Supramolecular motifs in metal complexes of Schiff bases. Part 5.1 Zinc(II)-assisted self-assembly of some bis-N,N- and N,O-bidentate Schiff bases and chiral packing modes in solid state
Noboru Yoshida*a, Kazuhiko Ichikawaa and Motoo Shirob
aLaboratory of Molecular Functional Chemistry, Division of Material Science, Graduate School of Environmental Earth Science, Hokkaido University, Sapporo, 060-0810, Japan
bRigaku Corporation, Akishima, Tokyo, 196-8666, Japan
First published on UnassignedUnassigned23rd December 1999
Abstract
Zinc(II)-assisted self-assembly of a new bis-N,O-bidentate Schiff base ligand, (N-salicylidene-4,4′-diaminodiphenyl)methane (L16) with two chelating sites linked by a spacer group (-C6H4CH2C6H4-), afforded in high yield the double-helical dinuclear complex (L16 : ZnII = 2∶2). Single-crystal X-ray analyses demonstrated clearly that the two ZnII centers have a distorted tetrahedral (Td) coordination sphere with two-wrapped ligands. The analogous Schiff base, bis[4-(2-pyridylmethyleneaminophenyl]methane (L17) was also designed to self-assemble in the presence of metal ions, leading to a triple-helical dinuclear (L17∶ZnII = 3∶2) supramolecular motif. Each zinc ion has six-coordinate octahedral geometry with six nitrogens from three ligands. These unprecedented helical motifs in the solid state and solution seem to be induced by the geometrical preference for octahedral or tetrahedral coordination mode of the ZnII ion and the interligand π-stacking interactions between the spacer groups of L16 and L17. Electrospray mass spectrometry proved a very useful characterisational tool in detecting the distribution of supramolecular species in solution. Use of a N,N![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) ′-bis(2-pyridylmethylene)-1,4-diaminobenzene (L35) with a rigid phenylene spacer in its center resulted in the one-dimensional zigzag polymeric structure ((L35∶ZnII)∞ = (1∶1)∞) where the multiple π-stacking interactions operate between the aromatic rings of linking ligands. Each zinc ion is octahedrally coordinated by two N,N-bidentate arms of two different ligands and two cis oxygens from N,N-dimethylformamide as coordinating solvent.
′-bis(2-pyridylmethylene)-1,4-diaminobenzene (L35) with a rigid phenylene spacer in its center resulted in the one-dimensional zigzag polymeric structure ((L35∶ZnII)∞ = (1∶1)∞) where the multiple π-stacking interactions operate between the aromatic rings of linking ligands. Each zinc ion is octahedrally coordinated by two N,N-bidentate arms of two different ligands and two cis oxygens from N,N-dimethylformamide as coordinating solvent.
Introduction
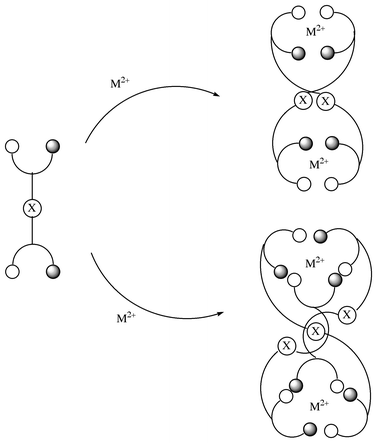
Recent synthetic advances using Schiff base ligands1–5 with aromatic spacer groups and other flexible bis-bidentate ligands6–15 as building blocks for metal-assisted self-assembly (metallosupramolecular chemistry) have demonstrated the requirements for the close and tunable control of the coordination sphere of the metal ion and the weak aromatic interactions between the spacer groups in the ligand. The mechanism and structure for the formation of metal-assisted self-assembled complexes are often impossible to predict in advance and many subtle factors such as π–π interactions,2,16 templated-anions,17–19 trapped-cations,20 deprotonation of the site remote from the metal center7b,21 and the preferred coordination geometry of metal ion22 can influence the final product.Achiral bridged Schiff bases, the previously designed series of bis-bidentate ligands shown in Scheme 1, have been shown to spontaneously wrap around copper(II) ion to create a self-assembled helical multinuclear complex in the solid state.1 Formation of some helical multinuclear supramolecular motifs has been explained on the basis of self-assembly processes involving π stacking of the spacer group (X) with free rotational freedom and a flexible coordination mode for tetrahedral and square-planar geometries of the CuII ion.
 | ||
| Scheme 1 | ||
Zinc(II) ion has been widely found in several zinc-containing metalloenzymes such as zinc-peptidases,23 human carbonic anhydrase,24 and alkalinephosphatase.25 The coordination mode for octahedral or tetrahedral geometries of zinc(II) ion may affect the formation and mechanism of the self-assembly processes of bis-bidentate Schiff bases. Furthermore, the CH⋯π interactions, which have recently received much attention in supramolecular architectures, have also been important in a variety of molecular phenomena at physical, chemical, and biological levels.26 We report here a one-pot self-assembly synthesis of several zinc(II) complexes utilizing weak CH⋯π and π⋯π interactions of a novel type of Schiff base ligand, L16, L17, and L35 as shown in Scheme 2. The variations in (a) metal-binding site from pyridylmethylene N,N moiety to salicylidene O,N one (L17→L16) and (b) spacer group from diphenylmethylene -C6H4CH2C6H4- to phenylene -C6H4- (L17→ L35) may produce some zinc(II) complexes with unprecedented supramolecular motifs.
 | ||
| Scheme 2 (a) Change in the coordination site from N,N bidentate to N,O bidentate. (b) Change into the shorter spacer group. | ||
Experimental
Preparation of Schiff base ligands, L17, L16, and L35, and their ZnII complexes
Ligand L17 was synthesized by usual Schiff-base condensation of bis(4-aminophenyl)methane and pyridine-2-aldehyde.2 To a stirred solution of bis(4-aminophenyl)methane (6.09 g, 0.0307 mol) in ethanol (300 ml) at room temperature was added dropwise an ethanolic solution of pyridine-2-aldehyde (6.58 g, 0.0615 mol). After the addition was complete, the reaction mixture was heated to 70–80 °C. The resultant precipitate was filtered off, washed with ethanol, and dried in air to afford 9.0 g (78%) of pale-yellow crystalline product. Calcd. for C25H20N4: C, 79.76; H, 5.35; N, 14.88; found: C, 79.59; H, 5.43; N, 14.93%. 1H-NMR (400 MHz, DMF-d7 (99.5%), 25 °C, internal ref. TMS): δ 8.74 (2H, ddd, J = 4.9, 2.0, 0.9, pyridyl H6), 8.65 (2H, s, -CH![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) N-), 8.21 (2H, ddd, J = 7.8, 2.0, 1.0, pyridyl H3), 7.98 (2H, td, J = 7.8, 1.5, pyridyl H4), 7.54 (2H, ddd, J = 7.5, 5.0, 1.4, pyridyl H5), 7.36 and 7.40 (8H, dd, J = 7.8, 1.5, aminophenyl), 4.09 (2H, s, -CH2-).
