Flow injection potentiometric determination of pipazethate hydrochloride
N. T. Abdel-Ghani*, A. F. Shoukry and R. M. El Nashar
Chemistry Department, Faculty of Science, Cairo University, Giza, Egypt
First published on 15th December 2000
Abstract
New plastic membrane electrodes for pipazethate hydrochloride based on pipazethatium phosphotungstate, pipazethatium phosphomolybdate and a mixture of the two were prepared. The electrodes were fully characterized in terms of composition, life span, pH and temperature and were then applied to the potentiometric determination of the pipazethate ion in its pure state and pharmaceutical preparations under batch and flow injection conditions. The selectivity of the electrodes towards many inorganic cations, sugars and amino acids was also tested.
Introduction
Pipazethate hydrochloride (PiCl), 10H-pyrido[3,2-b][1,4]benzothiadiazine-10-carboxylic acid 2-(2-piperidinoethoxy)ethyl ester,1 is a bronchodilator that suppresses irritative and spasmodic cough by inhibiting the excitability of the cough center and the peripheral neural receptors in the respiratory passage. The response to the drug takes about 10–20 min and lasts for 4–6 h.Pipazethate has been determined using a limited number of techniques including spectrophotometry,2,3 TLC4 and HPLC.5 Although there is an increased demand for chemical surveillance in the environmental, medical and many other industries which has created the need for highly sensitive, selective, precise and inexpensive techniques, no ion-selective electrode (ISE) has been constructed for the determination of pipazethate. In this study, plastic membrane electrodes for Pi cation were constructed based on the incorporation of pipazethatium phosphotungstate (Pi-PTA), pipazethatium phosphomolybdate (Pi-PMA) or a mixture of the two (Pi-PTA/PMA) ion exchanger(s) in poly(vinyl chloride) (PVC) membranes plasticized with dioctylphthalate (DOP). The electrodes were fully characterized under batch conditions and then used to determine the drug both in batch and with a flow injection technique, which is considered a very efficient way of improving the performance characteristics of ISEs for various reasons, including the following: (i) the permanent liquid stream has a conditioning effect on the sensor membrane, leading to better sensitivity and stability and increased reproducibility of the e.m.f. readings; (ii) the liquid junction and streaming potentials are stable; (iii) the way in which the sample is presented to the ISE is more closely defined and reproducible under flow-through conditions than in static (batch) measurements; (iv) the streaming of solution is beneficial in that it reduces the diffusion layer thickness and hence shortens the response time; and (v) the sample is not influenced by the electrode itself as any release from the membrane (e.g., dissolution) is transported downstream.6 The selectivity of the electrodes was also tested in batch and FIA procedures and compared with each other.
Experimental
Reagents
All reagents used for the preparation of solutions were of analytical-reagent grade. Doubly distilled water was used for preparing solutions and as a flow stream in FIA measurements. The carrier and reagent solutions were de-gassed by means of vacuum suction. Sample solutions used for injections were freshly prepared prior to measurements. Pure grade pipazethate hydrochloride and its pharmaceutical preparations (Selgon, tablets 20 mg per tablet and drops 40 mg ml−1) were provided by the Egyptian International Pharmaceutical Industries Company (EIPICO), (10th of Ramadan City, Egypt).Apparatus
Potentiometric measurements in the batch mode were carried out with a Schott-Gerate (Hofheim, Germany) CG 820 pH-meter and pMX microprocessor pH/ion meter (WTW, Weilheim, Germany). A Techne (Cambridge, UK) Model C-100 circulator thermostat was used to control the temperature of the test solutions. A WTW packed saturated calomel electrode (SCE) was used as an external reference electrode. The electrochemical system may be represented as follows:| Ag|AgCl|filling solution|membrane|testsolution∥KCl salt bridge∥SCE | (1) |
A wall-jet cell, providing a low dead volume, fast response, good wash characteristics, ease of construction and compatibility with electrodes of various shapes and sizes, was used in flow measurements, where a Perspex cup with axially positioned inlet polypropylene tubing was mounted at the sensing surface of the electrode body. The optimized distance between the nozzle and the sensing surface of the electrode was 5 mm; this provides the minimum thickness of the diffusion layer and consequently a fast response.7 The ISE with flow cup, reference electrode (SCE) and the outlet tube were placed in a beaker, where the level of solution was kept 1 cm above the electrode surface.
