Chirality control of a Cu(I)·(phenanthroline)2 complex by a sugar–boronic acid interaction. A preliminary step toward the total chain helicity control by a chain-end sugar-binding
Received
(in Cambridge, UK)
21st June 1999
, Accepted 1st November 1999
First published on 8th March 2000
Abstract
Compounds 1a and 1b which have a 1,10-phenanthroline moiety, were synthesized in order to form a helical structure in the presence of Cu(I), and a boronic acid moiety with which to bind a saccharide at the chain end. When saccharides were added, the Cu(I) complexes (as 1a2·Cu(I) and 1b2·Cu(I)) gave the CD-active species reflecting the absolute configurational structure of saccharides. Thus, the P versus M helicity of the complexes can be controlled by the boronic acid–saccharide interaction. The results show that the terminal boronic acid group is useful to create the chiral helical structure and the total helicity is governed by the chirality of the boronic acid-bound saccharide.
Introduction
Helical complexes have been of great interest as examples of self-assembled supramolecular structures in artificial systems and as models for DNA and RNA structures in nature.1 One important characteristic which differentiates the helical structure from other supramolecular structures is the ‘chirality’ generated by the twisting direction along the one-dimensional chain.2–15 In general, the ‘chirality’ is created by the introduction of chiral substituents into ‘helicates’. Meanwhile, Hamilton et al.16 have demonstrated that self-assembled helical metal complexes with terminal hydrogen-bonding sites are useful for the recognition of dicarboxylic acid guests. The results imply that the host–guest-type interaction at the helical chain end may crucially control the twisting direction of the total chain helicity. Recently, it has been shown that boronic acid–saccharide covalent interactions, which form readily and reversibly in aqueous media, represent an important alternative binding force for the recognition of saccharides and related molecular species.17–30 Here, it occurred to us that the chirality of the helical metal complexes created from helicates bearing a terminal boronic acid group may be reversibly controlled by the boronic acid–saccharide interaction. If this working hypothesis is correct, it follows that a saccharide library, containing abundant chirality resources, would be useful to create a variety of helical structures. As the first step to test this intriguing working hypothesis, we designed compounds 1a and 1b which have a 1,10-phenanthroline moiety with which to constitute the helical metal complex and an o-aminomethylphenyl boronic acid moiety with which to bind saccharides at the helical chain end. Compound 1a was mainly used for the absorption and CD spectroscopic studies whereas compound 1b was used for the 1H NMR spectroscopic studies because the p-anisyl group gives well-split peaks which are useful as a marker to detect configurational changes. Interestingly, we have found that, as shown in Scheme 1, added saccharides can influence the equilibrium between plus (P) and minus (M) enantiomers, reflecting their absolute configurational structure.31
 |
| | Scheme 1 | |
Results and discussion
Molecular design and synthesis of ligands (1a and 1b)
To test the working hypothesis we chose the chiral induction in a Cu(I)·(1,10-phenanthroline) complex. Examination with CPK molecular models and computational tools suggested that introduction of a boronic acid group into the 9-position of 1,10-phenanthroline via a phenyl group would satisfy the requirements: that is, the metal-binding 1,10-phenanthroline site is moderately insulated by the phenyl group from the saccharide-binding boronic acid site whereas the chirality in the Cu(I) complex seems sterically controllable by the boronic acid–saccharide interaction. The o-aminomethyl group interacts intramolecularly with the boronic acid group and stabilises the saccharide complex.9,10 Thus, compounds 1a (mp 174–179 °C) and 1b (mp 179–183 °C) were synthesised from 2-phenyl-9-p-tolyl-1,10-phenanthroline (2a) and 2-p-anisyl-9-p-tolyl-1,10-phenanthroline (2b), respectively, according to Scheme 2 and identified by 1H NMR and IR spectral evidence and elemental analyses (see Experimental section).
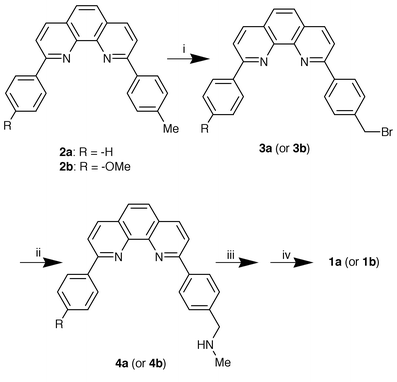 |
| | Scheme 2
Reagents and conditions (yield): i, NBS, AIBN, CCl4, reflux; ii, MeNH2, CCl4 (62%, calculated from 2a or 46%, calculated from 2b); iii, K2CO3, MeCN, reflux; iv, H2O (46%, calculated from 4a or 34%, calculated from 4b).
| |
Fig. 1 shows the absorption spectral change induced by the Cu(I) addition (added as [Cu(MeCN)4]ClO4). The λmax at 309 nm decreased while that at 439 nm increased (as shown in an inserted figure) with tight isosbestic points at 364 and 292 nm. The plots of the absorbances against [Cu(I)]/[1a] (Fig. 2) afforded a clear break-point at 0.5, indicating that the complex consists of one Cu(I) and two 1a ligands (as illustrated in Scheme 1). In the subsequent CD measurements we enhanced the concentrations up to [1a] = 0.200 mmol dm−3 and [Cu(I)] = 0.100 mmol dm−3 because the CD spectral change was not so sensitive as the absorption spectral change. Judging from the sharp break-point in Fig. 2, one can assume that 1a and Cu(I) are fully converted to the 1a2·Cu(I) complex under these CD measurement conditions.
 |
| | Fig. 1 Absorption spectral change of 1a (0.100 mmol dm−3) with increasing Cu(I) concentration.
| |
![Plots of absorbance versus [Cu(I)]/[1a]: •; 439.0 nm, ○; 309.0 nm.](/image/article/2000/P2/a904936c/a904936c-f2.gif) |
| | Fig. 2 Plots of absorbance versus [Cu(I)]/[1a]: •; 439.0 nm, ○; 309.0 nm.
