Binding, release, and functionalization of CO2 at a nucleophilic oxo anion complex of titanium†
Jared S.
Silvia
and
Christopher C.
Cummins
*
Department of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA, USA. E-mail: ccummins@mit.edu; Fax: +1 617 253 7670; Tel: +1 617 253 5332
First published on 26th May 2011
Abstract
The titanium oxo anion complex [(Et2O)2Li][OTi(N[tBu]3,5-Me2C6H3)3] ([(Et2O)2Li][1]) reacts with CO2 in diethyl ether to form the carbonate complex ([Li][O2COTi(N[tBu]3,5-Me2C6H3)3])6 ([Li][2]). The solid-state structure of complex [Li][2] is a hexamer with a hexagonal prismatic core comprised of six lithium cations bridged by the carbonate functionality. In the monomeric subunits of [Li][2], the carbonate ligand is bound κ1- to the titanium metal center and pseudo κ2- to the lithium countercation. The hexameric structure persists in benzene solutions as determined by 1H DOSY NMR techniques. The binding of CO2 in complex [Li][2] is reversible and can be effected by the introduction of the lithium sequestration reagent 12-crown-4 to diethyl ether solutions of [Li][2]. Complex [Li][2] is readily functionalized with Me3SiOS(O)2CF3 to yield the silyl carbonate complex Me3SiOC(O)OTi(N[tBu]3,5-Me2C6H3)3 (3), the solid-state structure of which is presented. Functionalization with pivaloyl chloride results in the rapid loss of CO2 and formation of the pivalate complex tBuC(O)OTi(N[tBu]3,5-Me2C6H3)3 (4).
Introduction
The reversible reaction of CO2 with metal oxides has been an area of investigation by chemists for over a century.1 Such research has received renewed interest in recent years for possible application in carbon capture and sequestration strategies.2 Furthermore, metal oxides containing lithium have been shown to be very effective at absorbing CO2,3 with lithium silicates and zirconates having received considerable attention for their ability to bind CO2 reversibly at elevated temperatures.4 In addition to sorption studies, the activation of CO2 with photoactive metal oxides such as titania to give reduced carbon fragments has been a growing area of research.5 In most cases, characterization of the CO2 bound species is limited to IR spectroscopy,6thermogravimetric analysis, and powder X-ray diffraction studies,4 thus limiting a detailed understanding of the bonding and structures in these systems. The development of a homogeneous system that may mimic the interaction of CO2 with charged metal oxides and could be studied using a greater variety of spectroscopic and structural techniques would complement the existing body of work and provide a greater understanding of the processes at play.Presented here are the results of studies focused on the reactivity of CO2 with the terminal oxide anion complex [(Et2O)2Li][OTi(N[tBu]Ar)3] ([(Et2O)2Li][1], Ar = 3,5-Me2C6H3). Generated from the deprotonation of a titanium(IV) formate complex with concomitant generation of CO,7 complex [(Et2O)2Li][1] is one of the few examples of an anionic terminal oxo complex of titanium.8,9 In addition to the clear parallels with CO2 absorption on metal oxides, inspiration for studying the reaction of CO2 with [(Et2O)2Li][1] came from discoveries within our group. Previous studies involving the reactivity of CO2 with the terminal nitride anion complexes [Na][NV(N[tBu]Ar)3] and [Na][NNb(N[tBu]Ar)3] had shown that CO2 binding to the terminal nitride ligand was rapid and irreversible, and in the case of niobium, activated the CO2 for eventual deoxygenation and conversion to CO.10,11 Given the electronic similarities between [(Et2O)2Li][1] and the terminal nitride anions, it was of interest to compare the binding of CO2 to [(Et2O)2Li][1] with these closely related systems, and perhaps discover a new system that would be capable of mediating the conversion of CO2 to CO.
We report herein that CO2 reacts readily with [(Et2O)2Li][1] in diethyl ether to yield the carbonate complex ([Li][O2COTi(N[tBu]Ar)3])6 ([Li][2]). The solid-state structure of [Li][2] was determined using single-crystal X-ray diffraction methods. Although stable under vacuum when dissolved in diethyl ether or in the solid state, complex [Li][2] readily liberates CO2 when dissolved in the presence of the lithium sequestration reagent 12-crown-4. Furthermore, complex [Li][2] readily reacts with electrophiles resulting in simple functionalization or CO2 extrusion.