N-), 8.21 (2H, ddd, J = 7.8, 2.0, 1.0, pyridyl H3), 7.98 (2H, td, J = 7.8, 1.5, pyridyl H4), 7.54 (2H, ddd, J = 7.5, 5.0, 1.4, pyridyl H5), 7.36 and 7.40 (8H, dd, J = 7.8, 1.5, aminophenyl), 4.09 (2H, s, -CH2-).Ligand L16 was synthesized in 90% yield by heating ethanolic solutions containing bis(4-aminophenyl)methane (1 equiv.) and salicylaldehyde (2 equiv.). Data for ligand L16: fluorescent yellow powder, 90% yield (Calcd. for C27H22N2O2; C, 79.78; H, 5.45; N, 6.89. Found: C, 79.78; H, 5.65; N, 6.89%). 1H-NMR (400 MHz, CDCl3, 25 °C, internal ref. TMS): δ 13.29 (2H, ddd, OH⋯H), 8.62 (2H, s, -CHN-), 7.38 (2H, d, J = 7.3, sal-ad H6), 7.37 (2H, td, sal-ad H4), 7.27 and 7.23 (8H, ddd, bis(amino)phenyl), 7.02 (2H, d, J = 7.8, sal-ad H3), 6.94 (2H, td, J = 7.8, 1.0, sal-ad H5), 4.04 (2H, s, -CH2-).
Ligand L35 was synthesized by the same condensation of 1,4-phenylenediamine and pyridine-2-aldehyde and obtained as dark orange crystals, yield 70% (Calcd. for C18H14N4: C, 75.5; H, 4.92; N, 19.56. Found: C, 75.36; H, 5.06; N, 19.25%). 1H-NMR (400 MHz, CDCl3, 30 °C, internal ref. TMS): δ 8.73 (2H, ddd, J = 4.9, 1.5, 1.0, pyridyl H6), 8.67 (2H, s, -CHN-), 8.22 (2H, ddd, J = 7.8, 1.3, 1.0, pyridyl H3), 7.83 (2H, td, J = 7.3, 1.5, pyridyl H4), 7.38 (2H, ddd (overlapped with diaminobenzene), pyridyl H5), 7.38 (4H, s, diaminobenzene).
Synthesis and characterization of the zinc(II) complexes of L17 and L16 used in this study were carried out by the usual synthetic method.2,3 For example, a mixture of L17 (0.64 g, 1.7 mmol) and Zn(ClO4)2·6H2O (0.33 g, 1.7 mmol) in CH3OH (50 ml) was stirred at room temperature for several hours. The resultant pale-yellow precipitate was filtered off and air-dried. The yields of this material were ca. 50–60%, but were not optimised. Pale-yellow crystals were obtained by slow diffusion of diethyl ether in the DMF–MeCN solution (Found: C, 53.62; H, 3.97; N, 10.50; Cl, 7.84%. Calcd. for C75H60N12O16Cl4Zn2·DMF·2MeCN: C, 54.32; H, 4.05; N, 11.58; Cl, 7.82%).
Reaction of L16 (3 equiv.) with Zn(ClO4)2·6H2O (2 equiv.) and K2CO3 (2 equiv.) in methanol at room temperature affords a fluorescent yellow solid (Found: C, 53.69; H, 4.05; N, 4.62%. Calcd. for C54H50N4O16Cl2Zn2: C, 53.48; H, 4.15; N, 4.62%) which is soluble in CHCl3. Fluorescent-yellow crystals were obtained by slow diffusion of diethyl ether into the CHCl3 solution of the metal complex.
Reaction of a methanol solution of 1 equiv. of the ligand L35 with Zn(ClO4)2·6H2O (1 equiv.) at room temperature leads to the deep orange solids with L35∶ZnII = 2∶2 and 3∶2 ratio as judged by elemental analysis. The yield was in the range 60–70%. (Calcd. forC54H42N12O16Cl4Zn2 as 3∶2 complex: C, 46.74; H, 3.05; N, 12.11; Cl, 10.22 and C36H28N8O16Cl4Zn2 as 2∶2 complex: C, 39.26; H, 2.56; N, 10.17; Cl, 12.87. Found: C, 42.48; H, 3.40; N, 11.04; Cl, 10.07%). X-Ray quality crystals were grown by slow diffusion of diethyl ether into a DMF solution of the crude material.
Single crystal X-ray structure analysis
Single crystals suitable for X-ray analysis of 1 ([Zn2(L17)3](ClO4)2), 2 ([Zn2(L16 − 2H)2]) and 3 ([ZnIIL35]·(ClO4)2) were obtained from DMF–MeCN/ether, CHCl3/ether, and DMF/ether, respectively. Precise data collection and crystal parameters for 1, 2 and 3 are summarized in Table 1. CCDC reference number 188/193. The structure was solved by direct method27 and expanded using Fourier techniques.28 The non-hydrogen atoms were refined anisotropically. Hydrogen atoms were included but not refined. All calculations were performed using the teXsan29 crystallographic software package of Molecular Structure Coorporation.
| 1·DMF·2CH3CN | 2·2CHCl3 | 3·3DMF | |
|---|---|---|---|
| a R = Σ ¶Fo| − |Fc||/Σ/Fo|.b Rw = [Σω(|Fo| − |Fc|)2/Σω|Fo|2]1/2, ω = 1/σ2(Fo) = [σ2(Fo) + p2(Fo)2/4]−1. | |||
| Formula | C82H73N15O17Cl4Zn2 | C56H42N4O4Cl6Zn2 | C27H35N7O11Cl2Zn |
| M | 1813.14 | 1178.45 | 769.90 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic |
| Space group | C2/c (no. 15) | P21/c (no. 14) | P22/a (no. 14) |
| Unit cell dimensions | |||
| a/Å | 54.89(2) | 18.263(4) | 13.472(2) |
| b/Å | 13.833(2) | 16.089(3) | 18.812(3) |
| c/Å | 22.01(1) | 19.160(4) | 14.538(4) |
| β/° | 95.40(3) | 109.22(3) | 106.17(1) |
| U/Å3 | 16639.9 | 5316(2) | 3539(2) |
| Z | 8 | 4 | 4 |
| T/K | 298(2) | 298(1) | 173(1) |
| μ(Mo-Kα)/cm−1 | 7.82 | 12.54 | 9.09 |
| No. of reflections measured | 10268 | 7883 | 5297 |
| Residuals: R,aRwb | 0.061, 0.084 | 0.077, 0.095 | 0.052, 0.075 |
Results and discussion
UV/VIS titrations in the formation of ZnII complexes with L16, L17, and L35
The UV/VIS spectrum for L16 is quite sensitive to the acidity of the solution and the presence of CuII ion. Fig. 1(a) represents the change in the UV/VIS spectrum of L16 upon addition of ZnII ion in ethanol. Four isosbestic points are observed at λ = 289, 314.5, 370, and 454 nm. Upon increasing ZnII concentration, there is a decrease in absorbance of the ligand π–π* (CHN) band at 345 nm and emergence of a new peak at 394 nm due to deprotonation of the phenol group and the ZnII coordination. A molar ratio plot at 345 and 394 nm demonstrates the formation of a ZnII∶L16 = 1∶1 ratio complex, as judged by the clear inflection at [ZnII]/[L16] = 1.![UV/VIS spectral changes of ligands L16 (a), L17 (b) and L35 (c) in ethanol upon addition of Zn(CH3COO)2·2H2O. [L16] = [L17] = [L35] = 6.50 × 10−5 mol dm−3.](/image/article/2000/P2/a908041d/a908041d-f1.gif) | ||
| Fig. 1 UV/VIS spectral changes of ligands L16 (a), L17 (b) and L35 (c) in ethanol upon addition of Zn(CH3COO)2·2H2O. [L16] = [L17] = [L35] = 6.50 × 10−5 mol dm−3. | ||
A smaller UV/VIS change (Fig. 1(b)) is observed in the case of the L17–Zn2+ system and an isosbestic point is not observed under similar conditions. An inflection at [ZnII]/[L17] = ca. 0.66 (λobs = 330.5 nm) may suggest the formation of ZnII∶L17 = 2∶3 complex. UV/VIS titration (Fig. 1(c)) was performed also in the L35/Zn2+ system. Since L35 has a considerably delocalized π-conjugate system, the π–π* band due to the coordination of the ZnII ion shows a large red shift up to 450 nm. The magnitude of this red-shift on coordination is approximately 100 nm, suggesting that the induced π-conjugation in the five-membered chelate rings occurs effectively through the spacer in the L35–Zn2+ system. The result is a deepening in colour from colourless to orange–yellow. Some isosbestic points at 281.5 and 395 nm and a molar ratio plot suggest the formation of ZnII∶L35 = 2∶3 complex. It is interesting to note that the UV/VIS absorption change at higher ZnII concentration of [ZnII]/[L35] > ca. 0.6 reverses both at λ = 366 and 441 nm.