| |
Stoichiometry of the 1a2·Cu(I)·D-glucose complex
In order to estimate the chiral twisting ability of monosaccharides for the 1a2·Cu(I) complex we primarily tested D-glucose as a representative monosaccharide. When D-glucose was added to the 1a2·Cu(I) complex solution, the CD bands appeared gradually at 250–350 nm region and 400–600 nm region (MLCT region: Fig. 3). This slow CD appearance implies that D-glucose is bound to the boronic acid groups rather slowly in the organic medium and/or D-glucose-induced conversion of one enantiomer to other occurs slowly. The appearance of the CD bands means that one enantiomer of the ternary complex has become in excess of the other. A plot of the CD intensity vs. time revealed that the CD intensity becomes constant after 6–7 h. In the following experiments, therefore, we measured the final CD spectra after 12 h. The continuous variation plots of the CD intensity vs. [1a2·Cu(I)]/([1a2·Cu(I)] + [D-glucose]) provided a maximum at 0.5, indicating that the ternary complex consists of 1∶1 stoichiometric 1a2·Cu(I) and D-glucose (Fig. 4). This finding was also supported by mass spectrometry (ESI-MS). To a MeCN–MeOH = 1∶1 (v/v) solution containing the 1a2·Cu(I) complex (1.00 × 10−4 mol dm−3) was added D-glucose (5.00 × 10−4 or 7.50 × 10−4 mol dm−3) and the solution was incubated at 25 °C for 12 h. This solution was subjected to the ESI-MS measurement. One strong peak (m/z = 1189) and several weak accompanying peaks with Δm/z = 1.0 were observed. The results indicate that the major species under the ESI-MS measurement conditions is [1a2·Cu(I)·D-glucose]+. These spectroscopic data consistently support the complexation mode as illustrated in Scheme 1: that is, the 12·Cu(I) complex binds one D-glucose molecule with two boronic acid–diol interactions to form a macrocyclic structure.
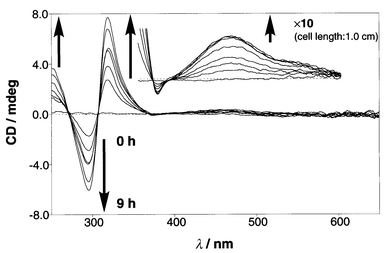 |
| | Fig. 3 Time dependence of the CD appearance after the addition of D-glucose (0.100 mmol dm−3) to the solution containing 1a2·Cu(I) (0.100 mmol dm−3): cell length 0.1 cm; (inserted) cell length 1.0 cm.
| |
![Continuous variation plots: the [1a2·Cu(I)] + [D-glucose] concentration was maintained constant (0.200 mmol dm−3): •; 463.0 nm, ○; 318.5 nm.](/image/article/2000/P2/a904936c/a904936c-f4.gif) |
| | Fig. 4 Continuous variation plots: the [1a2·Cu(I)] + [D-glucose] concentration was maintained constant (0.200 mmol dm−3): •; 463.0 nm, ○; 318.5 nm.
| |
Possible correlation between the saccharide structure and the CD sign
The CD spectra were measured as a function of the D- and L-glucose concentrations while the concentration of 1a2·Cu(I) was maintained constant (1.00 × 10−4 mol dm−3). The CD spectra changed with a few tight isosbestic points. As expected, the CD spectra obtained in the presence of L-glucose were symmetrical to those obtained in the presence of D-glucose (Fig. 5). Plots of the CD intensities at 319 and 464 nm vs. the D- and L-glucose concentrations are shown in Fig. 6. From analysis of the plots in Fig. 6 by computational curve fitting one can estimate the binding constant (Kb) to be 4800 ± 400 dm3 mol−1 for both D- and L-glucose.32
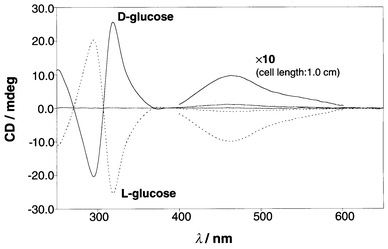 |
| | Fig. 5 CD spectra of 1a2·Cu(I) (0.100 mmol dm−3) in the presence of D- or L-glucose (7.50 mmol dm−3).
| |
![Plots of CD intensities (319 and 464 nm) vs. D- and l-glucose concentrations: [1a2·Cu(I)] = 0.100 mmol dm−3, D-glucose (•; 319 nm, ○; 464 nm), l-glucose (▲; 319 nm, △; 464 nm).](/image/article/2000/P2/a904936c/a904936c-f6.gif) |
| | Fig. 6 Plots of CD intensities (319 and 464 nm) vs. D- and L-glucose concentrations: [1a2·Cu(I)] = 0.100 mmol dm−3, D-glucose (•; 319 nm, ○; 464 nm), L-glucose (▲; 319 nm, △; 464 nm).
| |
Fig. 5 indicates that at the MLCT region (400–600 nm) D-glucose gives the positive CD sign whereas L-glucose gives the negative CD sign. The past CD studies on chiral bipyridine-based helicate–Cu(I) complexes have established that the positive CD sign is generated from the P-isomer whereas the negative CD sign is generated from the M-isomer:6,7,9 that is, D-glucose twists the 1a2·Cu(I) complex into the P chirality motif (clockwise direction around the central axis connecting Cu(I) with glucose) whereas L-glucose twists it into the M chirality motif (anti-clockwise direction around the central axis connecting Cu(I) with glucose). The exciton-coupling CD bands at around 300 nm reflect the π–π* transition in the phenanthroline moieties. Examination of the CD sign reveals that the positive exciton-coupling interaction is observed for D-glucose whereas the negative exciton-coupling interaction is observed for L-glucose (Fig. 5): that is, P-isomer and M-isomer in the MLCT region are always correlated with the positive exciton-coupling interaction and the negative exciton-coupling interaction, respectively, in the phenanthroline moieties.
To obtain a possible correlation between the saccharide structure and the CD sign we measured the CD spectra of 1a2·Cu(I) for seven D-saccharides in addition to D-glucose. The structures are illustrated in Scheme 3 (mainly, as their pyranose forms).
