Shape-shifting in contorted dibenzotetrathienocoronenes†
Chien-Yang
Chiu
,
Bumjung
Kim
,
Alon A.
Gorodetsky
,
Wesley
Sattler
,
Sujun
Wei
,
Aaron
Sattler
,
Michael
Steigerwald
and
Colin
Nuckolls
*
Department of Chemistry, Columbia University, New York, New York 10027, USA. E-mail: cn37@columbia.edu
First published on 18th May 2011
Abstract
We detail a general method for the synthesis of dibenzotetrathienocoronenes and elucidate their solid state structures in crystals and co-crystals. The contorted dibenzotetrathienocoronene (c-DBTTC) is a tetrathiophene-fused version of the previously studied contorted hexabenzocoronenes (c-HBC). The synthesis detailed here is simple and provides easy access to this important class of materials. We have found that these materials display molecular flexibility and tunable supramolecular self-assembly properties in the solid state by shifting molecular conformations between two different conformations. Depending on the conditions under which a c-DBTTC-containing material crystallizes, the c-DBTTC adopts either the “up-down” or the “butterfly” conformation. When grown from the vapor phase, crystals of the unsubstituted c-DBTTC show the molecule only in the up-down conformation, and it packs into dense crystals containing columnar arrays with close intracolumnar packing. The packing is controlled by the inherent molecular corrugation of the three-dimensional core and sulfur–sulfur interactions. When grown as co-crystals with electron acceptors from solution, the butyl-substituted c-DBTTC either adopts the butterfly conformation when the electron acceptor is small enough to be completely enveloped (TCNQ) or the up-down conformation when the electron acceptor is relatively large (C60). When grown from organic solvent crystals of the butyl-substituted c-DBTTC contain molecules of the solvent as the only guest, and we observe both conformations of the c-DBTTC. Controlling the supramolecular structure is the key to developing these materials for electronic applications.
Introduction
This study describes a simple, high-yield route to a wide variety of contorted dibenzotetrathienocoronenes (e.g., c-DBTTC, 1) and their unusual ability to modify their shapes in response to their environment. Although thiophene-fused acenes have been extensively studied and used as high performance organic field effect transistors (OFET)1–4 and organic photovoltaics (OPV),5thiophene-fused coronene materials have caught sparse attention.6 The c-DBTTC is a tetrathiophene-fused version of the previously studied contorted hexabenzocoronenes (c-HBC, 2). c-HBCs consist of six 4-carbon annulations at the periphery of the coronene core, leading to steric congestion between adjacent benzo groups and yielding a non-planar motif7 that can be further elaborated into molecular bowls8,9 and hemispheres.10c-HBCs consist of three fused interpenetrating pentacenes and possess a doubly concave shape. This type of distorted geometry enables alkoxy-substituted c-HBCs to be miscible with common solvents while maintaining effective π–π stacking interactions in the solid state.7 Substituted c-HBCs self-assemble from solution into nanowires7,11 that have carrier mobilities as high as ∼ 1 cm2 V−1 s (using carbon nanotube as the nanocontacts12) and exhibit one-dimensional photoconductivity.13,14 Furthermore, unsubstituted c-HBC forms a shape-complementary complex with C60, affording an intimate donor/acceptor interface and good performance in solar cells.15 In our recent disclosure of the first member of the c-DBTTC family, we reported that photovoltaic cells based on this electron donor display power conversion efficiencies of >2%.16 Here we