Preparation of the ion exchangers
The ion exchangers, pipazethatium phosphotungstate (Pi3-PTA) (buff powder) and pipazethatium phosphomolybdate (Pi3-PMA) (yellow powder), were prepared by the addition of 150 ml of 10−2 mol l−1 PiCl solution to 50 ml of 10−2 mol l−1 of each of phosphotungstic acid or phosphomolybdic acid, respectively. The precipitates were filtered, washed thoroughly with distilled water until chloride free and air-dried. The chemical composition of the precipitates was identified and confirmed by elemental analysis (C, H, N, S, P and metal); the results are given in Table 1.| Pi-PTA | Pi-PMA | |||
|---|---|---|---|---|
| Element | Found | Calculated | Found | Calculated |
| C | 18.49 | 18.53 | 26.67 | 26.70 |
| H | 1.98 | 1.91 | 2.79 | 2.75 |
| N | 3.11 | 3.09 | 4.46 | 4.45 |
| S | 2.37 | 2.35 | 3.41 | 3.39 |
| P | 0.72 | 0.76 | 1.12 | 1.09 |
| Metal | 54.21 | 54.13 | 40.71 | 40.69 |
Preparation of the electrodes
The electrodes were constructed as described previously.8 The membrane composition was studied by varying the percentages (w/w) of the ion exchanger(s), PVC and DOP until optimum composition that exhibits the best performance characteristics is reached. The membranes were prepared by dissolving the required amounts of PVC, ion-exchanger(s) and the plasticizer (DOP) in a 5.0 cm diameter Petri dish containing 10.0 ml of tetrahydrofuran (THF) (the total weight of ingredients was 0.35 g), the components being well mixed to ensure homogeneity of the membranes. The Petri dish was then covered with a filter-paper and left to dry in air. To obtain uniform membrane thickness, the amount of THF was kept constant and its evaporation was fixed for 24 h.An 8 mm diameter disk was cut out from the prepared membrane and glued using PVC–THF paste to the polished end of a plastic cap attached to a glass tube. The electrode body was filled with a solution that was 10−2 mol l−1 with respect to both NaCl and PiCl and preconditioned by soaking in 10−3 mol l−1 PiCl solution.
Results and discussion
Optimization of the ISE response in batch conditions
The best performances were obtained by using compositions containing 5.0% Pi-PTA, 47.5% PVC and 47.5% DOP, 7.0% Pi-PMA, 46.5% PVC and 46.5% DOP or 3.0% Pi-PTA, 3.0% Pi-PMA, 47.0% PVC and 47.0% DOP, resulting in slopes of 59.0, 57.5 and 58.7 mV per concentration decade after minimum pre-soak times of 0.5, 1 and 0.5 h for Pi-PTA, Pi-PMA and Pi-PTA/PMA, respectively. The usable concentration range for the three electrodes was 6.3 × 10−5–0.1 mol l−1. The above optimum compositions were used to prepare membrane electrodes for all further investigations.
It is noteworthy that the life span of the membrane containing mixed ion exchangers is much longer than those containing an individual ion exchanger. This is most probably due to some sort of physical interaction of the two ion exchangers within the PVC network. It may also be correlated with the diffusion and partition coefficients of both the ion exchangers and the plasticizer.10,11 Naturally, it was noted that in all cases, electrodes which had been kept dry in a closed vessel and stored in a refrigerator showed nearly constant slope values and the same response properties extending to several months. Hence it is recommended that unused electrodes be kept dry in closed vessels in a refrigerator in order to extend their life spans substantially.
The regeneration of the electrodes was tried simply by reforming the ion exchanger(s) on the external gel layer of the membrane,12 and this was successfully achieved by soaking the exhausted electrode for 24 h in a solution that was 10−2 mol l−1 in PTA, PMA or in both PTA and PMA followed by soaking for 1, 2 and 2 h in 10−2 mol l−1 PiCl solution for Pi-PTA, Pi-PMA and Pi-PTA/PMA, respectively. The slopes of the exhausted electrodes increased from 46.5, 46.0 and 45.0 to 55.0, 53.3 and 55.0 for Pi-PTA, Pi-PMA and Pi-PTA/PMA electrodes, respectively, as a result of the regeneration process.
It was found that the life span of the regenerated electrode was limited to 2 h of continuous soaking owing to the ease of leaching of the lipophilic salts from the gel layer at the electrode surface compared with those which are attached homogeneously to the PVC network through the solvent mediator.