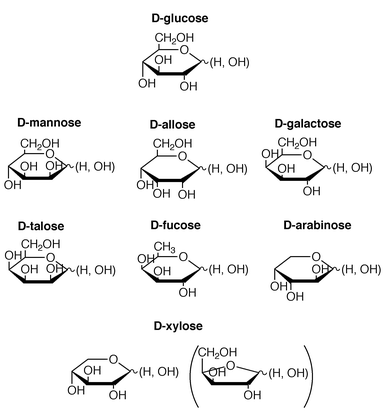 |
| | Scheme 3 | |
Firstly, we compared the CD spectra of D-mannose, D-allose and D-galactose which are classified into epimers of D-glucose. The typical concentration-dependent CD spectra and the typical plots of the CD intensity at the exciton-coupling band versus [D-saccharide] are shown in Figs. 7 and 8. Examination of these figures, together with that of Figs. 5 and 6, reveals that (i) D-mannose and D-galactose give the positive CD sign for the MLCT region and the positive exciton-coupling band for the phenanthroline region like D-glucose whereas D-allose gives the negative CD sign for the MLCT region and the negative exciton-coupling band for the phenanthroline region like L-glucose, (ii) the plus–minus in the MLCT region is always correlated with the plus–minus in the phenanthroline π–π* transition region and (iii) the absolute CD intensities in the MLCT region and the phenanthroline region both appear in the order of D-glucose > D-galactose > D-mannose > D-allose. Most interesting is the finding that the CD signs for D-allose are inverted from those for other three saccharides.
The spectroscopic data are summarised in Table 1. Eight saccharides tested herein all afforded the CD-active species with 1a2·Cu(I) and the plus–minus in the MLCT region is correlated with the plus–minus in the phenanthroline π–π* transition region (except D-talose, which did not give a clear exiton-coupling band). Very interestingly, we noticed that the plus–minus in the MLCT region is predictable from the absolute configuration of the 3-OH: when the 3-OH group is ‘up’, the MLCT region becomes plus whereas when it is ‘down’, the MLCT region becomes minus. The origin of this intriguing correlation is not yet explicable. We now consider that when the 1,2-diol group forms a complex with the boronic acid group,21,22 the absolute configuration of the neighboring 3-OH affects the twisting direction of the 1a2·Cu(I)·saccharide complexes. On the other hand, 1-methyl-α-D-glucopyranoside, in which the 1-OH group useful for the complexation is protected by the methyl group, was CD-silent. This suggests an idea that as already reported in related system,21,22,25 the 1,2-diol in monosaccharides acts as an essential boronic acid-binding site.
Table 1 CD spectroscopic data obtained in the presence of various monosaccharides![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) a
a
| |
|
|
|
|
Structure of monosaccharide |
| Monosaccharide |
π–π* bandbλmax or min/nm (CDmax or min intensity/mdeg) |
MLCT bandcλmax or min/nm (CDmax or min intensity/mdeg) |
Helicity |
1,2-diol |
3-OH |
|
[1a2·Cu(I)] = 0.100 mmol dm−3, [monosaccharide] = 7.50 mmol dm−3 ([D-galactose] = 1.50 mmol dm−3 [D-mannose] = 3.00 mmol dm−3), MeOH–MeCN = 1∶1 (v/v), 25 °C.
Cell length: 0.1 cm.
Cell length: 1.0 cm.
|
|
D-Glucose |
319.0 |
295.0 |
463.5 |
P |
down |
up |
| |
(25.61) |
(−20.34) |
(10.23) |
|
|
|
|
D-Fucose |
322.0 |
299.5 |
480.5 |
P |
down |
up |
| |
(5.25) |
(−3.38) |
(2.31) |
|
|
|
|
D-Galactose |
321.5 |
290.0 |
498.0 |
P |
down |
up |
| |
(3.06) |
(−1.97) |
(1.38) |
|
|
|
|
D-Xylose |
318.0 |
297.5 |
458.0 |
P |
down |
up |
| |
(1.85) |
(−0.51) |
(0.64) |
|
|
|
|
D-Mannose |
319.0 |
296.0 |
463.5 |
P |
up |
up |
| |
(0.89) |
(−0.86) |
(0.50) |
|
|
|
D-Talose |
317.5 |
511.5 |
P |
up |
up |
| |
(1.01) |
(0.56) |
|
|
|
D-Arabinose |
319.5 |
295.0 |
490.5 |
M |
up |
down |
| |
(−2.12) |
(1.51) |
(−1.03) |
|
|
|
|
D-Allose |
319.5 |
290.0 |
491.0 |
M |
down |
down |
| |
(−1.00) |
(0.56) |
(−0.47) |
|
|
| |
|
|
|
|
|
| Methyl-α-D-glucoside |
CD silent |
CD silent |
|
|
|
As a summary of the foregoing findings, one can now regard that the chirality in D-saccharides is transmitted to the helicity in the metal complex.
Estimation of the binding constants (Kb)
From plots of the CD intensity at the π–π* transition band against [D-saccharide] (Fig. 8) the Kb values were estimated assuming the formation of 1a2·Cu(I)∶D-saccharide = 1∶1 complexes. The band for D-talose was too weak to estimate the Kb accurately. The plot for D-mannose was biphasic. It has been established through the CD spectroscopic studies that the complex becomes CD-active only when the saccharide is recognised by the host at least at two points, forming a 1∶1 stoichiometric cyclic structure whereas it becomes CD-silent when it is converted into a 1∶2 host/saccharide noncyclic structure in the high saccharide concentration region.22,23,25,33,34 This plot suggests, therefore, a shift of a 1a2·Cu(I)∶D-mannose = 1∶1 cyclic complex to a 1a2·Cu(I)∶D-saccharide = 1∶2 noncyclic complex (CD-silent)25 with increasing D-mannose concentration.22–25 Hence, both the Kb for the 1∶1 complex with seven saccharides and the Kb′ for the 1∶2 complex with D-mannose were estimated by analysing these plots with a nonlinear least-squares method.32–34 The results are summarised in Table 2.
Table 2 Association constants for saccharides with 1a2·Cu(I)a
It is seen from Table 2 that (i) the Kb values appear in the order of D-glucose > D-mannose > D-xylose > D-arabinose > D-galactose > D-fucose > D-allose, (ii) six D-saccharides (except D-mannnose) show a single saturation curve, indicating that the cyclic 1a2·Cu(I)∶D-mannose = 1∶1 complex is more stable than the noncyclic 1a2·Cu(I)∶D-saccharide = 1∶2 complex and (iii) only in the binding of D-mannose, the formation of the noncyclic 1∶2 complex can compete with that of the cyclic 1∶1 complex although the Kb is still greater by 14-fold than the Kb′. The order of the Kb values is approximately coincident with the order of the CD intensity (D-glucose > D-galactose > D-mannose > D-allose: vide supra). This implies that the more stable is the cyclic 1∶1 complex, the more stronger becomes the CD intensity. On the other hand, it is not clear yet why the Kb′ for the D-mannose complex is exceptionally large compared with others in spite of the large Kb next to D-glucose.