Mass spectra of ZnII complexes with L16, L17, and L35 complexes
Positive electrospray ionization mass spectrometry (ESI-MS) of the L16–ZnII complex in MeOH (trace CHCl3) shows the presence of several aggregated species (Fig. 2). The fairly weak peak at m/z = 1879.1 could be assignable to the tetranuclear mono-protonated species, [Zn4(L16 − 2H)4]H+, (4∶4)+. Two peaks corresponding to the binuclear mono-protonated [Zn2(L16 − 2H)2]H+, (2∶2)+ species and trinuclear mono-protonated [Zn3(L16 − 2H)3]H+, (3∶3)+ species are strongly observed at m/z = 941.0 and 1408.9, respectively. The experimental isotope profiles were in excellent agreement with the theoretical isotope distributions expected for the [Zn3(L16 − 2H)3]H+.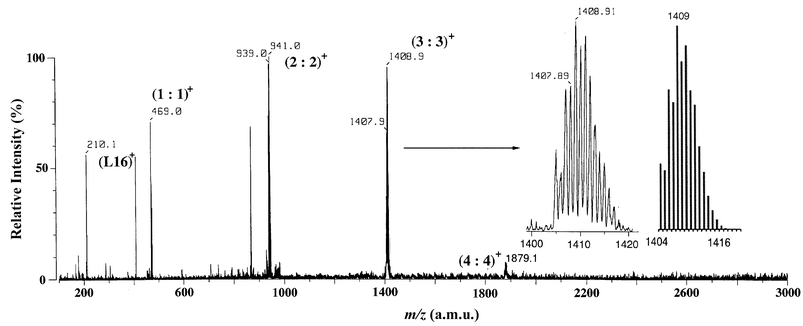 | ||
| Fig. 2 ESI mass spectrum of a methanol solution of 2. | ||
The solution behaviour of the L17–ZnII complex was analyzed by two types of mass spectrometry, ESI-MS and fast atom bombardment mass spectrometry (FAB-MS). FAB-MS of the L17–ZnII complex is very complicated but informative on the successive loss of ClO4− or HClO4 from neutral species, 1 ([Zn2(L17)3](ClO4)2) (Fig. 3). The FAB-MS (3-nitrobenzyl alcohol as a matrix in DMF as solvent) shown in Fig. 3 displays several characteristic peaks for (ZnII∶L17)n+ = (1∶1)+, (1∶2)+, (2∶2)+, and (2∶3)+ species (minus ClO4− or HClO4), where M1∶1, M1∶2, M2∶2, and M2∶3 denote the species [ZnL17](ClO4)2, [Zn(L17)2](ClO4)2, [Zn2(L17)2](ClO4)4, and [Zn2(L17)3](ClO4)4, respectively. The peaks for higher aggregates [M2∶3 − ClO4 − (HClO4)2]+, [M2∶3 − ClO4 − HClO4]+, and [M2∶3 − ClO4]+ can be detected at m/z = 1358.0, 1458.0, and 1559.0, respectively.
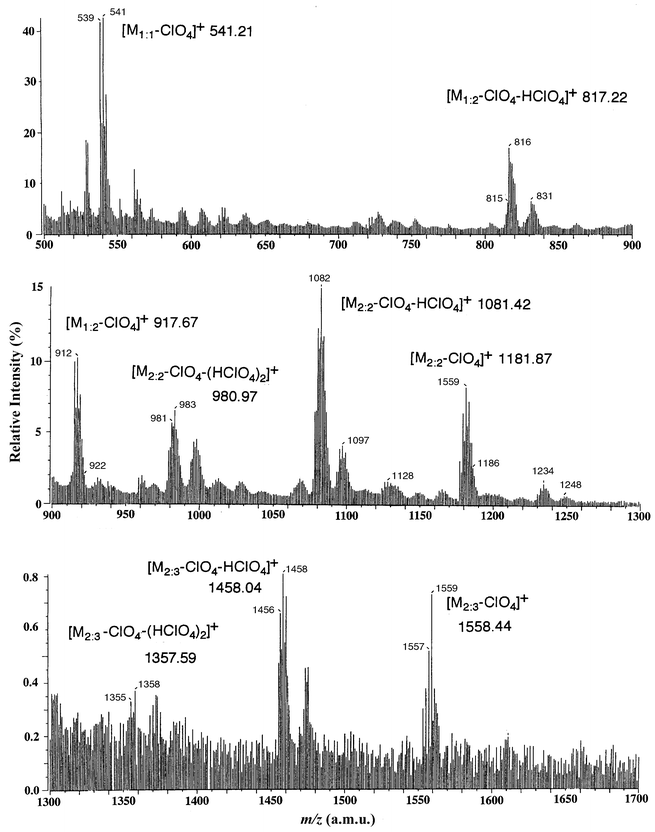 | ||
| Fig. 3 FAB mass spectrum for 1. | ||
ESI-MS of the L17–ZnII complex in MeOH is significantly different from the FAB-MS observed in the same complex (Fig. 4). The most important peak corresponding to the dinuclear-triple motif of crystal structure 1 (vide infra) is [M2∶3 − ClO4]+ at m/z = 1558.6. It is noted that minor 1∶1 complexes that are solvated by several MeOH molecules ([M1∶1(MeOH)2 − ClO4]+ and [M1∶1(MeOH)3 − ClO4]+) are clearly detected at m/z = 606.0 and 636.1.
