monoester hydrolysis.9 The most impressive mAb, OB2-48F8, was studied in greater detail since it degraded 4 to the products 3, 5, 6, 7 and 8 (Fig. 1) with the best overall rate estimated semi-quantitatively from the sum of product concentrations measured by HPLC. Thereafter, the antibody was freshly prepared and extensively purified by protein G affinity chromatography followed by MonoQ ion exchange chromatography. Notably, the antibody specific activity increased upon purification, as required. Also, the Fab fragment of OB2-48F8 was prepared, purified, and displayed a similar activity compared to the whole IgG. Hence, we ruled out contamination by esterases as a source of the deoligomerization reaction.
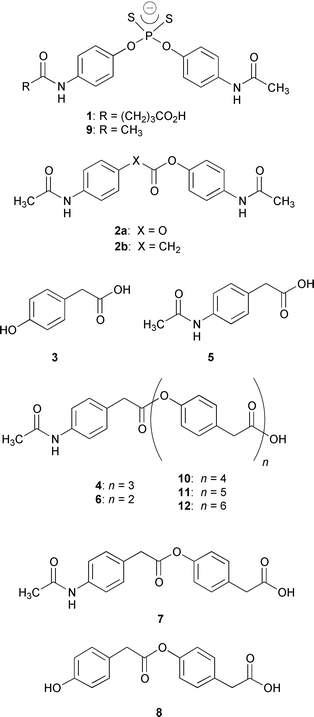 | ||
| Fig. 1 Structures of hapten 1, inhibitor 9, carbonate 2a, oligoesters 4, 6, 10–12, simple esters and products derived from hydrolysis, 2b, 3, 5, 7 and 8. | ||
Due to the solubility limitations of 4, the trimer 6 was used as a substrate to quantitatively obtain an assessment of antibody activity. The upper limit of solubility of 6 in the above buffer–cosolvent system was 750 μM and allowed a full kinetic analysis of the formation of 3 and 7 by OB2-48F8 (kcat = 2.2 × 10−2 min−1, Km = 580 μM, kcat/kuncat = 1.5 × 103). The phosphorodithioate 9 was a competitive inhibitor (Ki = 6.2 μM) for this route of trimer degradation.11,12 Concurrently, the antibody also catalyzed the cleavage of 6 to subunits 5 and 8 (kcat = 1.5 × 10−3 min−1, Km = 520 μM, kcat/kuncat = 1.2 × 102). The observed activity was interesting in the light of the haptenic structure for which conventional wisdom would predict a preferred hydrolysis to yield 5 and 8. We also tested the antibody-catalyzed hydrolysis of 2b,9 the ester most congruent to 1 and 9, as well as the cleavage of the two primary ester products 7 and 8. OB2-48F8 catalyzed the hydrolysis of 2b (kcat = 1.0 × 10−2 min−1, Km = 289 μM, kcat/kuncat = 3.3 × 102) and was competitively inhibited by 9 (Ki = 16.5 μM). The antibody also cleaved 7 (kcat = 1.7 × 10−3 min−1, Km = 695 μM, kcat/kuncat = 1.1 × 102) and 8 (kcat = 2.0 × 10−3 min−1, Km = 940 μM, kcat/kuncat = 90). Thus, mAb OB2-48F8 was capable of completely degrading 6 to its component monomeric building blocks. Unexpectedly, the trimer 6 was a better substrate than ester 2b for reasons that remain unclear. Additional studies employing the novel phosphorodithioate moiety might reveal unusual features with regard to the correlation between hapten and substrate structures.
Finally, to investigate the capabilities of OB2-48F8 for degrading larger oligomers approaching ‘real’ polymers, we synthesized polyesters 10–12 that contained additional repeating units (Fig. 1). The pentamer 10 (MW 729) was sparingly soluble (less than 30 μM) in our aqueous buffer conditions, however using a 500 μM heterogeneous mixture of 10 degradation still occurred into subunit structures (Fig. 2). As detailed in Fig. 2, there was an initial increase in the dimers 7 and 8 followed by a decrease as monomers 3 and 5 increased. This was rationalized based on the above cleavage preferences and consecutive-reaction kinetics. Over a one month period near neutral pH at ambient temperature in the presence of antibody, approximately 200 mM of 3 was formed from degraded 10. Without antibody, less than 3% occurred. In a similar fashion, OB2-48F8 also broke down polyesters 11 (MW 863) and 12 (MW 997) that were virtually insoluble under our assay conditions, yet cleavage occurred at multiple sites to yield suboligomers as primary products (data not shown). Significantly, the antibody was stable for more than a month under heterogeneous conditions while the progress of these reactions was being monitored.13 All the observations and data confirmed that the antibody performed oligomer degradations by ‘multimer’ processing using nonregioselective, kinetically biased endo-cleavage, rather than a stepwise deoligomerization through cleavage of monomers from a terminus.