Results and discussion
Synthesis of the titanium carbonate complex [Li][2]
Treatment of complex [(Et2O)2Li][1] in diethyl ether with one equivalent of CO2 produced a gradual colour change from pale yellow to bright orange over the course of minutes.‡ Following removal of solvent, the product was isolated as a bright yellow powder in 69% yield upon washing the residual solids with n-hexane (Scheme 1). The solids have minimal solubility in n-hexane and n-pentane, but are sufficiently soluble in benzene to allow for NMR spectroscopic characterization. The 1H NMR spectrum indicates that the product is isolated free of ethereal solvents in contrast with complex [(Et2O)2Li][1]. The IR spectrum of the material contains a very broad absorbance at 1590 cm−1, overlapping with the stretching modes of the aryl groups on the anilide ligands and precluding definitive assignment of a carbonate moiety. Further confirmation for the uptake of CO2 by complex [(Et2O)2Li][1] was achieved through a 13C labelling study. After treatment of complex [(Et2O)2Li][1] with 13CO2, the 13C NMR spectrum of the reaction mixture contains an intense resonance at 160 ppm, characteristic of carbonates.12–14![Synthesis of [Li][2].](/image/article/2011/SC/c1sc00215e/c1sc00215e-s1.gif) | ||
| Scheme 1 Synthesis of [Li][2]. | ||
Definitive structural assignment of [Li][2] was achieved by single-crystal X-ray diffraction methods (Fig. 1). Suitable crystals of [Li][2] were grown by slowly concentrating benzene solutions at room temperature. In the solid state, [Li][2] exists as a hexamer with a hexagonal prismatic core comprised of the six lithium countercations bridged by oxygen atoms derived from the carbonate ligand (Fig. 2). Such laddering of carboxylates and heterocarboxylates is a common structural motif.15 The carbonate ligand adopts a κ1-coordination mode to the titanium center and a pseudo κ2-coordination mode to the lithium ion. The titanium–oxygen interatomic distance (1.849(2) Å) is significantly longer than the titanium–oxygen distance found in [(Et2O)2Li][1] (1.712(2) Å). This lengthening is likely the result of a significant decrease in the π bonding between the titanium and oxygen atom as supported by DFT calculations (vide infra). The titanium–oxygen π bond is anti-bonding with respect to the titanium–anilide σ bonds, and as expected, the titanium–anilide distance is shorter in [Li][2] (1.930(3) Å) than in [(Et2O)2Li][1] (1.990(4) Å). The titanium–oxygen–carbon angle is also notable in its near linearity (174.7(2)°). A search of the Cambridge Structural Database revealed only one other carbonate complex with a comparable titanium–oxygen–carbon angle, the bimetallic carbonate complex [Cp*2Ti]2(μ-κ1:κ2-CO3) (175.43°).16 In this case the steric bulk of the Cp* ligand forces the titanium metal centers apart, preventing κ2:κ2 coordination. In the case of [Li][2], the linearity of the carbonate linkage is likely imposed by the steric bulk of the ligands and the propensity to chelate to the lithium countercation. Other examples of CO2 binding to a titanyl moiety include equilibrium formation of κ2-carbonato ligands through cycloaddition reactions.17,18
![Solid-state structure of the asymmetric unit of [Li][2] · (C6H6)1.33 with thermal ellipsoids at 50% probability and hydrogen atoms and interstitial benzene omitted for clarity. Selected distances (Å) and angles (°): Ti1–O4 1.849(2), O4–C41 1.319(3), C41–O411 1.232(3), C41–O412 1.292(3), O411–Li1 1.911(5), O412–Li1 2.257(5), O411–C41–O412 122.1(2), Ti1–O41–C41 174.7(2).20](/image/article/2011/SC/c1sc00215e/c1sc00215e-f1.gif) | ||
| Fig. 1 Solid-state structure of the asymmetric unit of [Li][2] · (C6H6)1.33 with thermal ellipsoids at 50% probability and hydrogen atoms and interstitial benzene omitted for clarity. Selected distances (Å) and angles (°): Ti1–O4 1.849(2), O4–C41 1.319(3), C41–O411 1.232(3), C41–O412 1.292(3), O411–Li1 1.911(5), O412–Li1 2.257(5), O411–C41–O412 122.1(2), Ti1–O41–C41 174.7(2).20 | ||