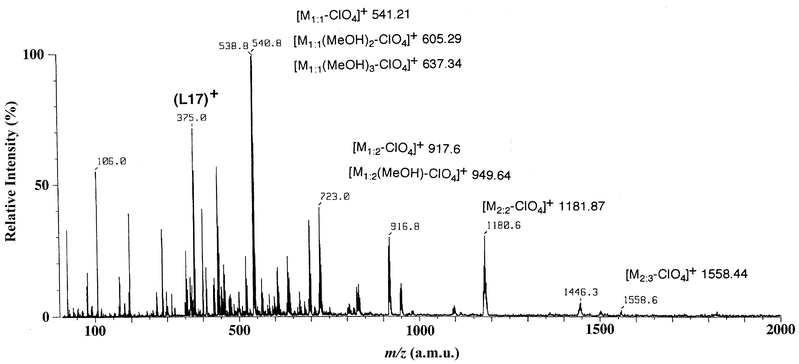 | ||
| Fig. 4 ESI mass spectrum of a methanol solution of 1. | ||
Fig. 5 shows the ESI-MS of the L35–ZnII complex in methanol. The peaks at m/z = 349.2, 451.2, and 637.5 could be assigned to lower aggregates [M1∶1 − ClO4 − HClO4]+, [M1∶1 − ClO4]+, and [M1∶2 − ClO4 − HClO4]+. Higher aggregates such as ZnII∶L35 = 2∶2 and 2∶3 complexes are not observed in this system.
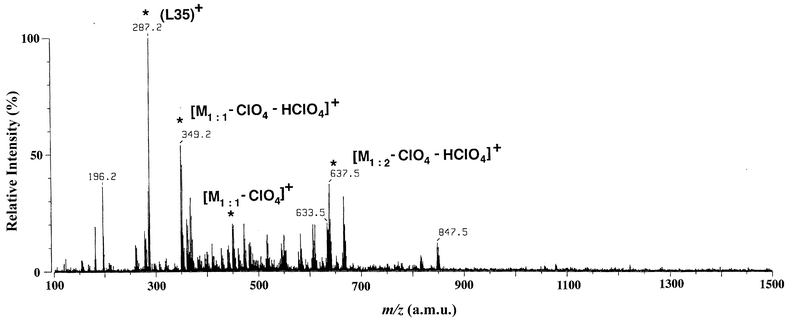 | ||
| Fig. 5 ESI mass spectrum of a methanol solution of 3. | ||
X-Ray crystal structure of L17–ZnII complex, 1
Table 1 lists relevant X-ray data for the crystal parameters of 1. As expected, the formation of a dinuclear structure (ZnII∶L17 = 2∶3) with a triple-helical motif is confirmed as shown in Fig. 6. The complex cation [(ZnII)2(L17)3]4+ contains two ZnII ions and three wrapped ligands L17. Selected bond lengths and angles for 1 are presented in Table 2.| Zn(1)–N(1) | 2.158(6) | Zn(1)–N(2) | 2.195(5) |
| Zn(1)–N(5) | 2.186(7) | Zn(1)–N(6) | 2.193(6) |
| Zn(1)–N(9) | 2.154(6) | Zn(1)–N(10) | 2.176(6) |
| Zn(2)–N(3) | 2.174(6) | Zn(2)–N(4) | 2.132(7) |
| Zn(2)–N(7) | 2.247(6) | Zn(2)–N(8) | 2.167(6) |
| Zn(2)–N(11) | 2.200(6) | Zn(2)–N(12) | 2.187(6) |
| C(6)–N(2) | 1.290(9) | C(20)–N(3) | 1.264(9) |
| C(31)–N(6) | 1.265(9) | C(45)–N(7) | 1.270(9) |
| C(56)–N(10) | 1.268(8) | C(70)–N(11) | 1.277(8) |
| N(1)–Zn(1)–N(2) | 76.7(2) | N(1)–Zn(1)–N(5) | 91.3(3) |
| N(1)–Zn(1)–N(6) | 164.7(2) | N(1)–Zn(1)–N(9) | 98.4(2) |
| N(1)–Zn(1)–N(10) | 94.5(2) | N(2)–Zn(1)–N(5) | 87.6(2) |
| N(2)–Zn(1)–N(6) | 93.5(2) | N(2)–Zn(1)–N(9) | 173.6(2) |
| N(2)–Zn(1)–N(10) | 99.9(2) | N(5)–Zn(1)–N(6) | 76.5(2) |
| N(5)–Zn(1)–N(9) | 96.7(2) | N(5)–Zn(1)–N(10) | 171.4(2) |
| N(6)–Zn(1)–N(9) | 92.1(2) | N(6)–Zn(1)–N(10) | 98.7(2) |
| N(9)–Zn(1)–N(10) | 76.2(2) | ||
| N(3)–Zn(2)–N(4) | 77.3(3) | N(3)–Zn(2)–N(7) | 96.0(2) |
| N(3)–Zn(2)–N(8) | 170.1(2) | N(3)–Zn(2)–N(11) | 100.6(2) |
| N(3)–Zn(2)–N(12) | 97.9(2) | N(4)–Zn(2)–N(7) | 93.2(2) |
| N(4)–Zn(2)–N(8) | 97.1(3) | N(4)–Zn(2)–N(11) | 173.3(2) |
| N(4)–Zn(2)–N(12) | 98.4(2) | N(7)–Zn(2)–N(8) | 76.1(2) |
| N(7)–Zn(2)–N(11) | 93.4(2) | N(7)–Zn(2)–N(12) | 163.6(2) |
| N(8)–Zn(2)–N(11) | 85.8(2) | N(8)–Zn(2)–N(12) | 90.9(2) |
| N(11)–Zn(2)–N(12) | 75.4(2) | ||
| Zn(1)–N(1)–C(1) | 128.6(5) | Zn(1)–N(1)–C(5) | 114.5(5) |
| Zn(1)–N(2)–C(6) | 111.3(5) | Zn(1)–N(2)–C(7) | 127.6(5) |
| Zn(1)–N(5)–C(26) | 130.0(6) | Zn(1)–N(5)–C(30) | 112.7(5) |
| Zn(1)–N(6)–C(31) | 113.2(5) | Zn(1)–N(6)–C(32) | 129.3(5) |
| Zn(1)–N(9)–C(51) | 126.2(6) | Zn(1)–N(9)–C(55) | 114.3(4) |
| Zn(1)–N(10)–C(56) | 114.2(4) | Zn(1)–N(10)–C(57) | 127.4(4) |
| Zn(2)–N(3)–C(17) | 127.5(5) | Zn(2)–N(3)–C(20) | 112.6(6) |
| Zn(2)–N(4)–C(21) | 114.1(6) | Zn(2)–N(4)–C(25) | 128.4(6) |
| Zn(2)–N(7)–C(42) | 130.3(5) | Zn(2)–N(7)–C(45) | 111.9(5) |
| Zn(2)–N(8)–C(46) | 114.4(4) | Zn(2)–N(8)–C(50) | 128.2(6) |
| Zn(2)–N(11)–C(67) | 125.0(4) | Zn(2)–N(11)–C(70) | 114.0(5) |
| Zn(2)–N(12)–C(71) | 114.3(5) | Zn(2)–N(12)–C(75) | 127.5(5) |
| C(10)–C(13)–C(14) | 114.7(7) | C(35)–C(38)–C(39) | 114.5(7) |
| C(60)–C(63)–C(64) | 114.7(6) |
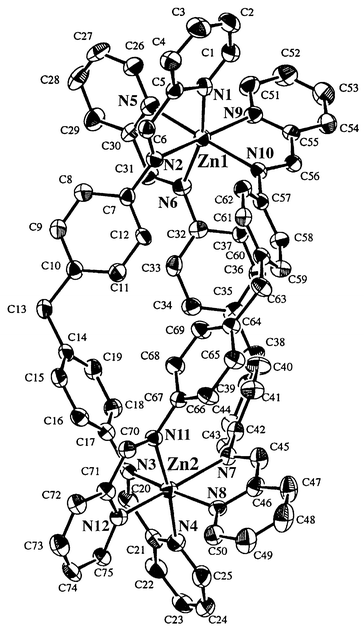 | ||
| Fig. 6 X-ray crystal structure for 1. Perchlorate ions are omitted for clarity. | ||