detail the general method for synthesis of this important class of materials and elucidate their structures in the solid state in crystals and co-crystals. At the molecular level the partial replacement of the benzo groups in the c-HBCs with thiophenes gives a more flexible structure that allows the molecule to shift its shape and sample multiple conformations. The c-DBTTCs adopt unusual supramolecular ensembles whose structure is dictated by the molecular conformation, π-stacking, and sulfur–sulfur interactions. Such versatility will allow these electronically active organic components to adapt their structures to accommodate the demands of multi-dimensional and hierarchically assembled devices.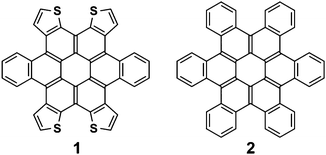
Results and discussion
Design and synthesis
c-DBTTC molecules comprise two anthradithiophene units and one pentacene unit that share the central six-membered ring. We draw inspiration here from the anthradithiophenes, where the thiophene moieties are utilized to enhance π–π stacking and crystallinity.2,5 However, in all previous syntheses of anthradithiophenes (other than the one example of c-DBTTC16), they have been prepared as inseparable mixtures of cis and trans isomers (with respect to the locations of the sulfur atoms).2,5,17 We detail here the synthesis of anthradithiophenes in which this regiochemistry is controlled. In principle, this modification is important because the location of the sulfur atoms is critical in the self-assembly and electronic behavior of functional thiophene derivatives.2,17Our synthesis of this family of polycyclic aromatic hydrocarbons is much more efficient than our original method. That previously reported 11-step synthesis of c-HBC utilized two Barton-Kellogg olefination reactions to furnish the c-HBC 2 in 16% total yield from the 6,13-pentacenequinone precursor (Scheme 1A).7,8 Not only is this previous process laborious, but it also cannot be applied to many substrates due to the inability to prepare the requisite diaryl diazomethanes. These limitations have forced us to develop the alternative synthetic strategy detailed here (shown in Scheme 1B). The key step in this reaction sequence is the preparation of 1,1,8,8-tetrabromobisolefin 3 from 6,13-pentacenequinonevia the Ramirez reaction using triphenylphosphine and carbon tetrabromide.18,19 Subsequently, the Suzuki-Miyaura coupling reaction between 3 and phenyl boronic acid yields the bisolefin skeleton.20,21 The fully cyclized product (c-HBC) is in turn furnished via a Katz-modified Mallory photocyclization in quantitative yield.7,22 The overall yield for this 3-step synthesis is greater than 80%.
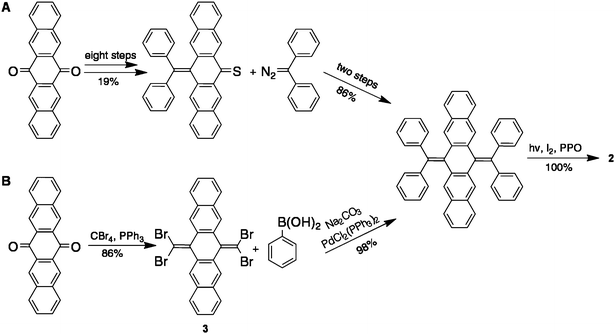 | ||
| Scheme 1 Comparison of (A) Barton-Kellogg reaction and (B) Ramirez reaction pathways for the synthesis of 2. | ||
Application of this synthetic pathway for the synthesis of c-DBTTC derivatives (Scheme 2) is not only high-yielding (Table 1) but also modular and flexible because of the wide range of 5-substituted-2-thiopheneboronic acids and esters (e.g., 4a–g) that are either commercially available or easily prepared through a borylation of the corresponding thiophenes.23 We are able to readily incorporate not only alkyl and perfluoroalkyl chains but also a variety of functional groups, larger pi-systems, and other heterocycles on the periphery of the c-DBTTC framework. Table 1 summarizes the c-DBTTC derivatives and the corresponding yields.
 | ||
| Scheme 2 Synthetic pathway to c-DBTTC derivatives 1a–g. key: (a) 3, Na2CO3, PdCl2(PPh3)2, THF/H2O, 70 °C. (b) Propylene oxide, I2, hν. | ||
| Boronic Ester | Bisolefin | c-DBTTC |
|---|---|---|
| 4a | 3a (98%) | 1a (92%) |