| Electrode | Temperature/°C | Slope/mV per decade | E°/mVa |
|---|---|---|---|
| a E° is the standard electrode potential versus the normal hydrogen electrode (NHE). | |||
| Pi-PTA | 25 | 59.0 | 53 |
| 30 | 59.5 | 58 | |
| 40 | 60.8 | 64 | |
| 50 | 62.5 | 85 | |
| 60 | 64.8 | 100 | |
| 70 | 67.5 | 114 | |
| Pi-PMA | 25 | 57.5 | 37 |
| 30 | 59.0 | 43 | |
| 40 | 61.5 | 55 | |
| 50 | 62.8 | 60 | |
| 60 | 64.8 | 70 | |
| 70 | 67.0 | 84 | |
| Pi-PTA/PMA | 25 | 58.7 | 88 |
| 30 | 60.5 | 92 | |
| 40 | 62.5 | 96 | |
| 50 | 64.5 | 105 | |
| 60 | 66.3 | 124 | |
| 70 | 68.3 | 138 | |
For the determination of the isothermal coefficients (dE°/dt) of the electrodes, the standard electrode potentials (E°) versus the normal hydrogen electrode (NHE) at the different temperatures were obtained from the calibration graphs as the intercepts at pPi = 0 (after subtracting the values of the standard electrode potential of the SCE at these temperatures) and plotted versust − 25, where t is the temperature of the test solution in °C. A straight line plot is obtained according to the following equation:13
| E° = E°(25) + (dE°/dt) (t − 25) | (2) |
Optimization of FIA response
The flow injection measurements were carried out in a two-line configuration. The sample was injected into a distilled water stream, which was then merged with another stream of distilled water. In both lines the same tubing size was used, offering the same flow rate. The connector of the two streams was connected to the detector by a 50 cm tube of 0.4 mm id. Fig. 1 shows the configuration of the system used in measurements. The dispersion coefficient was found to be 1.7, i.e., limited dispersion that aids optimum sensitivity and fast response of the electrodes.14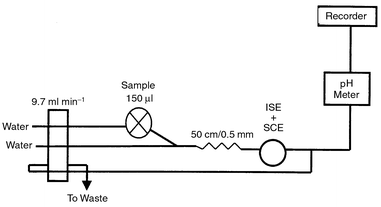 | ||
| Fig. 1 Schematic diagram of the flow injection system used in measurements. | ||
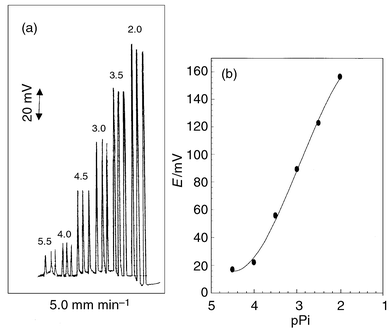 | ||
| Fig. 2 Recordings (a) and calibration graph (b) for Pi-PTA/PMA electrode under optimum FIA conditions. | ||
Electrode response in FIA
In potentiometric detection, the electrode potential depends on the activity of the main ion sensed. This is considered to be a principle advantage of this detection method; also in flow measurements the dependence is semi-logarithmic over a wide analyte activity range according to the Nickolsky–Eisenman equation. However, the main unfavorable feature of this detection method is the slow response of electrode potential to concentration change and this is pronounced when low concentrations are measured and depends on the state of the membrane surface at the interface with the measured solution.17,18 This slow response is a fairly good reason for the super-Nernstian sensitivities obtained in FIA measurements using the investigated electrodes at different flow rates. An increase in the slope of the calibration plots in FIA was observed compared with batch measurements, where potential is measured under conditions very close to the equilibrium at the membrane–solution interface.19The slopes of the calibration graphs ranged from 64.5 to 72.5, from 64.0 to 71.5 and from 65.5 to 73.7 compared with 59.0, 57.5 and 58.7 mV per concentration decade for Pi-PTA, Pi-PMA and Pi-PTA/PMA, respectively. The variation of the calibration graph slope with flow rate is shown in Fig. 3.
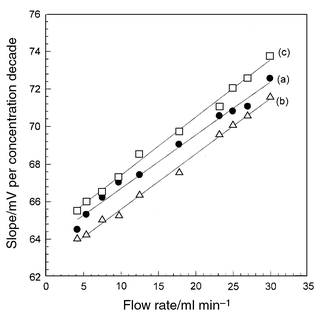 | ||
| Fig. 3 Variation of calibration graph slope for Pi-PTA (a), Pi-PMA (b) and Pi-PTA/PMA (c) electrodes with flow rate. | ||
The usable concentration range of the electrodes in FIA is 6.3 × 10−4–1.0 × 10−2 mol l−1, which is a slightly lower than that in the batch mode, mainly owing to the difference in the dispersion coefficients in the two techniques.
Effect of pH
The effect of the pH of the test solution on the electrode potentials was studied in batch and FIA measurements. In batch measurements, the variation in potential with pH change was followed by the addition of small volumes of HCl and NaOH (0.1–1.0 mol l−1), whereas in FIA, a series of solutions of 10−2 mol l−1 PiCl concentration and with pH ranging from 1.0 to 12.0 were injected into the flow stream and the peak heights, representing variation of potential response with pH, were measured.It is evident that the electrodes do not respond to pH changes in the range 2.0–9.0 in both batch and FIA conditions. Nevertheless, at pH values lower than these values, the potential decreases gradually, which can be related to interference of hydronium ion, while the decrease that takes place at pH >9.0 can most probably be attributed to the formation of the free pipazethate base in the solution or the ionization of the hydroxyl group in such high alkaline pH media, leading to a decrease in the concentration of pipazethatium cation.