The ZnII ions (Zn(1) and Zn(2)) are six-coordinated, being bound by three pyridine nitrogens (py) and three azomethine nitrogens (CHN) from three L17 ligands (L17(1)/C(1)–C(25), N(1)–N(4), L17(2)/C(26)–C(50), N(5)–N(8) and L17(3)/C(51)–C(75), N(9)–N(12)). The lengths of the Zn–N(py) bonds [2.154(6)–2.186(7) Å] are somewhat shorter than those of Zn–N(CHN) [2.176(6)–2.195 Å]. A slight trans influence is observed in the longer Zn(1)–N(5) (2.186(7) Å) and shorter Zn(1)–N(10) (2.176(6) Å). A pseudo-octahedral geometry around Zn(1) is given as judged by the N–Zn–N bond angles N(py)–Zn(1)–N(py), N(py)–Zn(1)–N(CHN) and N(CHN)–Zn(1)–N(CHN). The coordination geometry around Zn(2) is almost the same as that of Zn(1). The interatomic Zn(1)⋯Zn(2) separation is found to be 11.431 Å. Interestingly, a chiral cavity, which could accommodate small ions such as halide ion, is observed between the three spacer groups. The ClO4− ions are too large to be included at the center of the cavity.
The Corey–Pauling–Koltun (CPK) model for [(ZnII)2(L17)3]4+ shown in Fig. 7 illustrates the multiple π–π and CH–π aromatic interactions, which operate particularly in some interactive moieties between pyridine–spacer groups (2.8–3.4 Å) and spacer–spacer groups (2.9–3.1 Å), and which create the dinuclear triple-helical structure. The flexibility around the methine group of the spacer (-C6H4CH2C6H4-) may allow suitable rotational motion of the aromatic rings and CH–π interactions, thereby tuning the triple-helical structure of 1.
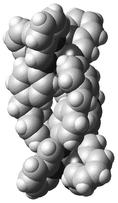 | ||
| Fig. 7 Space-filling representation of 1. Perchlorate ions are omitted for clarity. | ||
X-Ray crystal structure of L16–ZnII complex, 2
The X-ray crystal structure of [(ZnII)2(L16)2]·2CHCl3 (2·2CHCl3) shows clearly the dinuclear double-helical structure for the neutral complex 2 (Fig. 8). Two ZnII ions (Zn(1) and Zn(2)) and two L16 ligand molecules (L16(1)/C(1)–C(27), N(1), N(3), O(1) and O(3) and L16(2)/C(28)–C(54), N(2), N(4), O(2) and O(4)) which are wrapped around the Zn–Zn axis are involved in this supramolecular motif. Table 3 shows the selected bond lengths and angles for 2.| Zn(1)–O(1) | 1.895(6) | Zn(1)–O(2) | 1.907(6) |
| Zn(1)–N(1) | 2.019(6) | Zn(1)–N(2) | 2.030(6) |
| Zn(2)–O(3) | 1.895(5) | Zn(2)–O(4) | 1.894(6) |
| Zn(2)–N(3) | 2.020(6) | Zn(2)–N(4) | 2.019(7) |
| O(1)–C(1) | 1.328(10) | O(2)–C(28) | 1.32(1) |
| O(3)–C(27) | 1.322(9) | O(4)–C(54) | 1.340(10) |
| N(1)–C(7) | 1.310(10) | N(2)–C(34) | 1.297(10) |
| N(3)–C(21) | 1.30(1) | N(4)–C(48) | 1.298(9) |
| O(1)–Zn(1)–O(2) | 111.0(3) | O(1)–Zn(1)–N(1) | 95.5(2) |
| O(1)–Zn(1)–N(2) | 129.1(3) | O(2)–Zn(1)–N(1) | 126.8(3) |
| O(2)–Zn(1)–N(2) | 95.0(2) | N(1)–Zn(1)–N(2) | 102.9(2) |
| O(3)–Zn(2)–O(4) | 112.6(3) | O(3)–Zn(2)–N(3) | 96.0(2) |
| O(3)–Zn(2)–N(4) | 119.3(3) | O(4)–Zn(2)–N(3) | 129.9(3) |
| O(4)–Zn(2)–N(4) | 96.3(2) | N(3)–Zn(2)–N(4) | 104.5(2) |
| Zn(1)–O(1)–C(1) | 126.1(6) | Zn(1)–O(2)–C(28) | 127.6(5) |
| Zn(2)–O(3)–C(27) | 126.6(5) | Zn(2)–O(4)–C(54) | 122.5(6) |
| Zn(1)–N(1)–C(7) | 119.3(5) | Zn(1)–N(1)–C(3) | 120.8(5) |
| C(7)–N(1)–C(8) | 119.7(6) | Zn(1)–N(2)–C(34) | 121.3(5) |
| Zn(1)–N(2)–C(35) | 117.9(5) | C(34)–N(2)–C(35) | 119.1(6) |
| Zn(2)–N(3)–C(18) | 118.2(5) | Zn(2)–N(3)–C(21) | 120.4(5) |
| C(18)–N(3)–C(21) | 120.5(7) | Zn(2)–N(4)–C(45) | 120.3(5) |
| Zn(2)–N(4)–C(48) | 119.2(6) | ||
| C(11)–C(14)–C(15) | 116.2(7) | C(38)–C(41)–C(42) | 115.2(6) |
 | ||
| Fig. 8 X-ray crystal structure for 2 (a) and space-filling representation of 2 (b). | ||
The ZnII ions are four-coordinated with two phenolate oxygens and two azomethine nitrogens. The two Zn–O bonds are similar (1.895(6) and 1.907(6) Å) and the Zn–N (azomethine) bond 2.019(6)–2.030(6) Å appreciably shorter than the corresponding one in the L17–ZnII complex (2.174(6)–2.247(6) Å). The coordination environment is pseudo-tetrahedral as judged by the N–Zn–N, O–Zn–O and N–Zn–O bond angles. The Zn(1)⋯Zn(2) separation (11.725 Å) is longer than that of the L17–ZnII complex (11.431 Å).