Why does 1a2·Cu(I) generally tend to form P-isomers with D-monosaccharides (except D-arabinose and D-allose)? In the present system, it is not yet clear which form of furanose vs. pyranose is immobilised by the 1a2·Cu(I) complex.21,24,35 We thus energy-minimised the structure of the ternary 1a2·Cu(I)∶
D-glucose complex using the ESFF forcefield with molecular mechanics (see Experimental section) assuming both the furanose form and the pyranose form for the D-glucose moiety.36 For the 1a2·Cu(I)∶D-glucofuranose complex, there was no significant difference between the P-isomer and the M-isomer. On the other hand, the energy-minimised structure for the P-isomer of 1a2·Cu(I)∶D-glucopyranose is more or less symmetrical whereas that for the M-isomer is sterically distorted (Fig. 9). Careful examination of the D-glucopyranose moiety reveals that (i) in the Newman projection of the 1C–2C bond, the P-isomer can adopt an energetically-favourable staggered conformation whereas the M-isomer is enforced to adopt an energetically-unfavourable eclipsed conformation and (ii) the pyranose ring in the P-isomer is a chair form whereas that in the M-isomer is a twisted boat form. These lines of conformational difference should be effective to stabilise the P-isomer in preference to the M-isomer.
 |
| | Fig. 9 Energy-minimised structures for the P-isomer and the M-isomer for 1a2·Cu(I)·D-glucopyranose.
| |
On the ‘optical purity’ of the 1b2·Cu(I)·D-glucose complex
The 1a2·Cu(I) complex is an interconvertible racemic mixture of the P- and M-isomer. The complexation with the D-saccharide results in a diastereomeric mixture of 1a2·Cu(I)·D-saccharide. Since the P∶M ratio is changed by the D-saccharide binding, we tried to estimate the ‘optical purity’ of the P- or M-isomer induced by the binding of the D-saccharide. It seemed to us that 1H NMR spectroscopy is the sole tool able to solve this problem. To obtain clear 1H NMR spectra we chose D-glucose, as this gave the largest Kb value.37 As shown in the CD spectral measurements, the reaction rate for D-glucose binding is fairly slow. We therefore left the samples for 6 h at room temperature and then started the 1H NMR measurements.
Basically, the 1a2·Cu(I)·D-glucose complex involves two chiral centers, one in the (phenanthroline)2·Cu(I) moiety (P versus M) and the other in the bound D-glucose moiety. As a result, it provides a diastereomeric mixture, which should give the split 1H NMR peaks. In the 1H NMR spectrum of the 1a2·Cu(I)·D-glucose complex, however, we could find no such sharp peak that would be conveniently useful as a marker for peak splitting. We thus synthesised 1b, expecting that the p-anisyl group would be useful as a marker. Compound 1b showed complexation properties (as in Figs. 1 and 2) similar to 1a and the 1b2·Cu(I) complex showed binding properties for D-glucose (as in Figs. 3, 4 and 5) similar to the 1a2·Cu(I) complex. The 1∶1 stoichiometry between 1b2·Cu(I) and D-glucose was corroborated not only by a Job plot (as in Fig. 5) but also by ESI-MS spectrometry (m/z = 1250, which is assignable to [1b2·Cu(I)·D-glucose]+). From plots of the CD intensity at 322 nm and 459 nm vs. [D-glucose] the Kb was estimated to be 3900 ± 200 dm3 mol−1.
As shown in Fig. 10, the ortho-protons in the anisole moieties appeared as a doublet peak due to the coupling with the meta-protons in the absence of D-glucose. In the presence of D-glucose it changed into a pair of doublets. This change can be explained in two different ways: that is, (i) as mentioned above, the racemic mixture of 1b2·Cu(I) becomes a diastereomeric mixture after binding of D-glucose (Rationale A) or (ii) binding of D-glucose changes the interconvertible racemic mixture into the P-isomer of 1b2·Cu(I)·D-glucose and the two anisole moieties become inequivalent because of the unsymmetrical structure induced by the bound D-glucose (Rationale B). We believe that this splitting pattern should be attributed to Rationale B because (i) strong CD spectra were observed for both 1a2·Cu(I)·D-glucose and 1b2·Cu(I)·D-glucose, the θ values (1.03 × 104 deg cm2 dmol−1 at 464 nm and 1.06 × 104 deg cm2 dmol−1 at 459 nm, respectively) of which are comparable with those of optically-pure, bipyridine-based helicate–Cu(I) complexes (ca. 1.5 × 104 deg cm2 dmol−1 at 450–475 nm,6 1.73 × 104 deg cm2 dmol−1 at 479.3 nm9), (ii) large [α]25D values were observed for 1a2·Cu(I)·D-glucose and 1b2· Cu(I)·D-glucose [+2520 and +1300°, respectively: c = 0.048 g/100 ml (1a2·Cu(I)·D-glucose), 0.062 g/100 ml (1b2·Cu(I)·
D-glucose), solvent: MeCN–MeOH = 1∶1 (v/v)], which are sufficiently greater than those of optically-pure, bipyridine-based helicate–Cu(I) complex (+361,6 +501°9) and (iii) the absorption spectral change which reflects the binding of D-glucose to 1a2·Cu(I) or 1b2·Cu(I) was completed in a few minutes after the D-glucose addition38 whereas the CD spectral change which reflects the P–M isomerisation took several hours. Points (i) and (ii) clearly rule out the possibility that 1b2·Cu(I)·D-glucose is a diastereomeric mixture. Point (iii) is observable only when the P–M isomerisation proceeds within the 1a2·Cu(I)·D-glucose and 1b2·Cu(I)·D-glucose complexes. These lines of evidence clearly support the view that the 1b2· Cu(I)·D-glucose complex is not the diastereomeric mixture (Rationale A) but the P-isomer with the high optical purity. Judging from the sensitivity of the present 600 MHz 1H NMR apparatus, 5% of the isomer should be easily detectable (if it exists). We thus believe that the optical purity is higher than 95%.
![1H NMR spectra [600 MHz, CD3CN–CD3OD = 1∶1 (v/v)] of 1b2·Cu(I) (5.00 × 10−3 mol dm−3) at −15 °C in the absence (A) and the presence (B) of D-glucose (12.5 × 10−3 mol dm−3).](/image/article/2000/P2/a904936c/a904936c-f10.gif) |
| | Fig. 10
1H NMR spectra [600 MHz, CD3CN–CD3OD = 1∶1 (v/v)] of 1b2·Cu(I) (5.00 × 10−3 mol dm−3) at −15 °C in the absence (A) and the presence (B) of D-glucose (12.5 × 10−3 mol dm−3).
| |
Experimental
Materials
2-Phenyl-9-p-tolyl-1,10-phenanthroline (2a) and 2-anisyl-9-p-tolyl-1,10-phenanthroline (2b) were prepared according to the method of Goodman et al.3 and identified by IR and 1H NMR spectral evidence and elemental analysis.