Selectivity of the electrodes
It was shown earlier for solid state membrane electrodes that the apparent selectivity coefficient measured under transient flow injection conditions may differ significantly from that measured under batch conditions.20–23 This is interpreted by the difference in the time of interaction of interferents with the membrane surface. This difference increases with increase in the interaction of the interferent with the membrane in comparison with the main sensed ion, and also the interference process is highly dependent on the rate of diffusion and the exchange reaction of the interfering ion.24 Therefore, in FIA measurements, where the sample remains in contact with the electrode for a short period of time , the apparent selectivity should be different from that found in batch conditions.The influence of some inorganic cations, sugars and amino acids on the
Pi electrodes was investigated. Under FIA conditions, the values of
selectivity coefficients were calculated based on potential values measured
at the top of peaks for the same concentrations of the drug and the
interferent, whereas under batch conditions the separate solution method
was applied and a high concentration of the interferent ion was used
(10−2 mol l−1) to ensure that there is no
interference if lower concentrations than this are present. Although there
are many precautions that must be taken into consideration when using this
method in the determination of the selectivity coefficients, especially in
the case of solutions of differently charged ions, it is still the simplest
way to evaluate the degree of interference that might be taking place and
is used to perform measurements in important biological samples such as
blood25 as it is simple and easy to
perform. In this work, the matched potential and the mixed solution
methods, which are time consuming owing to the need to prepare many
solutions and to perform many steps, were used only for determining the
degree of tolerance towards sugars and amino acids and as a confirmation
when −log 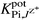 was <3.5.
was <3.5.
The selectivity coefficients 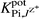 of the electrodes (Table 3), reflect a very high selectivity of the
investigated electrodes for the pipazethatium cation. The mechanism of
selectivity is mainly based on the stereospecificity and electrostatic
environment and it is dependent on how much fitting is present between the
locations of the lipophilicity sites in the two competing species in the
bathing solution side and those present in the receptor of the ion
exchanger.26 The inorganic cations do not
interfere because of differences in ionic size, mobility and permeability.
Also, the smaller the energy of hydration of the cation, the greater is the
response of the membrane. The electrodes exhibit good tolerance towards
sugars, amino acids and urea.
of the electrodes (Table 3), reflect a very high selectivity of the
investigated electrodes for the pipazethatium cation. The mechanism of
selectivity is mainly based on the stereospecificity and electrostatic
environment and it is dependent on how much fitting is present between the
locations of the lipophilicity sites in the two competing species in the
bathing solution side and those present in the receptor of the ion
exchanger.26 The inorganic cations do not
interfere because of differences in ionic size, mobility and permeability.
Also, the smaller the energy of hydration of the cation, the greater is the
response of the membrane. The electrodes exhibit good tolerance towards
sugars, amino acids and urea.
| −log
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Pi-PTA | Pi-PMA | Pi-PTA/PMA | |||||||
| Batch | Batch | Batch | |||||||
| Interferent | SSMa | MSMb | FIA | SSMa | MSMb | FIA | SSMa | MSMb | FIA |