The aromatic rings in the spacer groups are almost parallel to each other (dihedral angle 19.6–21.9°). Effective π–π stacking is observed between these aromatic rings (3.4–3.9 Å). The flexible methine group of the spacer group (-C6H4CH2C6H4-) and the phenolate coordinating group would lead to the double-helical motif. It is noted that two CHCl3 molecules per complex are trapped within the right-hand cleft. These solvent molecules are held by weak C(H)⋯Cl hydrogen bonds (3.3–3.9 Å). In the crystal lattice, the left-hand side of aromatic rings of the complex are efficiently stacked with those of another complex molecule.
X-Ray crystal structure of L35–ZnII complex, 3
Recrystallization from DMF–diethyl ether afforded orange blocks of 3·3DMF. The X-ray crystal structure (Fig. 9 and Table 4) of 3 shows a one-dimensional polynuclear structure as [ZnIIL35(DMF)2]n·(DMF)n·(ClO4)2n. The short spacer group (-C6H4-) of L35 prevents the structure from wrapping around the ZnII ions as observed in L17–ZnII and L16–ZnII. Each zinc ion is six-coordinated with four nitrogens from two L35 molecules. Two remaining coordination sites are occupied by two O-bound DMF molecules. Their Zn–O(DMF) bonds are 2.082 and 2.045 Å, respectively. It is noted that few DMF complexes with first-row transition metals have been crystallographically characterized, as pointed out by Ward et al.30 The Zn–N(CHN) [Zn(1)–N(2) 2.267 and Zn(1)–N(4) 2.238 Å] are appreciably longer than the Zn–N(py) bonds [Zn(1)–N(1) 2.143 and Zn(1)–N(3) 2.141 Å]. The coordination geometry at each ZnII center is pseudo-octahedral with bond angle in the range of O–Zn–O 92.5°, O–Zn–N 87.5–166.9° and N–Zn–N 76.5–161.5°.
| Zn(1)–O(1) | 2.082(3) | Zn(1)–O(2) | 2.045(4) |
| Zn(1)–N(1) | 2.143(5) | Zn(1)–N(2) | 2.267(4) |
| Zn(1)–N(3) | 2.141(5) | Zn(1)–N(4) | 2.238(4) |
| N(2)–C(6) | 1.276(7) | N(4)–C(18) | 1.274(7) |
| O(1)–Zn(1)–O(2) | 92.5(1) | O(1)–Zn(1)–N(1) | 92.2(2) |
| O(1)–Zn(1)–N(2) | 164.7(2) | O(1)–Zn(1)–N(3) | 100.7(1) |
| O(1)–Zn(1)–N(4) | 87.5(1) | O(1)–Zn(1)–N(1) | 102.2(2) |
| O(2)–Zn(1)–N(2) | 98.6(1) | O(2)–Zn(1)–N(3) | 90.6(2) |
| O(2)–Zn(1)–N(4) | 166.9(2) | N(1)–Zn(1)–N(2) | 75.2(2) |
| N(1)–Zn(1)–N(3) | 161.5(1) | N(1)–Zn(1)–N(4) | 90.9(2) |
| N(2)–Zn(1)–N(3) | 89.8(2) | N(2)–Zn(1)–N(4) | 84.1(1) |
| N(3)–Zn(1)–N(4) | 76.5(2) | ||
| Zn(1)–O(1)–C(19) | 124.7(3) | Zn(1)–O(2)–C(22) | 128.6(4) |
| Zn(1)–N(1)–C(1) | 125.9(3) | Zn(1)–N(1)–C(5) | 115.3(4) |
| Zn(1)–N(1)–C(5) | 115.3(4) | Zn(1)–N(2)–C(6) | 110.6(3) |
| Zn(1)–N(2)–C(7) | 125.9(3) | C(6)–N(2)–C(13) | 120.9(4) |
| Zn(1)–N(3)–C(13) | 126.9(4) | Zn(1)–N(3)–C(17) | 114.9(4) |
| Zn(1)–N(4)–C(10*) | 128.2(3) | Zn(1)–N(4)–C(18) | 111.5(3) |
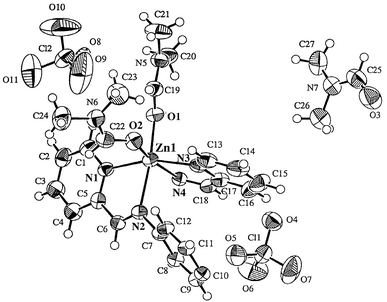 | ||
| Fig. 9 X-ray crystal structure for 3·3DMF. | ||
Chirality in packing mode
In the solid state of the L17–ZnII complex, it is noted that the ClO4− distribution around the complex cation is unsymmetrical. Three counter ClO4− ions are located at the periphery of [(ZnII)2(L17)3]4+ cation [Zn(1)⋯ClO4− (6.307 and 6.800 Å) and Zn(2)⋯ClO4− (7.579 Å)] while one ClO4− ion is situated far from the complex cation to fill the lattice space [Zn(1)⋯ClO4− and Zn(2)⋯ClO4− (12.814 and 12.365 Å)]. In addition, two acetonitrile molecules and one DMF molecule are placed in the pocket formed by the wrapped conformation of three L17 ligands.The [(ZnII)2(L17)3]4+ cations have Δ–Δ or Λ–Λ configuration with the ZnII ions. A view from the a–c plane in the crystal packing of 1 is presented in Fig. 10(a). The Δ–Δ and Λ–Λ optical isomers of 1 are alternately arrayed in the a–c plane. Four ZnII ions from two [(ZnII)2(L17)3]4+ cations along the a-axis are situated in the same plane and form a rhombic arrangement with the sum of interior angles 360° (11.431 × 14.459 Å, 47.3 and 132.7°). The other interesting projection of the crystal packing diagram in the a–b plane is two intersected Δ–Δ or Λ–Λ isomers with the bite angle 56.6°.