| 4b (63%) | 3b (87%) | 1b (89%) |
| 4c (88%) | 3c (98%) | 1c (91%) |
| 4d (86%) | 3d (93%) | 1d (86%) |
| 4e (78%) | 3e (73%) | 1e (92%) |
| 4f (56%) | 3f (56%) | 1f (86%) |
| 4g | 3g (53%) | 1g (89%) |
Structure of c-DBTTC
We investigated the differences between the derivatives using UV-vis absorbance spectroscopy (Fig. 1), cyclic voltammetry, and theoretical calculations. The newfound ability to vary the substituents grants some control over the HOMO–LUMO gap (summarized in Table 2). We see that replacement of alkyl chains (1c) with perfluoroalkyl chains (1f) causes a blue shift in UV-vis absorbance. When we append four more thiophene rings into the polyaromatic system (1g), we decrease the HOMO/LUMO gap and red-shift the UV-vis absorbance. Oxidation of the peripheral thiophene rings of 1c into sulfones (1h)24 also reduces the energy gap and lowers both HOMO and LUMO energies. The UV-vis absorbance of 1h extends out past 500 nm.| DBTTC | λ max (nm, eV) | E g,optical a (eV) | E HOMO, Echem (eV) | E LUMO, Echem (eV) | E g, Echem (eV) | E HOMO,DFT b (eV) | E LUMO,DFT b (eV) | E g,DFT b (eV) |
|---|---|---|---|---|---|---|---|---|
| a Optical HOMO–LUMO gaps determined from the onset of lowest-energy visible absorption band. The onset is defined as the intersection between the baseline and a tangent line that touches the point of inflection.25 b The energies were calculated with the up-down conformation of c-DBTTC. c The compound is too insoluble in regular solvent for measurement. | ||||||||
| 1c | 371(3.3) | 2.50 | −5.1 | −2.3 | 2.8 | −5.1 | −1.9 | 3.2 |
| 1f c | 351(3.5) | 2.44 | — | — | — | −6.0 | −2.8 | 3.2 |
| 1g | 392(3.1) | 2.21 | −4.9 | −2.6 | 2.3 | −5.2 | −2.3 | 2.9 |
| 1h c | 400(3.1) | 2.13 | — | — | — | −6.3 | −3.5 | 2.8 |
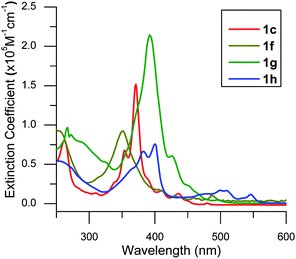 | ||
| Fig. 1 UV-visible absorption spectra of c-DBTTC derivatives (c = 1 × 10−5 M in CH2Cl2). | ||
We further explored the molecular structure of c-DBTTC with DFT modeling (Fig. 2). These calculations reveal that c-DBTTC is flatter than its c-HBC cousin (1) due to reduced steric interactions among the smaller thiophene rings at the periphery. Moreover, the DFT calculations show that the c-DBTTC can adopt at least three distinct but energetically similar conformations. The first two of these predicted conformations are isoenergetic, with the circumferential rings adopting an up-down-up-down-up-down [hereafter referred to as up-down (UD)] conformation and an up-down-down-up-down-down [hereafter referred to as the butterfly (BF)] conformation. (Fig. 2A, B). In the butterfly conformer, the pentacene subunit forms the body of the butterfly while two anthradithiophenes form the wings. The third molecular conformation is predicted to be ∼4 kcal mol−1 higher in energy, with the benzo groups adopting an up-down-down-twist-up-down (U-D-D-T-U-D) arrangement (Fig. 2C).
 | ||
| Fig. 2 Side and top views of three DFT-optimized conformations for c-DBTTC 1a. Hydrogen atoms have been deleted to clarify the view. Black = carbon, yellow = sulfur. | ||
We grew crystals of the c-DBTTC derivatives to investigate which conformation was preferred in the solid-state. We obtained crystals of the simplest derivative 1a from the gas phase using horizontal physical vapor transport crystallization.26 In this crystal, 1a adopts only the up-down (UD) conformation, similar to those of the c-HBCs, (Fig. 3); the splay angle between two proximal thiophenes is ∼16° and that between thiophene and benzene is ∼34°. As predicted from the DFT modeling the overall molecule is flattened relative to the c-HBC (2) due to the reduced steric interactions at the periphery of the molecule.