| a SSM: separate solution method. b MSM: mixed (matched) solution method. | |||||||||
| Na+ | 2.87 | 3.01 | 2.92 | 3.12 | 3.24 | 3.18 | 3.25 | 3.28 | 3.37 |
| K+ | 3.10 | 3.37 | 0.98 | 3.26 | 3.42 | 1.10 | 3.42 | 3.51 | 1.21 |
| Li+ | 3.19 | 3.34 | 3.23 | 3.31 | 3.45 | 3.39 | 3.38 | 3.48 | 3.41 |
| NH• | 3.25 | 3.37 | 3.34 | 3.33 | 3.54 | 3.43 | 3.42 | 3.57 | 3.46 |
| Mg2+ | 3.78 | — | 3.89 | 3.86 | — | 4.08 | 3.74 | — | 3.83 |
| Ca2+ | 3.91 | — | 4.17 | 3.76 | — | 3.98 | 3.89 | — | 4.16 |
| Sr2+ | 4.23 | — | 4.35 | 4.37 | — | 4.52 | 4.34 | — | 4.49 |
| Ba2+ | 4.56 | — | 4.64 | 4.47 | — | 4.58 | 4.65 | —- | 4.76 |
| Mn2+ | 4.63 | — | 4.79 | 4.74 | — | 4.94 | 4.85 | — | 4.98 |
| Co2+ | 4.55 | — | 4.71 | 4.58 | — | 4.65 | 4.87 | — | 4.97 |
| Ni2+ | 4.46 | — | 4.54 | 4.59 | — | 4.67 | 4.64 | — | 4.73 |
| Cu2+ | 4.87 | — | 4.95 | 4.76 | — | 4.87 | 4.88 | — | 5.00 |
| Zn2+ | 5.12 | — | 5.30 | 5.18 | — | 5.27 | 5.29 | — | 5.36 |
| Pb2+ | 5.43 | — | 5.52 | 5.37 | — | 5.45 | 5.49 | — | 5.59 |
| Cd2+ | 5.23 | — | 5.35 | 5.29 | — | 5.34 | 5.33 | — | 5.41 |
| Cr3+ | 5.48 | — | 5.58 | 5.52 | — | 5.67 | 5.61 | — | 5.70 |
| Fe3+ | 5.55 | — | 5.69 | 5.37 | — | 5.50 | 5.49 | — | 5.56 |
| Al3+ | 5.64 | — | 5.75 | 5.58 | — | 5.68 | 5.67 | — | 5.75 |
| Glycine | — | 4.52 | 4.57 | — | 4.43 | 4.58 | — | 4.52 | 4.59 |
| L-Threonine | — | 4.38 | 4.46 | — | 4.27 | 4.51 | — | 4.38 | 4.52 |
| Alanine | — | 4.76 | 4.98 | — | 4.65 | 4.82 | — | 4.73 | 4.86 |
| Arginine | — | 5.24 | 5.39 | — | 5.01 | 5.28 | — | 5.18 | 5.31 |
| Glucose | — | 5.57 | 5.71 | — | 5.25 | 5.41 | — | 5.34 | 5.50 |
| Maltose | — | 5.62 | 5.80 | — | 5.32 | 5.52 | — | 5.29 | 5.47 |
| Lactose | — | 4.97 | 5.21 | — | 5.10 | 5.34 | — | 5.14 | 5.35 |
| Urea | — | 4.13 | 4.42 | — | 4.20 | 4.37 | — | 4.31 | 4.53 |

Comparing the selectivity coefficients obtained for the investigated electrodes under both batch and FIA conditions (Table 3), it is clear that in most cases, the electrodes were more selective in FIA than in batch measurement with the exception of K+, where the selectivity was significantly lower. This may be due to its smaller atomic size, so that it is more easily adsorbed on the electrode surface and it is more difficult to remove it by the flowing stream. In the case of other monovalent ions, the selectivity coefficients obtained were smaller to some extent (less than 0.5 decade) than the values obtained under batch conditions.
Analytical applications
PiCl was determined potentiometrically using the investigated electrodes under batch conditions by both the interpolation and standard additions methods.27 In the latter, small portions (0.1 ml) of standard 0.1 mol l−1 PiCl solution were added to 50 ml of water containing 1, 2, 10, 20, 110 or 220 mg of pure compound or their equivalents from pharmaceutical preparations. The change in millivolt readings was recorded after each addition and used to calculate the concentration of the PiCl sample solution.For sampling of tablets (Selgon, 20 mg per tablet), 20 tablets were ground together and appropriate weights of each were taken as samples. The required amount (1.0–200.0 mg) was dissolved in 0.01 mol l−1 HCl (about 0.1 ml mg−1 of pipazethate tablets) and diluted to 50 ml with distilled water. For drops (Selgon, 40 mg ml−1), the contents of three bottles were mixed and volumes equivalent to the required weights were taken and then used for the determination of the drug content by the standard additions method.
The results of the standard additions method given in Table 4 are in good agreement with those obtained from the official method1 (the latter is based on measuring the absorbance of the drug solution in 0.1 mol l−1 HCl at 252 nm). The mean recovery of the amounts taken (1.0–220.0 mg) ranged from 97.0 to 101.1% with RSD = 0.17–0.42%.