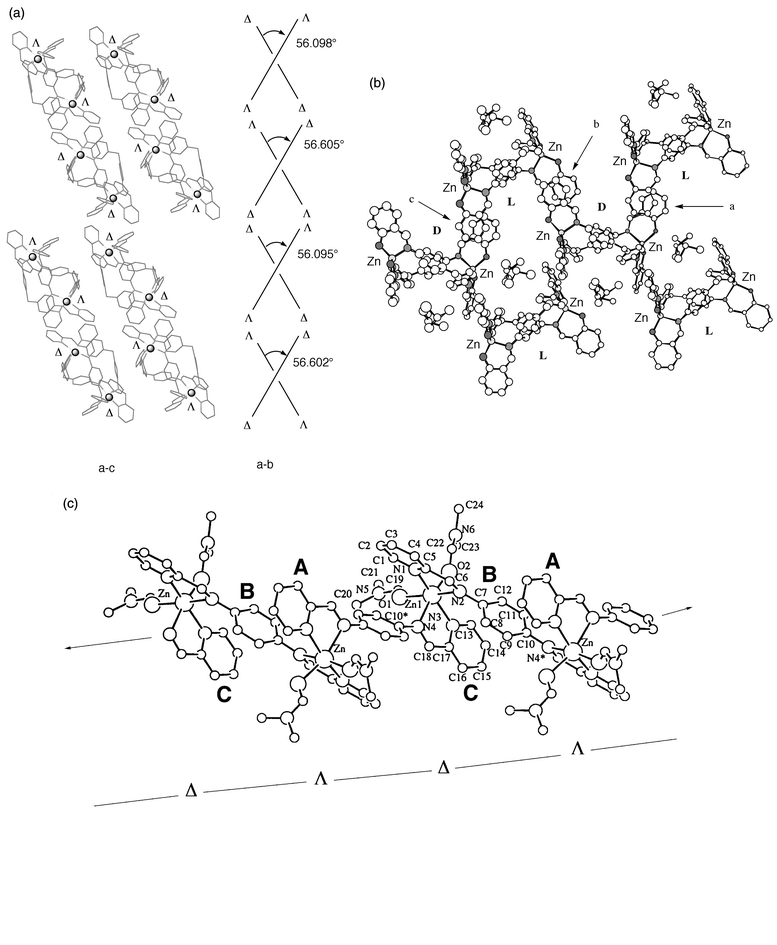 | ||
| Fig. 10 Chiral packing mode in the solid state of 1 (a), 2 (b) and 3 (c). | ||
A projection of the crystal packing of 2 in the b–c plane is presented in Fig. 10(b). Two layers, right-hand (D) and left-hand (L) helicates of the neutral complex 2 are observed in this plane. Sections of the aromatic rings depicted at a, b and c (dihedral angle 2°) have a face-to-face arrangement resulting in infinite π-stacking interactions (roughly the c axis in Fig. 10(b)) between adjacent D and L optical isomers of 2.
A polynuclear zigzag arrangement is observed in the crystal packing of 3 (Fig. 10(c)). In this arrangement, 1D chain with strong-conjugated π-system is formed in the alternating –Δ–Λ–Δ–Λ–Δ– chains. The Zn⋯Zn separation is 9.033 Å. Furthermore, duplicated π-stacking from overlap of aromatic rings in interligand py(A)-spacer(B)-py(C) sites are observed.
Solution structure of L17–ZnII and L16–ZnII complexes
The crystal structure of complex 1 shows the formation of dinuclear triple-helical [(ZnII)2(L17)3]4+ species, while ESI mass spectrum indicates the mixture of some aggregated species such as (ZnII∶L17)n+ = (1∶1)+, (1∶2)+, (2∶2)+, and (2∶3)+ species. If the L17–ZnII complex is to retain a (2∶3)+ structure in solution, the 1H NMR spectrum should exhibit one set of sharp signals. The 1H NMR spectra in DMF-d7 of complex 1 and a comparison with its ligand L17 are shown in Fig. 11.4, 1.](/image/article/2000/P2/a908041d/a908041d-f11.gif) | ||
| Fig. 11 1H NMR spectra of L17 and its ZnII complex [(ZnII)2(L17)3](ClO4)4, 1. | ||
The room temperature spectrum of 1 indicates peak broadening in all protons. This broadening may arise from instability of the complex or exchange on the NMR time scale between different species including ligand molecule. Sharper signals are obtained at − 20 °C. Since the exchange is fast on the 1H NMR time scale, we cannot determine precisely from these spectra alone the extent of ligand dissociation by integrating peaks associated with the zinc-bound and dissociated L17 ligand. In general, the protons of H(1) and H(2) of the py moiety tend to shift downfield (H(1)* and H(2)*) upon complexation. In particular, the azomethine proton H(5) shows much a larger downfield shift due to the -CH(5)N- coordination to ZnII ion. However, two sets of spacer group resonances at H(6)* and H(7)* show large upfield shifts. These pronounced upfield shifts may be attributed to the CH⋯π interaction between the spacer aromatic protons and py moiety, which is mentioned for the [(ZnII)2(L17)3]4+ complex in the solid-state.
In contrast to complex 1, the 1H NMR spectrum of 2 in CDCl3 at 25 °C is simple to interprete and sharply resolved into the ligand peak and complex peak (Fig. 12). The integrating ratio of peak is about 1∶2 (ligand∶complex). Upon complexation of L16 with ZnII ion, large upfield shift, due to the deprotonation of the phenolic group, is observed particularly in the protons of H(3) and H(5). The large upfield shift observed in the protons of spacer group (H(6)→H(6)* and H(7)→H(7)*) of 2 would be ascribed to the existence of CH⋯π interactions in solution.
![1H NMR spectra of L16 and its ZnII complex [(ZnII)2(L16)2], 2.](/image/article/2000/P2/a908041d/a908041d-f12.gif) | ||
| Fig. 12 1H NMR spectra of L16 and its ZnII complex [(ZnII)2(L16)2], 2. | ||
Conclusion
The series of ZnII complexes with L16, L17 and L35 have been found to show interesting variations of structure in the solid-state and solution. Three ZnII-assisted supramolecular motifs were presented using the bis-bidentate Schiff bases L16, L17 and L35, having flexible aromatic spacer groups. X-Ray crystal diffraction characterization revealed that the L16–ZnII, L17–ZnII and L35–ZnII complexes display dinuclear double-helical, dinuclear triple-helical and 1-D polynuclear architectures in the solid-state, depending on subtle differences of coordination site and spacer group in the ligand. UV/VIS, ESI-MS and 1H NMR data were also consistent with the formation of these species in solution. The coordinatively flexible ZnII ion and the aromatic–aromatic interactions between the flexible spacer group in the wrapped ligands would be a crucial factor for the formation of the supramolecular structure. Three types of chiral-linking mode have been generated for the crystal packing in the L16–ZnII, L17–ZnII and L35–ZnII complexes.References
- Part 4. N. Yoshida, H. Oshio and T. Ito, J. Chem. Soc., Perkin Trans. 2, 1999, 975 Search PubMed.
- N. Yoshida and K. Ichikawa, Chem. Commun., 1997, 1091 RSC; N. Yoshida, H. Oshio and T. Ito, Chem. Commun., 1998, 63 RSC.
- N. Yoshida, N. Ito and K. Ichikawa, J. Chem. Soc., Perkin Trans. 2, 1997, 2387 RSC.
- M. J. Hannon, C. L. Painting and N. W. Alcock, Chem. Commun., 1999, 2023 RSC; L. Douce, A. El-ghayoury, A. Skoulios and R. Ziessel, Chem. Commun., 1999, 2033 RSC; S. Brooker, P. G. Plieger, B. Moubaraki and K. S. Murray, Angew. Chem., Int. Ed., 1999, 38, 408 CrossRef CAS; M. J. Hannon, S. Bunce, A. J. Clarke and N. W. Alcock, Angew. Chem., Int. Ed., 1999, 38, 1277 CrossRef CAS; S. Brooker, R. J. Kelly and P. G. Plieger, Chem. Commun., 1998, 1079 RSC; P. K. Bowyer, K. A. Porter, A. D. Rae, A. C. Willis and S. B. Wild, Chem. Commun., 1998, 1153 RSC; M. J. Hannon, C. L. Painting, A. Jackson, J. Hamblin and W. Errington, Chem. Commun., 1997, 1807 RSC; P. Comba, A. Fath, G. Huttner and L. Zsolnai, Chem. Commun., 1996, 1885 RSC.