2-(![[hair space]](https://www.rsc.org/images/entities/b_char_200a.gif) p-Methylaminomethylphenyl)-9-phenyl-1,10-phenanthroline (4a)..
Compound 2a (2.22 g, 6.4 mmol) was treated with N-bromosuccinimide (NBS) (1.37 g, 7.7 mmol) and α,α′-azobisisobutyronitrile (0.14 g, 10 wt% of NBS) in CCl4 at the reflux temperature for 50 min. The progress of the reaction was followed by 1H NMR spectroscopy [250 MHz, CCl4–CDCl3 = 1∶1 (v/v)] with the disappearance of δCH3 2.47 ppm and the appearance of δCH2Br 4.60 ppm. After cooling, the insoluble materials were removed by filtration, the filtrate being dried over Na2SO4. This solution containing 3a was used for the synthesis of 4a without further purification. Into the CCl4 solution cooled with an ice-bath was introduced methylamine gas for 4 h. The progress of the reaction was followed by a TLC method [silica gel, CH2Cl2–MeOH = 10∶1 (v/v)]. After 4 h when the spot for 3a (Rf = 0.90) disappeared, the reaction was finished by the addition of aqueous 5% NaHCO3 solution. The mixture was stirred for one day and then the insoluble materials were removed by filtration. The CCl4 layer was washed three times with aqueous 5% NaHCO3 solution and then dried over Na2SO4. The solution was concentrated to dryness, the solid residue being further purified through chromatography [silica gel, column φ 2.5 × 17 cm, CH2Cl2–MeOH = 10∶1 (v/v)] to yield 4a, 1.55 g (62%, calculated from 2a), slightly yellow powder, mp 83–87 °C; 1H NMR (CDCl3, 600 MHz, 27 °C) δ 2.60 (3H, s, NH-CH3), 4.04 (2H, s, Ar-CH2-NH), 7.52 (1H, t (7.2 Hz), 9-phenyl-Hp), 7.60 (2H, d (7.6 Hz), 2-phenyl-Hm), 7.61 (2H, t (7.6 Hz), 9-phenyl-Hm), 7.70, 7.74 (2H, d × 2 (8.6 Hz), phen-H5,6), 7.98 (1H, d (8.3 Hz), phen-H3), 8.11 (1H, d (8.3 Hz), phen-H8), 8.23 (3H, d (7.9 Hz), phen-H4, 2-phenyl-Ho), 8.29 (1H, d (8.3 Hz), phen-H7), 8.36 (2H, d (7.6 Hz), 9-phenyl-Ho); Anal. Calcd. for C26H21N3: C, 83.18; H, 5.64; N, 11.19%. Found: C, 83.14; H, 6.06; N, 11.20%.
p-Methylaminomethylphenyl)-9-phenyl-1,10-phenanthroline (4a)..
Compound 2a (2.22 g, 6.4 mmol) was treated with N-bromosuccinimide (NBS) (1.37 g, 7.7 mmol) and α,α′-azobisisobutyronitrile (0.14 g, 10 wt% of NBS) in CCl4 at the reflux temperature for 50 min. The progress of the reaction was followed by 1H NMR spectroscopy [250 MHz, CCl4–CDCl3 = 1∶1 (v/v)] with the disappearance of δCH3 2.47 ppm and the appearance of δCH2Br 4.60 ppm. After cooling, the insoluble materials were removed by filtration, the filtrate being dried over Na2SO4. This solution containing 3a was used for the synthesis of 4a without further purification. Into the CCl4 solution cooled with an ice-bath was introduced methylamine gas for 4 h. The progress of the reaction was followed by a TLC method [silica gel, CH2Cl2–MeOH = 10∶1 (v/v)]. After 4 h when the spot for 3a (Rf = 0.90) disappeared, the reaction was finished by the addition of aqueous 5% NaHCO3 solution. The mixture was stirred for one day and then the insoluble materials were removed by filtration. The CCl4 layer was washed three times with aqueous 5% NaHCO3 solution and then dried over Na2SO4. The solution was concentrated to dryness, the solid residue being further purified through chromatography [silica gel, column φ 2.5 × 17 cm, CH2Cl2–MeOH = 10∶1 (v/v)] to yield 4a, 1.55 g (62%, calculated from 2a), slightly yellow powder, mp 83–87 °C; 1H NMR (CDCl3, 600 MHz, 27 °C) δ 2.60 (3H, s, NH-CH3), 4.04 (2H, s, Ar-CH2-NH), 7.52 (1H, t (7.2 Hz), 9-phenyl-Hp), 7.60 (2H, d (7.6 Hz), 2-phenyl-Hm), 7.61 (2H, t (7.6 Hz), 9-phenyl-Hm), 7.70, 7.74 (2H, d × 2 (8.6 Hz), phen-H5,6), 7.98 (1H, d (8.3 Hz), phen-H3), 8.11 (1H, d (8.3 Hz), phen-H8), 8.23 (3H, d (7.9 Hz), phen-H4, 2-phenyl-Ho), 8.29 (1H, d (8.3 Hz), phen-H7), 8.36 (2H, d (7.6 Hz), 9-phenyl-Ho); Anal. Calcd. for C26H21N3: C, 83.18; H, 5.64; N, 11.19%. Found: C, 83.14; H, 6.06; N, 11.20%.
1-{N-Methyl-N-[2-(dihydroxyboryl)phenylmethyl]aminomethyl}-4-(9-phenyl-1,10-phenanthrolin-2-yl)benzene (1a)..