 | ||
| Fig. 3 Structure of 1a grown from horizontal vapor phase transport. (A) Eclipsed interlayer packing with atoms perfectly overlapped with each other in distorted hexagonal columnar arrangement. (B) Intimate intermolecular sulfur–sulfur interaction with sulfur–sulfur distance 3.6 Å. (C) Side view of columnar packing with interlayer distance 3.3 Å and sulfur–sulfur distance between layers 3.8 Å. | ||
These crystals of 1a exhibit an unusual packing structure, however. The atoms of one of the molecules in a stack nearly eclipsed perfectly with the next nearest neighbors in the molecular stack (Fig. 3A). Desiraju and Gavezzotti have divided crystal structures of polyaromatic hydrocarbons into four main classes: herringbone, sandwich-herringbone, γ−structure and β−structure.27,28 The packing that we see in these crystals of c-DBTTC 1a does not belong to any of these types. These molecular stacks are arranged into a distorted hexagonal arrangement of columns in the solid state. We posit that the glue that holds the molecules together and enforces the UD conformer of 1a is the contact between the sulfur atoms.29,30 Within a stack the sulfur–sulfur distance is 3.8 Å (Fig. 3B) and between adjacent columns it is as small as 3.6 Å (Fig. 3C). However, if the structure were solely the result of sulfur–sulfur interactions, we would not see the perfectly parallel columnar packing.31 The other factor dictating such columnar packing is the molecular corrugation and three dimensional structure of the c-DBTTC molecules. Within these columns, the interlayer distance between the adjacent thiophenes (Fig. 3B) is very short (∼3.3 Å).32,33
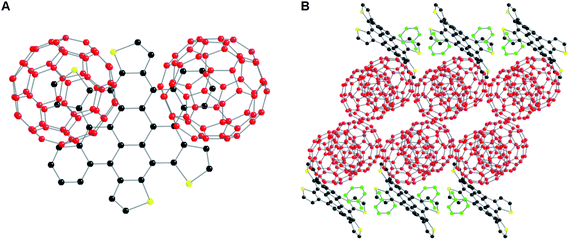
c-DBTTC, like other members of this contorted PAH family, is electron-rich, so to see whether the UD conformation dominated in other crystal forms as well, we grew crystals of the c-DBTTC in the presence of electron acceptors such as tetracyano-p-quinodimethane (TCNQ) and C60, expecting that donor–acceptor co-crystals would form.34 (Previously, we observed that the c-DBTTC is a p-type semiconducting material,16 and therefore we expect it to form complexes with these n-type semiconductors.) The co-crystals with TCNQ and C60 are shown in Fig. 4 and Fig. 5, respectively. We found exclusively the BF conformer in the co-crystal of 1b with TCNQ and exclusively the UD conformer in the co-crystal of 1b with C60.
 | ||
| Fig. 4 Crystal structure of c-DBTTC 1b with TCNQ. (A) Top view of this complex. One TCNQ molecule sits on the top of one thiophene ring of DBTTC molecule; (B) Columnar structure forms along (010) direction. Distance between c-DBTTC layer and TCNQ layer is 3.2 Å and DBTTC-DBTTC interlayer distance is 4.5 Å. (C) Viewing along (100) direction reveals that TCNQ-directed structure comprises c-DBTTC and TCNQ sheets. Blue dashed rectangle shows the unit cell in this co-crystal. Hydrogen atoms and butyl side chains have been removed in Fig. 4A and 4C for clarity. Black = carbon, yellow = sulfur, blue = nitrogen. | ||
 | ||
| Fig. 5 Crystal structure of 1b with C60 (red carbon atoms). (A) Top view of this complex. Two C60 molecules sit on the top of one benzo group and one thiophene; (B) A unit cell viewed from (010) direction shows the sandwich structure. Toluene molecules are shown in green color. Butyl groups and hydrogen atoms have been removed for clarity. | ||
The co-crystals of 1b and TCNQ were grown by slow evaporation of the solvent (p-xylene) from a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio of 1b and TCNQ to form black crystals. The stoichiometry of the molecular components in the co-crystal is 1:1. The TCNQ molecule sits over one of the thiophene rings of 1b (Fig. 4A). The organic complex forms an offset columnar structure with all of the c-DBTTC molecules adopting the butterfly conformation and enveloping the TCNQ (Fig. 4B). When viewed along (100) direction, the crystal features alternating layers of c-DBTTCs and TCNQs forming a sandwich structure with c-DBTTC layers as the bread (Fig. 4C), in which the c-DBTTC-TCNQ interplane distance is ∼3.2 Å. This distance is shorter than the DBTTC-DBTTC interplane distance of ∼4.5 Å. The presence of the TCNQ molecules forces the c-DBTTC molecules to adopt only one conformation and further directs them into the ordered sandwich-like structure, presumably through a charge transfer interaction.