![Hexagonal prismatic Li6O6 core of [Li][2] with thermal ellipsoids drawn at 50% probability and hydrogen atoms and interstitial benzene molecules omitted for clarity.20](/image/article/2011/SC/c1sc00215e/c1sc00215e-f2.gif) | ||
| Fig. 2 Hexagonal prismatic Li6O6 core of [Li][2] with thermal ellipsoids drawn at 50% probability and hydrogen atoms and interstitial benzene molecules omitted for clarity.20 | ||
To determine whether the hexameric structure observed in the solid state persists in solution, we investigated the diffusion properties of the complex using 1H DOSY NMR techniques. As was shown by Waldeck et al., the diffusion coefficients of two molecules in the same solvent are proportional to the cube root of the inverse ratio of the molecular weights (eqn (1)).19
 | (1) |
Two assumptions are made in deriving this relationship: 1) the Stokes–Einstein theory of diffusion holds for the molecules of interest, and 2) the molecules can be approximated as uniform spheres. For our purposes, we selected the complex [(12-crown-4)Li][OTi(N[tBu]Ar)]3 ([(12-crown-4)Li][1]) as an external reference.‡ The 12-crown-4 ensures that the complex remains monomeric in solution by satisfying the coordination sphere of lithium. A salt was selected to stay as consistent as possible to the nature of [Li][2]. The diffusion coefficient of [(12-crown-4)Li][1] was determined to be 6.6 × 10−10 m2s−1 and the diffusion coefficient of [Li][2] was determined to be 3.8 × 10−10 m2s−1. Using the relationship from eqn (1) and the molecular weight of [(12-crown-4)Li][1] of 775.68 g mol−1, the calculated value of the molecular weight of [Li][2] is 4064 g mol−1. This value is very close to the expected value of 3862 g mol−1 for the hexamer [Li][2]. We take this measured value to provide strong evidence that the hexameric structure of [Li][2] observed in the solid state persists in solution in the absence of strongly coordinating agents.
Computational studies of [Li][2]
To gain insight into the electronic structure of [Li][2], DFT calculations were carried out. The large number of atoms associated with the hexameric structure led us to model [Li][2] as a monomer in the gas phase. The optimized geometry of the monomer [Li][2] contains a carbonato ligand in a κ1-titanium, κ2-lithium binding motif (Fig. 3). The titanium–oxygen distance (1.857 Å), titanium-bound oxygen–carbon distance (1.326 Å), and titanium–oxygen–carbon angle (174.34°) all agree well with crystallographically determined values. A vibrational mode analysis of the optimized geometry predicts the νOCO stretching mode to be observed at 1585 cm−1, in good agreement with the experimentally observed value of 1590 cm−1. Similarly, the chemical shift of the carbonate carbon was predicted to be 162 ppm, in good agreement with experimentally observed 160 ppm. The agreement with observed structural and spectroscopic values supports the notion that the simplified monomeric model is sufficiently similar to the hexamer for the purpose of our analysis. The HOMO of [Li][2] is dominated by the nitrogen lone pair orbitals of the anilide ligands. An analysis of the frontier molecular orbitals from the DFT calculations reveals no clear π-bonding interactions between the titanium and the bound oxygen atom. Earlier DFT computational studies of [(Et2O)2Li][1] had revealed a substantial degree of π-bonding between the titanium and oxo ligand.7 This change in the bonding agrees well with our earlier hypothesis based on the structural data. Bonding to CO2 likely results in a contraction and lowering in the energy of the p-orbitals at the bound oxygen, making the bonding interaction with the titanium much less favourable. Such effects on bonding have been observed previously when terminal oxos were functionalized with Lewis acids.21 This apparent weakening of the titanium–oxygen bond contrasts sharply with the analogous niobium nitride system. In that system, the short niobium–nitrogen interatomic distance persists in going from the terminal nitride to the N-bound carbamate complex and was taken as support for assigning the presence of a niobium nitrogen triple-bond both with and without CO2.11![Optimized geometry of [Li][2] as a monomer. Selected distances (Å) and angles (°): Ti1–O1 1.857, O1–C1 1.326, C1–O21 1.275, C1–O22 1.277, Ti1–O1–C1 174.3, O22–C1–O21 121.9.](/image/article/2011/SC/c1sc00215e/c1sc00215e-f3.gif) | ||