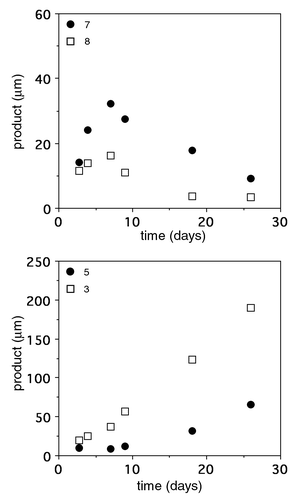 | ||
| Fig. 2 Reaction profiles showing products from the antibody-catalyzed hydrolysis of oligoester 10. Reactions were carried out in 50 mM PIPES, 50 mM NaCl, pH 6.7, 5% DMSO at 21 °C in the presence of 500 μM 10 and 20 μM OB2-48F8. The uncatalyzed reactions were carried out under the same conditions in the absence of antibody and the rates subtracted. | ||
In conclusion, a catalytic antibody has been discovered that degrades oligomeric esters. Our findings are of fundamental importance as now catalytic antibodies share another trait thought only to be associated with enzymes, the biodegradation of oligo and polymeric materials.1
Acknowledgements
We thank Dr Peter Wirsching for reviewing the manuscript. Financial support was provided by The National Institute of Health (GM 43858) and The Skaggs Institute of Chemical Biology.Notes and references
- A. C. Albertsson and S. Karlsson, Acta Polym., 1995, 46, 114 CrossRef CAS.
- M. P. Stevens, Polymer Chemistry, 2nd edition, Carl-Hanser Verlag, Munich, 1990. Search PubMed.
- U. Witt, M. Yamamoto, U. Seeliger, R.-J. Muller and V. Warzelhan, Angew. Chem., Int. Ed., 1999, 38, 1438 CrossRef CAS; S. Kobayashi, H. Uyama and T. Takamoto, Biomacromolecules, 2000, 1, 3 CrossRef CAS.
- E. C. King, A. J. Blacker and T. D. H. Bugg, Biomacromolecules, 2000, 1, 75 CrossRef CAS; K. Tabata, H. Abe and Y. Doi, Biomacromolecules, 2000, 1, 157 CrossRef CAS.
- J. J Jesudason, R. H. Marchessault and T. Saito, J. Environ. Polym. Degrad., 1993, 1, 89 Search PubMed.
- E. Rantze, I. Kleeberg, U. Witt, R.-J. Mueller and W.-D. Deckwer, Macromol. Symp., 1998, 130, 319 Search PubMed; U. Witt, R.-J. Mueller and W.-D. Deckwer, Macromol. Chem. Phys., 1996, 197, 1525 CrossRef CAS.
- H. N. Eisen, General Immunology, 3rd ed., J. B. Lippincott Company, Philadelphia, 1990. Search PubMed.
- K. D. Janda, C. G. Shelvin and C.-H. L. Lo, in Comprehensive Supramolecular Chemistry, Vol. 4: Supramolecular Reactivity and Transport: Bioorganic Systems, ed. J. L. Atwood, J. E. D. Davies, D. D. MacNicol, F. Vögtle, J.-L. Lehn, Y. Murakami, Elsevier, New York, 1996, p. 43 Search PubMed; P. Wentworth and K. D. Janda, Curr. Opin. Chem. Biol., 1998, 2, 138 Search PubMed.
- O. Brümmer, P. Wentworth, D. P. Weiner and K. D. Janda, Tetrahedron Lett., 1999, 40, 7307 CrossRef CAS.
- The synthesis of the compounds under discussion (4, 6–8 and 10–12) can be found in the supplementary information..
- Phosphorodithioate 9 is a tight-binding inhibitor of OB2-48F8. Ki determinations were therefore not made using the classical Lineweaver–Burk analysis over a range of inhibitor concentrations, since this is known to be subject to error for tight-binding inhibitors. Rather the method of Copeland et al. ( R. A. Copeland, D. Lombardo, J. Glannaras and C. P. Decicco, Bioorg. Med. Chem. Lett., 1995, 5, 1947 CrossRef CAS ) was employed, which allows for the accurate determination of the Ki values of tight-binding inhibitors..
- Symmetrical phosphorodithioate 9 was prepared starting from 4-acetamidophenol.9.
- Aliquots of sample were removed periodically and checked by ELISA for hapten binding..
Footnote |
| † Electronic supplementary information (ESI) available: experimental procedures are provided including kinetic assays, and full characterization (1H and 13C NMR, HRMS, HPLC traces) of the compounds under discussion (4, 6–8, 10–12). See http://www.rsc.org/suppdata/cc/b0/b008065i/ |
| This journal is © The Royal Society of Chemistry 2001 |