:2 molar ratio of 1b and TCNQ to form black crystals. The stoichiometry of the molecular components in the co-crystal is 1:1. The TCNQ molecule sits over one of the thiophene rings of 1b (Fig. 4A). The organic complex forms an offset columnar structure with all of the c-DBTTC molecules adopting the butterfly conformation and enveloping the TCNQ (Fig. 4B). When viewed along (100) direction, the crystal features alternating layers of c-DBTTCs and TCNQs forming a sandwich structure with c-DBTTC layers as the bread (Fig. 4C), in which the c-DBTTC-TCNQ interplane distance is ∼3.2 Å. This distance is shorter than the DBTTC-DBTTC interplane distance of ∼4.5 Å. The presence of the TCNQ molecules forces the c-DBTTC molecules to adopt only one conformation and further directs them into the ordered sandwich-like structure, presumably through a charge transfer interaction.
The co-crystal of 1b and C60 grows by slow diffusion of methanol into a toluene solution containing an equimolar mixture of 1b and C60 (Fig. 5). Despite the 1:1 stoichiometry in solution, the co-crystals have a 1:2 ratio of 1b to C60. Each DBTTC molecule is associated with two C60s, one C60 sits on top of a benzo group and the other C60 sits on a thienyl ring (Fig. 5A). Co-crystals of c-HBC show a similar packing with the fullerene nested in the concave face of the contorted aromatic in a ball-and-socket motif (Fig. 5B).15 Furthermore, the co-crystal in Fig. 5 may elucidate the molecular-level origin of the electronic interaction found in the interdigitated films of c-DBTTC and C60 in photovoltaic devices.16
With solvent as the only guest in the co-crystal of 1b, the molecule adopts both the up-down and butterfly forms (Fig. 6A–E). It is remarkable that both of the conformers are found within the same crystal. The presence of two different conformations from polycyclic aromatics within a single crystal is rare.35 In this structure, the c-DBTTC 1b shifts its shape to accommodate the solvent molecules. There is no significant sulfur–sulfur interaction within this structure as they are overwhelmed by the presence of the alkyl sidechains and the interactions with the solvent molecules.
 | ||
| Fig. 6 Crystal structures of 1b grown from toluene/hexane with two marked splay angles between aromatic rings. (A) Side view and (B) top view of “up-down” conformation; (C) side view and (D) top view of “butterfly” conformation. The orientation relative to the central ring for each peripheral ring is labeled; (E) Columnar packing of 4b including one toluene molecule. The column is composed of a repeating unit of two up-down (UD) and four butterfly (BF) conformations in the order of UD-BF-BF-UD-BF-BF. Butyl chains and hydrogen atoms have been removed to clarify the view. Black = carbon, yellow = sulfur. | ||
In the up-down conformation (Fig. 6A and 6B), the three interpenetrating subunits of the c-DBTTC adopt an armchair motif that is similar to the previously reported c-HBC derivatives.7 In the UD conformation, the splay angle is ∼31° between adjacent thiophenes and benzenes, but between adjacent thiophenes this angle is ∼17° due to a reduction in steric interactions at the periphery of the molecule. In addition, the ∼14.4° bend of the central pentacene moiety is different from the ∼12.0° bend of the anthradithiophene moiety, with both of these angles smaller than the ∼20° bend in previously reported c-HBC. Overall, relative to c-HBC, the up-down conformation is flattened. In the butterfly conformation (Fig. 6C), the pentacene and anthradithiophene subunits are now bent in the opposite directions but at angles similar to the ones in the up-down conformation. Here, the two adjacent thiophenes are nearly coplanar while the splay angle is ∼35° between the sterically encumbered thiophene and benzene moieties. These two different conformers of c-DBTTC then form together like a tessellation of tiles (Fig. 6E) into a column-like supramolecular structure consisting two up-down and four butterfly conformations. The void space between columns is filled with ordered toluene molecules (Fig. 6E). The presence of solvent molecules distorts the alignment of the molecular columns. The c-DBTTC is able to shift its shape and adopt multiple conformations to accommodate the complex packing arrangement of UD-BF-BF-UD-BF-BF.