Compound 4a (1.30 g, 3.5 mmol) and 2-(2-bromomethylphenyl)-1,3-dioxaborinane (1.15 g, 4.5 mmol) were treated in refluxing acetonitrile (100 ml) in the presence of K2CO3 (0.96 g, 6.9 mmol). The progress of the reaction was followed by a TLC method [silica gel, CH2Cl2–MeOH = 5∶1 (v/v)]. After 17 h when the spot for 1a (Rf = 0.45) disappeared, the reaction was finished. After cooling, the insoluble materials were filtered off, the filtrate being evaporated to dryness. The solid residue was taken with a mixture of dichloromethane and aqueous 5% NaHCO3 solution and the phase-separated mixture was stirred for 30 min. The organic layer was separated, washed with water and dried over Na2SO4. The solution was concentrated to dryness, the solid residue being further purified through chromatography [silica gel, column φ 2.5 × 15 cm, CH2Cl2–MeOH = 5∶1 (v/v)] to yield 1a, 0.93 g (46%), slightly yellow powder, mp 174–179 °C; IR (KBr) νB-O 1340 cm−1; 1H NMR (600 MHz, CDCl3, 27 °C) δ 2.19 (3H, s, N-CH3), 3.80, 3.83 (4H, s × 2, -CH2-NMe-CH2-), 7.25 (1H, d (6.7 Hz), borylphenyl-H6), 7.38 (2H, t (7.1 Hz), borylphenyl-H4,5), 7.50 (1H, t (8.2 Hz), 9-phenyl-Hp), 7.51 (2H, t (8.1 Hz), benzene-H2), 7.60 (2H, t (7.6 Hz), 9-phenyl-Hm), 7.80 (2H, s, phen-H5,6), 7.97 (1H, d (6.7 Hz), borylphenyl-H3), 8.13, 8.15 (2H, d × 2 (8.4 Hz), phen-H3,8), 8.32 (2H, d (8.3 Hz), 9-phenyl-Ho), 8.46 (4H, m, phen-H4,7, benzene-H3); Anal. Calcd. for C33H26N3BO + 0.3H2O: C, 79.91; H, 5.50; N, 8.31%. Found: C, 79.79; H, 5.40; N, 8.46%.
2-(p-Methylaminomethylphenyl)-9-p-anisyl-1,10-phenanthroline (4b)..
Compound 2b (4.11 g, 10.9 mmol) was treated with N-bromosuccinimide (NBS) (2.33 g, 13.1 mmol) and α,α′-azobisisobutyronitrile (0.23 g, 10 wt% of NBS) in CCl4 at the reflux temperature for 40 min. The progress of the reaction was followed by 1H NMR spectroscopy [250 MHz, CCl4–CDCl3 = 1∶1 (v/v)] with the disappearance of δCH3 2.47 ppm and the appearance of δCH2Br 4.59 ppm. After cooling, the insoluble materials were removed by filtration, the filtrate being dried over Na2SO4. This solution containing 3b was used for the synthesis of 4b without further purification. Into the CCl4 solution cooled with an ice-bath was introduced methylamine gas for 6 h. The progress of the reaction was followed by a TLC method [silica gel, toluene–MeOH = 10∶1 (v/v)]. After 4 h when the spot for 3b (Rf = 0.67) disappeared, the reaction was finished by the addition of aqueous 5% NaHCO3 solution. The mixture was stirred for one day and then the insoluble materials were removed by filtration. The CCl4 layer was washed three times with aqueous 5% NaHCO3 solution and then dried over Na2SO4. The solution was concentrated to dryness, the solid residue being further purified through chromatography [silica gel, column φ 5 × 8 cm, CHCl3–MeOH = 5∶1 (v/v)] to yield 4b, 1.99 g (46%, calculated from 2b), slightly yellow powder, mp 42–48 °C; 1H NMR (CDCl3, 250 MHz, 27 °C) δ 2.51 (3H, s, NH-CH3), 3.87 (2H, s, Ar-CH2-NH), 3.92 (3H, s, -OCH3), 7.11 (2H, d (8.8 Hz), 2-phenyl-Hm), 7.41 (2H, d (8.1 Hz), 9-phenyl-Hm), 7.76 (2H, s, phen-H5,6), 8.09 (1H, d (8.5 Hz), phen-H3), 8.13 (1H, d (9.8 Hz), phen-H8), 8.26 (1H, d (7.2 Hz), phen-H4), 8.29 (1H, d (8.3 Hz), phen-H7), 8.44 (4H, d (7.6 Hz), 2-phenyl-Hm, 9-phenyl-Ho); Anal. Calcd. for C27H23N3O + 0.4H2O: C, 78.57; H, 5.81; N, 10.18%. Found: C, 78.65; H, 5.70; N, 10.09%.
1-{N-Methyl-N-[2-(dihydroxyboryl)phenylmethyl]aminomethyl}-4-(9-p-anisyl-1,10-phenanthrolin-2-yl)benzene (1b)..
Compound 4b (1.80 g, 4.4 mmol) and 2-(2-bromomethylphenyl)-1,3-dioxaborinane (1.47 g, 5.8 mmol) were treated in refluxing acetonitrile (100 ml) in the presence of K2CO3 (1.23 g, 8.9 mmol). The progress of the reaction was followed by a TLC method [silica gel, CHCl3–MeOH = 5∶1 (v/v)]. After 6 h when the spot for 4b (Rf = 0.13) disappeared, the reaction was finished. After cooling, the insoluble materials were filtered off, the filtrate being evaporated to dryness. The solid residue was taken with a mixture of dichloromethane and aqueous 5% NaHCO3 solution and the phase-separated mixture was stirred for 30 min. The organic layer was separated, washed with water and dried over Na2SO4. The solution was concentrated to dryness, the solid residue being further purified through chromatography [silica gel, column φ 5 × 10 cm, CHCl3–MeOH = 5∶1 (v/v)] to yield 1b, 0.81 g (34%), slightly yellow powder, mp 178–183 °C; IR (KBr) νB-O 1344 cm−1; 1H NMR (600 MHz, CDCl3, 27 °C) δ 2.20 (3H, s, N-CH3), 3.69, 3.79 (4H, s × 2, -CH2-NMe-CH2-), 3.93 (3H, s, -OCH3), 7.13 (2H, d (8.5), 9-phenyl-Hm), 7.25 (1H, d (6.9 Hz), borylphenyl-H6), 7.38 (2H, t (7.1 Hz), borylphenyl-H4,5), 7.51 (2H, d (8.0 Hz), benzene-H2), 7.77 (2H, d × 2 (8.9 Hz), phen-H5,6), 7.97 (1H, d (6.4 Hz), borylphenyl-H3), 8.10, 8.12 (2H, d × 2 (8.4 Hz), phen-H3,8), 8.27 (2H, d (8.4 Hz), phen-H4,7), 8.43 (4H, d (8.0 Hz), phen-H4,7, 9-phenyl-Ho, benzene-H3); Anal. Calcd. for C34H30N3BO2 + 0.5H2O: C, 76.98; H, 5.51; N, 7.92%. Found: C, 77.12; H, 5.36; N, 7.94%.