- B. J. McNeils, L. C. Nathan and C. J. Clark, J. Chem. Soc., Dalton Trans., 1999, 1831 RSC; M.-L. Tong, X.-M. Chen, B. H. Ye and L.-N. Ji, Angew. Chem., Int. Ed., 1999, 38, 2237 CrossRef CAS.
- C. Bonnefous, N. Bellec and R. P. Thummel, Chem. Commun., 1999, 1243 RSC.
- (a) K. L. V. Mann, J. C. Jeffery, J. A. McCleverty, P. Thornton and M. D. Ward, J. Chem. Soc., Dalton Trans., 1998, 89 RSC; (b) J. C. Jeffery, P. L. Jones, K. L. V. Mann, E. Psillakis, J. A. McCleverty, M. D. Ward and C. M. White, Chem. Commun., 1997, 175 RSC.
- O. Mamula, A. von Zelewsky and G. Bernardinelli, Angew. Chem., Int. Ed., 1998, 37, 290 CrossRef.
- R. W. Saalfrank, N. Löw, B. Demleitner, D. Stalke and M. Teichert, Chem. Eur. J., 1998, 4, 1305 CrossRef CAS.
- D. C. Caulder, R. E. Powers, T. N. Parac and K. N. Raymond, Angew. Chem., Int. Ed., 1998, 37, 1840 CrossRef CAS.
- J. R. Farrell, C. A. Mirkin, I. A. Guzei, L. M. L.-Sands and A. L. Rheingold, Angew. Chem., Int. Ed., 1998, 37, 465 CrossRef CAS.
- M. Albrecht, Chem. Eur., 1997, 3, 1466 Search PubMed.
- V. A. Grillo, E. J. Seddon, C. M. Grant, G. Aromí, J. C. Bollinger, K. Folting and G. Christou, Chem. Commun., 1997, 1561 RSC; V. A. Grillo, M. J. Knapp, J. C. Bollinger, D. N. Hendrickson and G. Christou, Angew. Chem., Int. Ed. Engl., 1996, 35, 1818 CrossRef CAS.
- U. Velten and M. Rehahn, Chem. Commun., 1996, 2639 RSC.
- L. J. Charbonniére, M.-F. Gilet, K. Bernauer and A. F. Williams, Chem. Commun., 1996, 39 RSC.
- (a) J.-M. Lehn , Supramolecular Chemistry, VCH, Weinheim, 1995; Search PubMed; (b) D. Philp and J. F. Stoddart, Angew. Chem., Int. Ed. Engl., 1996, 35, 1154 CrossRef.
- B. Hasenknopf, J.-M. Lehn, N. Boumediene, E. Leize and A. V. Dorsselaer, Angew. Chem., Int. Ed., 1998, 37, 3265 CrossRef CAS; B. Hasenknopf, J.-M. Lehn, N. Boumediene, A. D.-Gervais, A. V. Dorsselaer and D. Fenske, J. Am. Chem. Soc., 1997, 119, 10956 CrossRef CAS; B. Hasenknopf, J.-M. Lehn, B. O. Kneisel, G. Baum and D. Fenske, Angew. Chem., Int. Ed. Engl., 1996, 35, 1838 CrossRef CAS.
- R. Vilar, D. M. P. Mingos, A. J. P. White and D. J. Williams, Chem. Commun., 1999, 229 RSC; J. S. Fleming, K. L. V. Mann, C.-A. Carraz, E. Psillakis, J. C. Jeffery, J. A. McCleverty and M. D. Ward, Angew. Chem., Int. Ed., 1998, 37, 1279 CrossRef CAS; D. A. McMorran and P. J. Steel, Angew. Chem., Int. Ed., 1998, 37, 3295 CrossRef CAS; P. L. Jones, J. C. Jeffery, J. A. McCleverty and M. D. Ward, Chem. Commun., 1997, 1361 RSC.
- R.-D. Schnebeck, E. Freisinger and B. Lippert, Angew. Chem., Int. Ed., 1999, 38, 168 CrossRef CAS; S. Mann, G. Huttner, L. Zsolnai and K. Heinze, Angew. Chem., Int. Ed. Engl., 1996, 35, 2808 CrossRef CAS.
- M. Albrecht, O. Blau and R. Fröhlich, Chem. Eur. J., 1999, 5, 48 CrossRef CAS; M. Albrecht, H. Röttele and P. Burger, Chem. Eur. J., 1996, 2, 1264 CrossRef CAS.
- M. Tadokoro, K. Isobe, H. Uekusa, Y. Ohashi, J. Toyoda, K. Tashiro and K. Nakasuji, Angew. Chem., Int. Ed., 1999, 38, 95 CrossRef CAS; H. Miyasaka, S. Okamura, T. Nkashima and N. Matsumoto, Inorg. Chem., 1997, 36, 4329 CrossRef CAS.
- A. Marquis-Rigault, A. Dupont-Gervas, A. V. Dorsselaer and J.-M. Lehn, Chem. Eur. J., 1996, 2, 1395 CrossRef CAS; C. R. Woods, M. Benaglia, F. Cozzi and J. S. Siegel, Angew. Chem., Int. Ed. Engl., 1996, 35, 1830 CrossRef CAS.
- D. C. Rees, M. Lewis and W. N. Lipscomb, J. Mol. Biol., 1983, 168, 367 CAS; D. R. Holland, A. C. Haustrath, D. Juers and B. W. Matthews, Protein Sci., 1995, 4, 1955 CrossRef CAS.
- S. K. Nair and D. W. Christianso, J. Am. Chem. Soc., 1991, 113, 9455 CrossRef CAS; D. N. Silverman and S. Lindskog, Acc. Chem. Res., 1988, 21, 30 CrossRef CAS.
- E. E. Kim and H. W. Wyckoff, J. Mol. Biol., 1991, 218, 449 CrossRef CAS.
- M. Nishio , M. Hirota and Y. Umezawa , The CH/π Interaction-Evidence, Nature and Consequences, Wiley-VCH, New York, 1998. Search PubMed.
- SIR92: A. Altomare , M. C. Burla , M. Camalli , M. Cascarano , C. Giacovazzo , A. Guagliardi and G. Polidori , J. Appl. Crystallogr., 27, 435 ( 1994). Search PubMed.
- DIRDIF94 (1994): P. T. Beurskens , G. Admiraal , G. Beurskens , W. P. Bosman , R. de Gelder , R. Israel and J. M. M. Smiths , The DIRDIF-94 program system, Technical Report of the Crystallography Laboratory, University of Nijmegen, The Netherlands. Search PubMed.
- teXsan: Crystal Structure Analysis Package, Molecular Structure Corporation ( 1985 and 1992). Search PubMed.
- D. A. Bardwell, J. C. Jeffery and M. D. Ward, J. Chem. Soc. Dalton Trans., 1995, 3071 RSC.
| This journal is © The Royal Society of Chemistry 2000 |