Conclusion
We have synthesized a new class of contorted heteroaromatic molecules, the dibenzotetrathienocoronenes (c-DBTTCs). Our general synthetic procedure is high yield (usually > 80%), requires only three steps under mild conditions and easily accommodates a high degree of functionality. We have found that these materials display molecular flexibility and tunable supramolecular self-assembly properties in the solid state by shifting molecular conformations between the up-down and butterfly conformations. The unsubstituted c-DBTTC, 1a, adopts solely the up-down conformation and packs into dense crystals containing columnar arrays with close intracolumnar packing. The packing in 1a is influenced by sulfur–sulfur interactions and the inherent molecular corrugation of the three-dimensional c-DBTTC core. In co-crystals with electron acceptors, the butyl-DBTTC (1b) either adopts the butterfly conformation when the electron acceptor is small enough to be completely enveloped (TCNQ) or the up-down conformation when the electron acceptor is relatively large (2 molecules of C60). In co-crystals of the butyl-DBTTC (1b), with only solvent as a guest, we observe both conformations.We have shown previously9 that the structurally complementary association of electron-donors with electron-acceptors can yield binary materials that are useful in organic photovoltaics. The new class of structurally adaptive (shape-shifting) contorted coronenes that we report here holds great potential for creating geometrically complementary interfaces and nanostructured films for applications in organic transistors and organic solar cells.
Acknowledgements
Portions of this work were also supported by the US Department of Energy, Office of Basic Energy Sciences as part of Re-Defining Photovoltaic Efficiency through Molecule-Scale Control, an Energy Frontier Research Center (DE-SC0001085) and is based in part upon work supported by the National Science Foundation under Grant No. (CHE0936923). The National Science Foundation (CHE-0619638) is thanked for acquisition of an X-ray diffractometer. This work was also supported by the Center on Functional Engineered Nano Architectonics (FENA) under award number 2009-NT-2048, subaward: UCLA 0160 SMB 959.References
- J. G. Laquindanum, H. E. Katz and A. J. Lovinger, J. Am. Chem. Soc., 1998, 120, 664–672 CrossRef CAS.
- J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS.
- O. D. Jurchescu, S. Subramanian, R. J. Kline, S. D. Hudson, J. E. Anthony, T. N. Jackson and D. J. Gundlach, Chem. Mater., 2008, 20, 6733–6737 CrossRef.
- P. Gao, D. Beckmann, H. N. Tsao, X. L. Feng, V. Enkelmann, M. Baumgarten, W. Pisula and K. Mullen, Adv. Mater., 2009, 21, 213 CrossRef CAS.
- M. T. Lloyd, A. C. Mayer, S. Subramanian, D. A. Mourey, D. J. Herman, A. V. Bapat, J. E. Anthony and G. G. Malliaras, J. Am. Chem. Soc., 2007, 129, 9144–9149 CrossRef.
- Z. T. Li, L. J. Zhi, N. T. Lucas and Z. H. Wang, Tetrahedron, 2009, 65, 3417–3424 CrossRef CAS.
- S. X. Xiao, M. Myers, Q. Miao, S. Sanaur, K. L. Pang, M. L. Steigerwald and C. Nuckolls, Angew. Chem., Int. Ed., 2005, 44, 7390–7394 CrossRef CAS.
- K. N. Plunkett, K. Godula, C. Nuckolls, N. Tremblay, A. C. Whalley and S. X. Xiao, Org. Lett., 2009, 11, 2225–2228 CrossRef CAS.
- A. C. Whalley, K. N. Plunkett, A. A. Gorodetsky, C. L. Schenck, C. Y. Chiu, M. L. Steigerwald and C. Nuckolls, Chem. Sci., 2011, 2, 132–135 RSC.
- K. T. Rim, M. Siaj, S. X. Xiao, M. Myers, V. D. Carpentier, L. Liu, C. C. Su, M. L. Steigerwald, M. S. Hybertsen, P. H. McBreen, G. W. Flynn and C. Nuckolls, Angew. Chem., Int. Ed., 2007, 46, 7891–7895 CrossRef CAS.
- S. X. Xiao, J. Y. Tang, T. Beetz, X. F. Guo, N. Tremblay, T. Siegrist, Y. M. Zhu, M. Steigerwald and C. Nuckolls, J. Am. Chem. Soc., 2006, 128, 10700–10701 CrossRef CAS.
- X. F. Guo, M. Myers, S. X. Xiao, M. Lefenfeld, R. Steiner, G. S. Tulevski, J. Y. Tang, J. Baumert, F. Leibfarth, J. T. Yardley, M. L. Steigerwald, P. Kim and C. Nuckolls, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11452–11456 CrossRef CAS.
- X. F. Guo, S. X. Xiao, M. Myers, Q. Miao, M. L. Steigerwald and C. Nuckolls, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 691–696 CrossRef CAS.