Miscellaneous
1H NMR, absorption spectra, optical rotation, ESI-MS spectra and CD spectra were measured with Bruker DMX 600, JASCO V-570 , HORIBA SEPA-300, Perseptive Mariner and JASCO J-720 WI, respectively. The energy minimisation of the 1a2·Cu(I)·D-glucose complexes was performed using the ESFF forcefield with molecular mechanics as implemented by Discover (MSI).
Acknowledgements
This work was supported by a Grant-in-Aid for COE Research ‘Design and Control of Advanced Molecular Assembly Systems’ from the Ministry of Education, Science and Culture, Japan (#08CE2005).
References
- For a recent comprehensive review see:
E. C. Constable
, in Comprehensive Supramolecular Chemistry, ed. J.-M. Lehn, Pergamon, Oxford, 1996, vol. 9, p. 213;
Search PubMed; A. F. Williams, Chem. Eur. J., 1997, 3, 15 Search PubMed; C. Piguet, G. Bernardinelli and G. Hopfgartner, Chem. Rev., 1997, 97, 2005 Search PubMed; A. E. Rowan and R. J. M. Nolte, Angew. Chem., Int. Ed. Engl., 1998, 37, 63 CrossRef CAS.
- C. J. Carrano and K. N. Raymond, J. Am. Chem. Soc., 1978, 100, 5371 CrossRef CAS; B. Kersting, M. Meyer, R. E. Powers and K. N. Raymond, J. Am. Chem. Soc., 1996, 118, 7221 CrossRef CAS.
- J. Libman, Y. Tor and A. Shanzer, J. Am. Chem. Soc., 1987, 109, 5880 CrossRef CAS; L. Zelikovich, J. Libman and A. Shanzer, Nature, 1995, 374, 790 CrossRef CAS; C. Bianchini, A. Meli, V. Patinec, V. Sernau and F. Vizza, J. Am. Chem. Soc., 1997, 119, 4945 CrossRef CAS.
- S. Christie, I. F. Fraser, A. McVitie and R. D. Peacock, Polyhedron, 1986, 5, 35 CrossRef CAS; P. Agaskar, F. A. Cotton, I. F. Fraser and R. D. Peacock, J. Am. Chem. Soc., 1984, 106, 1851 CrossRef CAS; P. A. Agasker, F. A. Cotton, I. F. Fraser, L. Manojlovic-Muir, K. W. Muir and R. D. Peacock, Inorg. Chem., 1986, 25, 2511 CrossRef.
- M. Gerards, Inorg. Chim. Acta, 1995, 229, 101 CrossRef CAS.
- W. Zarges, J. Hall and J.-M. Lehn, Helv. Chim. Acta, 1991, 74, 1843 CrossRef.
- E. C. Constable, T. Kulke, M. Neuburger and M. Zehnder, Chem. Commun., 1997, 489 RSC; G. Baum, E. C. Constable, D. Fenske and T. Kulke, Chem. Commun., 1997, 2043 RSC; G. Baum, E. C. Constable, D. Fenske, C. E. Housecroft and T. Kulke, Chem. Commun., 1998, 2659 RSC.
- C. Provent, S. Hewage, G. Brand, G. Bernardinelli, L. J. Charbonniere and A. F. Williams, Angew. Chem., Int. Ed. Engl., 1997, 36, 1287 CrossRef CAS.
- C. R. Woods, M. Benaglia, F. Cozzi and J. S. Siegel, Angew. Chem., Int. Ed. Engl., 1996, 35, 1830 CrossRef CAS; C. R. Woods, M. Benaglia, P. Blom, A. Fuchicello, F. Cozzi and J. S. Siegel, Polym. Prepr., 1996, 37, 480 Search PubMed.
- A. L. Airey, G. F. Swiegers, A. C. Willis and S. B. Wild, J. Chem. Soc., Chem. Commun., 1995, 695 RSC.
- T. Suzuki, H. Kotsuki, K. Isobe, N. Moriya, Y. Nakagawa and M. Ochi, Inorg. Chem., 1995, 34, 530 CrossRef CAS.
- J. F. Modder, G. van Koten, K. Vrieze and A. L. Spek, Angew. Chem., Int. Ed. Engl., 1989, 28, 1698 CrossRef; J. F. Modder, K. Vrieze, A. L. Spek, G. Challa and G. Van Koten, Inorg. Chem., 1992, 31, 1238 CrossRef CAS.
- Y. Dai, T. J. Katz and D. A. Nichols, Angew. Chem., Int. Ed. Engl., 1996, 35, 2109 CrossRef CAS.
- E. J. Enemark and T. D. P. Stack, Angew. Chem., Int. Ed. Engl., 1995, 34, 996 CrossRef CAS.
- M. Albrecht, Synlett., 1996, 565 CrossRef CAS.
- M. S. Goodman, A. D. Hamilton and J. Weiss, J. Am. Chem. Soc., 1995, 117, 8447 CrossRef CAS.
- J. Yoon and A. W. Czarnik, J. Am. Chem. Soc., 1992, 114, 5874 CrossRef CAS; L. K. Mohler and A. W. Czarnik, ibid., 1993, 115, 2998 Search PubMed.
- P. R. Westmark and B. D. Smith, J. Am. Chem. Soc., 1994, 116, 9343 CrossRef CAS and references cited therein..
- Y. Nagai, K. Kobayashi, H. Toi and Y. Aoyama, Bull. Chem. Soc. Jpn., 1993, 66, 2965 CAS.
- G. Wulff, S. Krieger, B. Kubneweg and A. Steigel, J. Am. Chem. Soc., 1994, 116, 409 CrossRef CAS and references cited therein..
- J. C. Norrild and H. Eggert, J. Am. Chem. Soc., 1995, 117, , 1479 CrossRef.
- For comprehensive reviews on boronic acid-based saccharide receptors, see: T. D. James, K. R. A. S. Sandanayake and S. Shinkai, Supramol. Chem., 1995, 6, 141 Search PubMed; T. D. James, P. Linnane and S. Shinkai, Chem. Commun., 1996, 281 Search PubMed; T. D. James, K. R. A. S. Sandanayake and S. Shinkai, Angew. Chem., Int. Ed. Engl., 1996, 35, 1911 RSC; K. R. A. S. Sandanayake, T. D. James and S. Shinkai, Pure Appl. Chem., 1996, 68, 1207 CrossRef; S. Shinkai and M. Takeuchi, Trends Anal. Chem., 1996, 15, 188 CrossRef.