| Fig. 3 Optimized geometry of [Li][2] as a monomer. Selected distances (Å) and angles (°): Ti1–O1 1.857, O1–C1 1.326, C1–O21 1.275, C1–O22 1.277, Ti1–O1–C1 174.3, O22–C1–O21 121.9. | ||
Releasing CO2 from [Li][2]
As previously mentioned, [Li][2] is isolated free of ethereal solvents and shows marked stability in the solid state under vacuum at room temperature. Furthermore, in the synthesis of [Li][2], the diethyl ether reaction mixture is dried under dynamic vacuum, and the NMR spectra of crude reaction mixtures reveal near quantitative formation of [Li][2] with no observable amounts of [(Et2O)2Li][1]. However, when attempts were made to recrystallize [Li][2] for X-ray diffraction studies, complex [(Et2O)2Li][1] would often be found as a contaminant or as the sole anilide containing species in the isolated material. Clearly, CO2 was being released slowly as solutions of [Li][2] were allowed to stand. Preliminary observations indicated that the choice of solvent played an important role in the resulting amount of [(Et2O)2Li][1] that was isolated from these crystallization experiments, with strongly coordinating ethereal solvents giving rise to significantly more oxo. Specifically, this effect was most pronounced when 12-crown-4 was added to solutions of [Li][2].Dissolving [Li][2] in diethyl ether containing one equivalent of 12-crown-4 followed by introduction of dynamic vacuum to concentrate the solution resulted in a colour change from deep orange to pale yellow in a matter of seconds. The IR spectrum of the material contained no absorbance that could be attributed to a carbonate linkage. The NMR spectrum of the resulting off-white powder revealed a single anilide containing species in solution, along with resonances attributable to 12-crown-4; the chemical shift of the signals agreed perfectly with the spectroscopic signature of [(12-crown-4)Li][1]. This result indicates that the binding affinity of CO2 to the oxo ligand is strongly dependent on the coordination environment of the countercation and perhaps on the ability of the complex to form the hexagonal prismatic core. The formation of multiple lithium-oxygen interactions in [Li][2] might be required to overcome the entropically disfavoured reaction of trapping CO2. Hence, it appears that the combination of the nucleophilicity of the oxo ligand and the electrophilicity of the lithium is critical for effective binding of CO2.
Functionalization of [Li][2]
Although the binding of CO2 in [Li][2] was chemically reversible, the persistence of the carbonate functionality in the absence of the strongly coordinating 12-crown-4 ether led us to investigate methods to functionalize [Li][2]. If suitable functionalization reagents could be discovered, developing a method for deoxygenating [Li][2] and eventually releasing CO might be possible. In this vein, functionalization of [Li][2] can be achieved using select electrophiles. Treatment of [Li][2] with one equivalent of trimethylsilyl triflate in diethyl ether produced a gradual colour change of bright orange to red with concomitant formation of a colourless precipitate. Analysis of the crude reaction mixture by NMR spectroscopy confirmed the formation of a single product assigned as the trimethylsilyl carbonate complex Me3SiOC(O)OTi(N[tBu]Ar)3 (3, Scheme 2). Complex 3 was isolated in crystalline form by storing saturated solutions of 3 in diethyl ether at −35 °C. The IR spectrum of the isolated material contains a strong absorbance at 1691 cm−1 that can be attributed to νCO of the carbonato ligand. The solid-state structure of 3 was determined by X-ray crystallographic methods (Fig. 4). The carbon–oxygen distances in the carbonato ligand are reasonable for the expected carbon–oxygen single and double bonds. The titanium–oxygen interatomic distance (1.857(2) Å) is not significantly longer than the titanium–oxygen distance found in [Li][2] (1.849(2) Å) indicating little change in the bonding interaction between the titanium and the oxygen. Complex 3 is stable under ambient conditions, but loses CO2 at elevated temperatures to give the siloxide complex Me3SiOTi(N[tBu]Ar)3.