- Y. S. Cohen, S. X. Xiao, M. L. Steigerwald, C. Nuckolls and C. R. Kagan, Nano Lett., 2006, 6, 2838–2841 CrossRef CAS.
- N. J. Tremblay, A. A. Gorodetsky, M. P. Cox, T. Schiros, B. Kim, R. Steiner, Z. Bullard, A. Sattler, W. Y. So, Y. Itoh, M. F. Toney, H. Ogasawara, A. P. Ramirez, I. Kymissis, M. L. Steigerwald and C. Nuckolls, ChemPhysChem, 2010, 11, 799–803 CrossRef CAS.
- A. A. Gorodetsky, C. Y. Chiu, T. Schiros, M. Palma, M. Cox, J. Zhang, W. Sattler, I. Kymissis, M. Steigerwald and C. Nuckolls, Angew. Chem., Int. Ed., 2010, 49, 7909–7912 CrossRef CAS.
- A. Mishra, C. Q. Ma and P. Bauerle, Chem. Rev., 2009, 109, 1141–1276 CrossRef CAS.
- R. Neidlein and M. Winter, Synthesis, 1998, 1362–1366 CrossRef CAS.
- F. Ramirez, N. Mckelvie and N. B. Desai, J. Am. Chem. Soc., 1962, 84, 1745 CrossRef CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- A. Bauer, M. W. Miller, S. F. Vice and S. W. McCombie, Synlett, 2001, 254–256 CAS.
- L. B. Liu, B. W. Yang, T. J. Katz and M. K. Poindexter, J. Org. Chem., 1991, 56, 3769–3775 CrossRef CAS.
- T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura, N. R. Anastasi and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 390–391 CrossRef CAS.
- 1h was prepared in quantitative yield from the reaction of 1c and mCPBA in CH2Cl2. Please see Supplementary Information† for reaction details.
- I. Kaur, W. L. Jia, R. P. Kopreski, S. Selvarasah, M. R. Dokmeci, C. Pramanik, N. E. McGruer and G. P. Miller, J. Am. Chem. Soc., 2008, 130, 16274–16286 CrossRef CAS.
- R. A. Laudise, C. Kloc, P. G. Simpkins and T. Siegrist, J. Cryst. Growth, 1998, 187, 449–454 CrossRef CAS.
- G. R. Desiraju and A. Gavezzotti, Acta Crystallogr., Sect. B: Struct. Sci., 1989, 45, 473–482 CrossRef.
- M. D. Watson, A. Fechtenkotter and K. Mullen, Chem. Rev., 2001, 101, 1267–1300 CrossRef CAS.
- Y. Mazaki and K. Kobayashi, J. Chem. Soc., Perkin Trans. 2, 1992, 761–764 RSC.
- W. Jiang, D. J. Hardy, J. C. Phillips, A. D. MacKerell, K. Schulten and B. Roux, J. Phys. Chem. Lett., 2011, 2, 87–92 Search PubMed.
- K. Y. Chernichenko, V. V. Sumerin, R. V. Shpanchenko, E. S. Balenkova and V. G. Nenajdenko, Angew. Chem., Int. Ed., 2006, 45, 7367–7370 CrossRef CAS.
- L. Q. Li, Q. X. Tang, H. X. Li, X. D. Yang, W. P. Hu, Y. B. Song, Z. G. Shuai, W. Xu, Y. Q. Liu and D. B. Zhu, Adv. Mater., 2007, 19, 2613 CrossRef CAS.
- V. Coropceanu, J. Cornil, D. A. da Silva, Y. Olivier, R. Silbey and J. L. Bredas, Chem. Rev., 2007, 107, 926–952 CrossRef CAS.
- E. Fischer, J. Larsen, J. B. Christensen, M. Fourmigue, H. G. Madsen and N. Harrit, J. Org. Chem., 1996, 61, 6997–7005 CrossRef CAS.
- M. A. Heinrich, J. Pflaum, A. K. Tripathi, W. Frey, M. L. Steigerwald and T. Siegrist, J. Phys. Chem. C, 2007, 111, 18878–18881 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The online supporting information contains details of the synthesis, 1H and 13C NMR, IR spectra, cyclic voltammetry, DFT calculations (including the PDB files), and crystallographic information files. CCDC reference numbers 817721–817724. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00156f |
| This journal is © The Royal Society of Chemistry 2011 |