| Pi-PTA | Pi-PMA | Pi-PTA/PMA | |||||
|---|---|---|---|---|---|---|---|
| Sample | Takena/mg | Recovery (%) | RSDb (%) | Recovery (%) | RSDb (%) | Recovery (%) | RSDb (%) |
| a Taken mg per 50 ml. b Five determinations. | |||||||
| Pure solutions | 1.0 | 98.3 | 0.26 | 97.9 | 0.16 | 98.4 | 0.29 |
| 2.00 | 98.7 | 0.22 | 98.3 | 0.34 | 98.9 | 0.24 | |
| 10.0 | 99.5 | 0.19 | 99.0 | 0.27 | 99.8 | 0.31 | |
| 20.0 | 100.5 | 0.17 | 99.8 | 0.35 | 100.1 | 0.20 | |
| 110.0 | 100.3 | 0.32 | 99.8 | 0.27 | 100.5 | 0.26 | |
| 220.0 | 100.8 | 0.21 | 100.4 | 0.24 | 100.3 | 0.33 | |
| Tablets (20 mg per tablet) | 1.0 | 97.0 | 0.31 | 97.4 | 0.30 | 97.8 | 0.25 |
| 2.00 | 97.9 | 0.24 | 98.6 | 0.32 | 98.3 | 0.34 | |
| 10.0 | 98.5 | 0.19 | 99.5 | 0.35 | 98.9 | 0.28 | |
| 20.0 | 99.6 | 0.24 | 100.3 | 0.23 | 99.7 | 0.26 | |
| 110.0 | 100.2 | 0.27 | 100.5 | 0.26 | 99.9 | 0.37 | |
| 220.0 | 100.5 | 0.18 | 100.6 | 0.20 | 100.5 | 0.24 | |
| Drops (40 mg ml−1) | 1.0 | 98.5 | 0.18 | 98.9 | 0.34 | 99.0 | 0.25 |
| 2.00 | 99.2 | 0.25 | 99.5 | 0.25 | 99.8 | 0.34 | |
| 10.0 | 100.5 | 0.31 | 100.4 | 0.31 | 100.2 | 0.25 | |
| 20.0 | 100.8 | 0.42 | 100.3 | 0.40 | 100.6 | 0.31 | |
| 110.0 | 101.0 | 0.35 | 100.5 | 0.36 | 100.5 | 0.20 | |
| 220.0 | 100.6 | 0.38 | 101.1 | 0.28 | 100.9 | 0.37 | |
Under FIA conditions, a series of solutions of different concentrations were prepared from the tablets and drops and the peak heights were measured at three flow rates (4.15, 9.70 and 23.25 ml min−1), then compared with those obtained from injecting a standard solution of the same concentration prepared from pure pipazethate hydrochloride. From the results obtained (Table 5), it clear that the flow rates did not affect the recovery values except for a rate of 4.15 ml min−1, where the electrodes needed a longer time to attain the baseline, and the recoveries were lower than those obtained under batch conditions. The mean recoveries for the amounts taken (1.0–220 mg) ranged from 95.3 to 99.0, from 97.9 to 101.3 and from 97.5 to 101.6% with RSDs of 0.21–0.52, 0.19–0.50 and 0.19–0.56% for flow rates of 4.15, 98.7 and 23.25 ml min−1, respectively.
| Pi-PTA | Pi-PMA | Pi-PTA/PMA | |||||
|---|---|---|---|---|---|---|---|
| Flow rate/ ml min−1 | Takena/mg | Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | Recovery (%) | RSD (%) |
| a (a) Tablets, (b) drops. | |||||||
| 4.15 | 1.0 (a) | 95.5 | 0.24 | 95.3 | 0.21 | 95.6 | 0.26 |
| (b) | 95.8 | 0.35 | 95.4 | 0.23 | 95.4 | 0.29 | |
| 2.0 (a) | 96.0 | 0.27 | 95.8 | 0.25 | 96.1 | 0.24 | |
| (b) | 95.9 | 0.28 | 96.2 | 0.27 | 96.5 | 0.28 | |
| 10.0 (a) | 96.4 | 0.34 | 96.2 | 0.34 | 96.7 | 0.31 | |
| (b) | 96.3 | 0.41 | 96.0 | 0.35 | 96.5 | 0.36 | |
| 20.0 (a) | 96.2 | 0.42 | 96.7 | 0.41 | 97.0 | 0.37 | |
| (b) | 96.5 | 0.36 | 96.4 | 0.37 | 96.8 | 0.38 | |
| 110.0 (a) | 97.8 | 0.48 | 97.3 | 0.52 | 97.5 | 0.39 | |
| (b) | 98.2 | 0.52 | 97.8 | 0.49 | 98.1 | 0.43 | |
| 220.0 (a) | 98.6 | 0.21 | 98.6 | 0.28 | 98.3 | 0.33 | |
| (b) | 99.0 | 0.28 | 98.5 | 0.34 | 98.7 | 0.39 | |
| 9.7 | 1.0 (a) | 98.9 | 0.20 | 97.9 | 0.21 | 98.7 | 0.41 |
| (b) | 98.7 | 0.25 | 98.4 | 0.27 | 98.5 | 0.47 | |
| 2.0 (a) | 99.6 | 0.24 | 98.6 | 0.24 | 98.9 | 0.37 | |
| (b) | 99.8 | 0.27 | 99.0 | 0.26 | 99.2 | 0.34 | |
| 10.0 (a) | 100.1 | 0.19 | 99.2 | 0.31 | 99.6 | 0.35 | |
| (b) | 99.8 | 0.23 | 99.7 | 0.37 | 99.7 | 0.42 | |
| 20.0 (a) | 100.2 | 0.31 | 100.0 | 0.41 | 100.1 | 0.43 | |
| (b) | 100.5 | 0.35 | 100.4 | 0.43 | 99.8 | 0.46 | |
| 110.0 (a) | 100.1 | 0.23 | 100.2 | 0.33 | 100.1 | 0.28 | |
| (b) | 100.3 | 0.25 | 100.8 | 0.35 | 100.5 | 0.27 | |
| 220.0 (a) | 100.6 | 0.30 | 100.7 | 0.27 | 100.9 | 0.50 | |
| (b) | 100.9 | 0.33 | 101.2 | 0.34 | 101.3 | 0.48 | |
| 23.25 | 1.0 (a) | 98.2 | 0.28 | 97.5 | 0.41 | 97.9 | 0.34 |
| (b) | 98.8 | 0.32 | 97.9 | 0.39 | 98.5 | 0.37 | |
| 2.0 (a) | 99.1 | 0.41 | 98.2 | 0.26 | 98.6 | 0.28 | |