‡ Thus, it is proposed that complex 3 is isolable for kinetic reasons (vide infra).![Functionalization reactions of [Li][2]: synthesis of 3 and 4.](/image/article/2011/SC/c1sc00215e/c1sc00215e-s2.gif) | ||
| Scheme 2 Functionalization reactions of [Li][2]: synthesis of 3 and 4. | ||
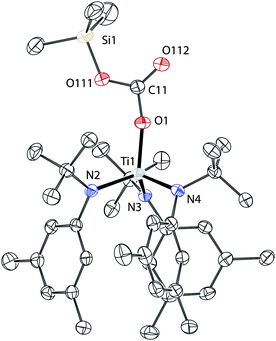 | ||
| Fig. 4 Solid-state structure of 3 with thermal ellipsoids at 50% probability and hydrogen atoms omitted for clarity. Selected distances (Å) and angles (°): Ti1–O1 1.857(2), O1–C11 1.317(3), C11–O111 1.336(3), C11–O112 1.197(3), Ti1–O1–C11 166.9(2), O1–C11–O111 111.2(2), O1–C11–O112 124.4(2).20 | ||
Knowing that functionalization of [Li][2] was possible, attempts to deoxygenate the complex were pursued. Building on precedent observed in deoxygenating the related niobium carbamate complex [Na][O2CNNb(N[tBu]Ar)3],11 treatment of [Li][2] with acylating reagents was investigated. Treatment of [Li][2] with tBuC(O)Cl in diethyl ether resulted in formation of a white precipitate and an observed colour change of yellow to orange. Analysis of the reaction mixture by proton NMR spectroscopy confirmed clean formation of a single product. The IR spectrum of the crude reaction mixture contained a strong absorbance at 1680 cm−1, characteristic of a νCO stretching mode for a carbonyl moiety. If the salt-elimination reaction had taken place with no further reaction, the expected anhydride moiety would have two IR active modes, symmetric and antisymmetric stretching.22 The single IR absorbance in the carbonyl region led us to interpret the reaction in terms of titanium pivalate complex tBuC(O)OTi(N[tBu]Ar)3 (4) formation with loss of CO2 (Scheme 2). This was confirmed through independent synthesis of 4 from the reaction of tBuC(O)Cl with [(Et2O)2Li][1], with identical spectroscopic signatures being observed in both reactions. It is not clear when the CO2 loss occurs in the reaction of [Li][2] with tBuC(O)Cl, but it is tempting to suggest that the expected salt elimination occurs to give an intermediate anhydride complex that then undergoes a rapid intramolecular rearrangement to lose CO2 (Scheme 2). This is consistent with the previously observed reaction of organic acid chlorides and acid anhydrides with the niobium complex [Na][O2CNNb(N[tBu]Ar)3] to give the five-coordinate complexes of the type (RC(O)O)(OCN)Nb(N[tBu]Ar)3.11 Although other possible mechanisms (e.g. bimolecular, insertion of carbonyl into the titanium-oxygen bond) cannot be ruled out, the proposed pathway is consistent with the data presented.
The rapid loss of CO2 upon acylation is rationalized by recognizing the ease of forming the proposed six-membered metallacyclic intermediate. Such a transition state is not possible in the case of silylation. Loss of CO2 from complex 3 likely proceeds through a four-membered metallacylic intermediate, if an intramolecular rearrangement is operative. Such a transition state would result in a greater kinetic barrier. Hence, the silylcarbonate complex 3 is isolable at ambient conditions, whereas the intermediate carbonic anhydride species is not.
Attempts to intercept the intermediate anhydride complex with an in situ reductant such as cobaltocene did not change the outcome of the reaction. In addition, oxygen abstraction reagents such as three-coordinate vanadium(III) complexes (e.g.V(N[tBu]Ar)3)23 show no reactivity with [Li][2]. Perhaps the hexameric structure and steric bulk of [Li][2] prevents such reagents from accessing the carbonate oxygens.