- K. Tsukagoshi and S. Shinkai, J. Org. Chem., 1991, 56, 4089 CrossRef CAS; Y. Shiomi, M. Saisho, K. Tsukagoshi and S. Shinkai, J. Chem. Soc., Perkin Trans. 1, 1993, 2111 RSC.
- T. D. James, K. R. A. S. Sandanayake and S. Shinkai, Angew. Chem., 1994, 106, 2287 CAS; T. D. James, K. R. A. S. Sandanayake and S. Shinkai, Angew. Chem., Int. Ed. Engl., 1994, 22, 2207 CrossRef; T. D. James, K. R. A. S. Sandanayake and S. Shinkai, J. Chem. Soc., Chem. Commun., 1994, 477 RSC.
- M. Takeuchi, T. Imada and S. Shinkai, J. Am. Chem. Soc., 1996, 118, 10658 CrossRef CAS; M. Takeuchi, T. Imada and S. Shinkai, Bull. Chem. Soc. Jpn., 1998, 71, 1117 CAS; M. Takeuchi, T. Mizuno, H. Shinmori, M. Nakashima and S. Shinkai, Tetrahedron, 1996, 52, 1195 CrossRef CAS; M. Takeuchi, S. Yoda, T. Imada and S. Shinkai, Tetrahedron, 1997, 53, 8335 CrossRef CAS; H. Shinmori, M. Takeuchi and S. Shinkai, J. Chem. Soc., Perkin Trans. 2, 1998, 847 RSC.
- T. Imada, H. Kijima, M. Takeuchi and S. Shinkai, Tetrahedron, 1996, 52, 2817 CrossRef CAS; M. Yamamoto, M. Takeuchi and S. Shinkai, Tetrahedron, 1998, 54, 3125 CrossRef CAS; M. Takeuchi, M. Taguchi, H. Shinmori and S. Shinkai, Bull. Chem. Soc. Jpn., 1996, 69, 2613 CAS.
- S. Arimori, M. Takeuchi and S. Shinkai, J. Am. Chem. Soc., 1996, 118, 245 CrossRef; T. Kimura and S. Shinkai, Chem. Lett., 1998, 1035 CrossRef CAS; T. Kimura, M. Takeuchi and S. Shinkai, Bull. Chem. Soc. Jpn., 1998, 71, 2197 CAS.
- M. Mikami and S. Shinkai, J. Chem. Soc., Chem. Commun., 1995, 153 RSC; M. Mikami and S. Shinkai, Chem. Lett., 1995, 603 CAS; M. Takeuchi, Y. Chin, T. Imada and S. Shinkai, Chem. Commun., 1996, 1867 RSC.
- T. Mizuno, M. Takeuchi, I. Hamachi, K. Nakashima and S. Shinkai, J. Chem. Soc., Perkin Trans. 2, 1998, 2281 RSC; G. Nuding, K. Nakashima, R. Iguchi, T. Ishi-i and S. Shinkai, Tetrahedron Lett., 1998, 39, 9473 CrossRef CAS.
- M. Takeuchi, K. Koumoto, M. Goto and S. Shinkai, Tetrahedron, 1996, 52, 12931 CrossRef CAS.
- Preliminary communication: M. Yamamoto, M. Takeuchi and S. Shinkai, Tetrahedron Lett., 1998, 39, 1189 Search PubMed.
- J. A. Nelder and R. Mead, Comput. J., 1965, 7, 308 Search PubMed; S. L. Morgan and S. N. Deming, Anal. Chem., 1974, 46, 1170 CrossRef CAS.
- T. D. James, K. R. A. S. Sandanayake, R. Iguchi and S. Shinkai, J. Am. Chem. Soc., 1995, 117, 8982 CrossRef CAS; T. D. James, K. R. A. S. Sandanayake and S. Shinkai, Nature, 1995, 374, 345 CrossRef CAS.
- K. Kondo, Y. Shiomi, M. Saisho, T. Harada and S. Shinkai, Tetrahedron, 1992, 48, 8239 CrossRef CAS; T. D. James and S. Shinkai, Chem. Commun., 1995, 1483 RSC.
- M. Bielecki, H. Eggert and J. C. Norrild, J. Chem. Soc., Perkin Trans. 2, 1999, 449 RSC.
- Here, we take only the α-anomer into consideration, because it has a 1,2-cis-diol group that can bind a boronic acid group by forming a cyclic ester, whereas the β-anomer, having a 1,2-trans diol group, cannot or finds it very difficult to form such a cyclic ester.Here, we take only the α-anomer into consideration, because it has a 1,2-cis-diol group that can bind a boronic acid group by forming a cyclic ester, whereas the β-anomer, having a 1,2-trans diol group, cannot or finds it very difficult to form such a cyclic ester..
- To simplify the 1H NMR spectral pattern C2-symmetrical saccharides such as D-threitol and D-mannitol-3,4-carbonate would be more suitable for the present system than C2-unsymmetrical D-glucose. Unfortunately, the affinity of these C2-symmetrical saccharides with the 1a2·Cu(I) complex was not as high and the resultant 1H NMR spectra were very complex because of overlap with uncomplexed saccharides..
- The concentrations used for the 1H NMR measurements ([1b2· Cu(I)] = 5.00 mmol dm−3 and [D-glucose] = 12.5 mmol dm−3 in Fig. 10) are much higher than those used for the CD measurements ([1a2·Cu(I)] = [D-glucose] = 0.100 mmol dm−3 in Fig. 3). This is why the equilibrium is attained much faster in 1H NMR measurements than in the CD measurements..
|
| This journal is © The Royal Society of Chemistry 2000 |
Click here to see how this site uses Cookies. View our privacy policy here. 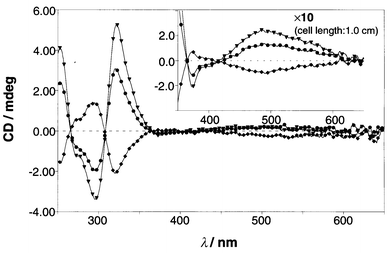
![Plots of the CD intensity vs. [saccharide]: ▼; D-fucose, •; D-galactose, ✦; D-arabinose, ■; D-mannose.](/image/article/2000/P2/a904936c/a904936c-f8.gif)