| (b) | 99.3 | 0.46 | 98.6 | 0.24 | 98.3 | 0.27 | |
| 10.0 (a) | 99.8 | 0.52 | 99.4 | 0.35 | 99.6 | 0.19 | |
| (b) | 99.6 | 0.54 | 99.7 | 0.37 | 99.9 | 0.23 | |
| 20.0 (a) | 100.0 | 0.41 | 100.5 | 0.46 | 100.4 | 0.43 | |
| (b) | 100.2 | 0.46 | 100.1 | 0.50 | 100.0 | 0.39 | |
| 110.0 (a) | 100.6 | 0.28 | 100.8 | 0.33 | 100.2 | 0.56 | |
| (b) | 100.8 | 0.32 | 101.0 | 0.38 | 100.6 | 0.53 | |
| 220.0 (a) | 100.9 | 0.25 | 101.3 | 0.43 | 100.8 | 0.27 | |
| (b) | 101.5 | 0.27 | 101.6 | 0.49 | 101.3 | 0.30 | |
The results were subjected to linear regression analysis (found values versus taken), using the computer program Excel-5, in order to establish whether the investigated electrodes exhibit any fixed or proportional bias. The slopes and intercepts of the regression lines did not differ significantly from the ideal values, revealing the absence of a systematic error during the measurements within the investigated concentration range. Also, they were compared with the results obtained from the official method,1 which is based on measuring the absorbance of the drug solution in 0.1 mol l−1 HCl at 252 nm, by applying F- and t-tests.28 The values obtained (Table 6), show that the present methods are of comparable precision to the official method and there is no significant difference between the mean values obtained by the two methods.
| Proposed method | |||||||
|---|---|---|---|---|---|---|---|
| Pi-PTA | Pi-PMA | Pi-PTA/PMA | |||||
| Official method | Batch | FIA | Batch | FIA | Batch | FIA | |
| a One-tailed critical F value. | |||||||
| Pure solutions— | |||||||
| x ± SE | 100.08 ± 0.120 | 99.98 ± 0.141 | 100.1 ± 0.141 | 99.95 ± 0.524 | 100.12 ± 0.211 | 100.03 ± 0.871 | 100.07 ± 0.621 |
| Probability | >0.01 | >0.01 | >0.01 | >0.01 | >0.01 | >0.01 | |
| Relative error (%) | 0.02 | 0.10 | 0.05 | 0.12 | 0.03 | 0.07 | |
| F3,3 value (9.27)a | 4.14 | 5.28 | 3.54 | 4.62 | 3.17 | 3.97 | |
| Selgon tablets (20 mg per tablet)— | |||||||
| x ± SE | 98.70 ± 0.524 | 98.95 ± 0.075 | 98.80 ± 0.411 | 99.48 ± 0.055 | 98.65 ± 0.072 | 99.46 ± 0.053 | 98.96 ± 0.097 |
| Probability | <0.05 | >0.01 | >0.01 | >0.01 | >0.01 | >0.01 | |
| Relative error (%) | 1.05 | 1.2 | 0.52 | 1.35 | 0.54 | 1.04 | |
| F3,3 value (9.27)a | 5.34 | 4.63 | 6.42 | 5.86 | 6.34 | 5 .47 | |
| Selgon drops (40 mg per ml−1)— | |||||||
| x ± SE | 100.5 ± 0.613 | 100.10 ± 0.084 | 100.38 ± 0.653 | 100.11 ± 0.125 | 100.28 ± 0.634 | 100.16 ± 0.726 | 100.28 ± 0.682 |
| Probability | <0.05 | >0.01 | <0.05 | >0.01 | >0.01 | >0.01 | |
| Relative error (%) | 0.10 | 0.38 | 0.11 | 0.28 | 0.16 | 0.28 | |
| F3,3 value (9.27)a | 3.87 | 5.52 | 5.48 | 5.40 | 5.38 | 5.617 | |
Conclusion
The application of the proposed method to the determination of pipazethate hydrochloride in its pure solutions and pharmaceutical preparations is characterized by a high degree of precision and accuracy when compared with the official method. Comparing the results obtained from Pi-PTA, Pi-PMA and Pi-PTA/PMA electrodes under batch and FIA conditions, it clear that the application of the electrodes in FIA increased considerably the selectivity of the electrodes towards the pipazethate ion and shortened the time needed for the determination of the drug in its pure state or in its pharmaceutical preparations. It is noteworthy also that the life span of the electrode containing the mixed ion exchanger is much longer than that of an electrode containing a single ion exchanger.Acknowledgement
The authors express their thanks to Professor Dr. Marek Trojanowicz, Head of the Laboratory of Flow Analysis and Chromatography, Chemistry Department, Warsaw University, Poland, for his help in the construction of the FIA system and for his valuable guiding comments.References
- Martindale, The Extra Pharmacopoeia, ed. J. E. F. Reynolds, Pharmaceutical Press, London, 30th edn., 1993p. 8140. Search PubMed.