Conclusions
In summary, we have observed chemically reversible binding of CO2 to an anionic terminal oxo complex of titanium. The resulting κ1-carbonate complex [Li][2] can be isolated in good yield as a bright yellow powder. The solid-state structure of [Li][2] consists of a hexameric unit with the lithium countercations bridged by the carbonate functionality. DFT calculations on the system corroborate the conclusion that the titanium-oxygen bond undergoes a substantial decrease in π-bonding character concomitant with CO2 binding. Further, spectroscopic signatures of [Li][2] agree well with DFT calculated values. The release of CO2 can be effected by introducing strong coordination reagents such as 12-crown-4 to solutions of [Li][2]. This reactivity contrasts sharply with the previously investigated vanadium and niobium nitride systems. In those systems, the binding of CO2 was found to be very robust and irreversible, even when the countercation was fully sequestered.10,11 The nitride complexes are expected to be more nucleophilic than the oxo complex, and this characteristic likely explains the observed difference in reactivity. Complex [Li][2] can be functionalized readily with Me3SiOTf to give the silyl carbonate complex 3, but acylation with pivaloyl chloride results in rapid loss of CO2 at ambient conditions to give the carboxylate complex 4.This work complements several previous studies focused on the nucleophilic activation of CO2. The binding of CO2 to [(Et2O)2Li][1] relies both on the nucleophilicity of the oxo ligand and on the acidity of the lithium countercation. This contrasts with the binding of CO2 by strong nucleophiles such as N-heterocyclic carbenes,24guanidines,25 and terminal nitrides,11 which is effective in the absence of an external Lewis acid. Instead, a better comparison for the reaction between [(Et2O)2Li][1] and CO2 might be the binding of CO2 by frustrated Lewis pairs,26 where cooperative interactions lead to a greater binding affinity. Our system also shows marked similarities to model complexes of carbonic anhydrase, whose operative mechanism involves nucleophilic attack of a hydroxide ligand on CO2.27 Of course, the major difference in our system is the need for strictly aprotic conditions due to the hydrolytic sensitivity of titanium-anilide linkages. Also in this vein, the binding of CO2 in transition metal alkoxide, hydroxide, and oxide complexes is known and is occasionally reversible,13,28–32 and the concept of cation dependent binding has been illustrated for systems involving the direct interaction of CO2 with electron-rich, low-coordinate metal centers.33,34 To contrast, our discovery provides a system for study where the binding affinity for CO2 can be externally modified (e.g., by altering coordination sphere of Li+). This work also builds upon the extensive literature precedent for the binding of CO2 by heterogeneous metal oxide systems, as mentioned in the introduction. We are currently expanding our investigations to understand the thermodynamic parameters of CO2 uptake by [(Et2O)2Li][1], to probe cation effects on binding affinity, and to explore alternative oxide platforms to determine if this CO2 binding modality can be generalized.
Acknowledgements
The authors acknowledge the NSF-CCI (CHE-0802907) for financial support, and Peter Müller for insightful discussions regarding the crystal structure refinement of [Li][2].Notes and references
- J. Johnston, J. Am. Chem. Soc., 1910, 32, 938–946 Search PubMed.
- J. Blamey, E. J. Anthony, J. Wang and P. S. Fennell, Prog. Energy Combust. Sci., 2010, 36, 260–279 CrossRef CAS.
- H. A. Mosqueda, C. Vazquez, P. Bosch and H. Pfeiffer, Chem. Mater., 2006, 18, 2307–2310 CrossRef CAS.
- B. N. Nair, R. P. Burwood, V. J. Goh, K. Nakagawa and T. Yamaguchi, Prog. Mater. Sci., 2009, 54, 511–541 CrossRef CAS.
- V. P. Indrakanti, J. D. Kubicki and H. H. Schobert, Energy Environ. Sci., 2009, 2, 745–758 RSC.
- G. Busca and V. Lorenzelli, Mater. Chem., 1982, 7, 89–126 Search PubMed.
- A. Mendiratta, J. S. Figueroa and C. C. Cummins, Chem. Commun., 2005, 3403–3405 RSC.
- W. W. Lukens, P. T. Matsunaga and R. A. Andersen, Organometallics, 1998, 17, 5240–5247 CrossRef CAS.
- S. De Angelis, E. Solari, C. Floriani, A. Chiesi-Villa and C. Rizzoli, Organometallics, 1995, 14, 4505–4512 CrossRef CAS.
- J. K. Brask, V. Durá-Vilá, P. L. Diaconescu and C. C. Cummins, Chem. Commun., 2002, 902–903 RSC.
- J. S. Silvia and C. C. Cummins, J. Am. Chem. Soc., 2010, 132, 2169–2171 CrossRef CAS.
- D. J. Darensbourg, K. M. Sanchez and A. L. Rheingold, J. Am. Chem. Soc., 1987, 109, 290–292 CrossRef CAS.
- D. J. Darensbourg, M. L. M. Jones and J. H. Reibenspies, Inorg. Chem., 1996, 35, 4406–4413 CrossRef CAS.
- D. J. Darensbourg, M. W. Holtcamp, G. E. Struck, M. S. Zimmer, S. A. Niezgoda, P. Rainey, J. B. Robertson, J. D. Draper and J. H. Reibenspies, J. Am. Chem. Soc., 1999, 121, 107–116 CrossRef CAS.