- S. S. Zarapker, R. V. Rele and V. M. Shah, Indian Drugs, 1987, 24, 445 Search PubMed.
- S. S. Zarapker, R. V. Rele and V. J. Doshi, Indian Drugs, 1987, 24, 560 Search PubMed.
- H. D. Revanasiddappa and P. G. Ramappa, Indian Drugs, 1995, 32, 73 Search PubMed.
- H. D. Revanasiddappa and P. G. Ramappa, Indian Drugs, 1995, 32, 534 Search PubMed.
- W. Frenzel, Analyst, 1988, 113, 1039 RSC.
- H. Gunasingham and B. Fleet, Anal. Chem., 1983, 55, 1409 CAS.
- A. Craggs, G. J. Moody and J. D. R. Thomas, J. Chem. Educ., 1974, 51, 541 Search PubMed.
- E. Linder, K. Toth and E. Pungor, Dynamic Characteristics of Ion-Selective Electrodes, CRC Press, Boca Raton, FL, 1988. Search PubMed.
- V. Oesh and W. Simon, Anal. Chem., 1980, 52, 692 CrossRef.
- R. D. Armstrong and G. Horvai, Electrochim. Acta, 1990, 35, 1 CrossRef CAS.
- A. F. Shoukry, S. S. Badway and Y. M. Issa, Anal. Chem., 1987, 59, 1078 CrossRef CAS.
- V. Oesch, D. Ammann and W. Simon, Clin. Chem., 1986, 32, 1148 Search PubMed.
- M. Trojanowicz and W. Matuszewski, Anal. Chim. Acta, 1982, 138, 71 CrossRef CAS.
- X. Yang, D. B. Hibbert and P. W. Alexander, Anal. Chim. Acta, 1998, 372, 387 CrossRef CAS.
- W. Frenzel and P. Bratter, Anal. Chim. Acta, 1986, 185, 187 CrossRef.
- W. E. Morf, E. Linder and W. Simon, Anal. Chem., 1975, 47, 1596 CrossRef CAS.
- A. Shatkay, Anal. Chem., 1976, 48, 1039 CrossRef CAS.
- L. Ilcheva, M. Trojanowicz and T. K. Vel Krawczyk, Fresenius’ Z. Anal. Chem., 1987, 328, 27 CAS.
- M. Trojanowicz and W. Matuszewski, Anal. Chim. Acta, 1983, 151, 77 CrossRef CAS.
- L. Ilcheva and K. Cammann, Fresenius’ Z. Anal. Chem., 1985, 322, 322.
- D. E. Davey, D. E. Mulcahy, G. R. O’Connel and R. S. Smart, Electroanalysis, 1995, 7, 461 CAS.
- L. K. Shpigun, O. V. Basanova and Yu. A. Zolotov, Sens. Actuators B, 1992, 10, 15 CrossRef.
- A. Hulanicki and A. Lewenstam, Anal. Chem., 1981, 53, 1401 CrossRef CAS.
- L. L. Antropov, Theoretical Electrochemistry, Mir, Moscow, 1977. Search PubMed.
- N. T. Abdel Ghani, M. S. Rizk and R. M. El-Nashar, Analyst, 2000, 125, 1129 RSC.
- E. Baumann, Anal. Chim. Acta, 1986, 42, 127 CrossRef.
- J. C. Miller and J. N. Miller, Statistics for Analytical Chemistry, Ellis Horwood, Chichester, 1984. Search PubMed.
| This journal is © The Royal Society of Chemistry 2001 |