- A. Downard and T. Chivers, Eur. J. Inorg. Chem., 2001, 2001, 2193–2201 Search PubMed.
- V. V. Burlakov, F. M. Dolgushin, A. I. Yanovsky, Y. T. Struchkov, V. B. Shur, U. Rosenthal and U. Thewalt, J. Organomet. Chem., 1996, 522, 241–247 Search PubMed.
- V. L. Goedken and J. A. Ladd, J. Chem. Soc., Chem. Commun., 1982, 142–144 RSC.
- C. E. Housmekerides, D. L. Ramage, C. M. Kretz, J. T. Shontz, R. S. Pilato, G. L. Geoffroy, A. L. Rheingold and B. S. Haggerty, Inorg. Chem., 1992, 31, 4453–4468 CrossRef.
- A. R. Waldeck, P. W. Kuchel, A. J. Lennon and B. E. Chapman, Prog. Nucl. Magn. Reson. Spectrosc., 1997, 30, 39–68 CrossRef CAS.
- A. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- F. Wolff, R. Choukroun, C. Lorber and B. Donnadieu, Eur. J. Inorg. Chem., 2003, 2003, 628–632 Search PubMed.
- J. B. Lambert, H. F. Shurvell, D. A. Lightner and R. G. Cooks, Organic Structural Spectroscopy, Prentice-Hall, Inc., Upper Saddle River, NJ, 1998 Search PubMed.
- M. G. Fickes, Ph.D. thesis, Massachusetts Institute of Technology, Cambridge, MA, 1998.
- H. A. Duong, T. N. Tekavec, A. M. Arif and J. Louie, Chem. Commun., 2004, 112–113 RSC.
- C. Villiers, J. P. Dognon, R. Pollet, P. Thuery and M. Ephritikhine, Angew. Chem., Int. Ed., 2010, 49, 3465–3468 CAS.
- C. M. Momming, E. Otten, G. Kehr, R. Frohlich, S. Grimme, D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2009, 48, 6643–6646 CrossRef.
- G. Parkin, Chem. Rev., 2004, 104, 699–767 CrossRef CAS.
- O. P. Lam, S. C. Bart, H. Kameo, F. W. Heinemann and K. Meyer, Chem. Commun., 2010, 46, 3137–3139 RSC.
- Y. Dussart, C. Harding, P. Dalgaard, C. McKenzie, R. Kadirvelraj, V. McKee and J. Nelson, J. Chem. Soc., Dalton Trans., 2002, 1704–1713 RSC.
- T. Tsuda, Y. Chujo and T. Saegusa, J. Am. Chem. Soc., 1980, 102, 431–433 CrossRef CAS.
- S. K. Mandal, D. M. Ho and M. Orchin, Organometallics, 1993, 12, 1714–1719 CrossRef CAS.
- A. M. Appel, R. Newell, D. L. DuBois and M. Rakowski DuBois, Inorg. Chem., 2005, 44, 3046–3056 CrossRef CAS.
- G. Fachinetti, C. Floriani and P. F. Zanazzi, J. Am. Chem. Soc., 1978, 100, 7405–7407 CrossRef CAS.
- M. H. Schmidt, G. M. Miskelly and N. S. Lewis, J. Am. Chem. Soc., 1990, 112, 3420–3426 CrossRef CAS.
- A. Mendiratta, C. C. Cummins, F. A. Cotton, S. A. Ibragimov, C. A. Murillo and D. Villagran, Inorg. Chem., 2006, 45, 4328–4330 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental, crystallographic, and computational details and spectroscopic data. CCDC [Li][2], 819846; 3, 819847. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00215e |
| ‡ If treated with an excess of CO2, [Li][2] reacts further to give other products. We suspect that the products are the result of CO2 insertion into the titanium-anilide bonds to give carbamate moieties. Such reactivity has been observed in titanium-anilide chemistry.35 Complex [(12-crown-4)Li][1] is synthesized by adding one equivalent of 12-crown-4 to solutions of [(Et2O)2Li][1] in diethyl ether. Thermal stability study of complex 3 was performed by heating a solution of 3 in C6D6 at 80 °C for 12 h in a flame-sealed NMR tube. See ESI for details.† |
| This journal is © The Royal Society of Chemistry 2011 |