The synthesis and mesomorphic properties of 4,4′′-dialkyl-2,2′,3- and 2,2′,3′-trifluoro-1,1′∶4′,1′′-terphenyls for high dielectric biaxiality ferroelectric liquid crystal mixtures
Margaret E. Glendenning, John W. Goodby, Michael Hird and Kenneth J. Toyne
Department of Chemistry, University of Hull, Hull, UK HU6 7RX
First published on UnassignedUnassigned23rd December 1999
Abstract
Two series of liquid crystalline terphenyls with terminal alkyl chains have been synthesised with three lateral fluoro substituents in close proximity to each other. Two terminal alkyl chains were employed to minimise viscosity, maximise solubility and generate low melting points. The first series of materials employs fluoro substituents at an outer-edge position to maximise the smectic phase C phase stability, whereas the second series has the fluoro substituents in the centre of the molecule in an attempt to minimise melting points, but surprisingly the melting points of the former compounds are much lower. The synthetic routes to all of the materials involved low-temperature lithiations to generate arylboronic acids which were then involved in sequential, selective palladium-catalysed cross-coupling reactions. Many of the materials produced have extremely low melting points and some exhibit the smectic C phase at room temperature. In general, the 2,2′,3- and 2,2′,3′-patterns of lateral fluoro substitution in 1,1′∶4′,1′′-terphenyls generate materials of low viscosity and high lateral dipole. In particular, many materials from the first series have low melting points and exhibit the smectic C phase, hence they show great promise for the formulation of ferroelectric mixtures with a high dielectric biaxiality which is very important in τV minimum driving schemes; whereas the materials from the second series are nematogens and hence could be useful for vertically aligned nematic devices.
Introduction
Ferroelectric mixtures for fast-switching displays are composed of achiral host materials which enables the fine-tuning of the mesomorphic behaviour, and allows the tailoring of many important physical properties, such as birefringence and particularly the requirement for a low viscosity.1–6 Such achiral host mixtures need to be doped with a chiral material which then confers chirality on the whole mixture; removal of the macroscopic helical structure then generates the required ferroelectric properties. Ideally, the quantity of the required chiral material should be as low as possible so as to minimise the viscosity, but must be sufficiently high to provide the necessary spontaneous polarisation.6 Ferroelectric mixtures for displays that operate in the τVmin mode are additionally required to have a high dielectric biaxiality, which is achieved through the use of components with a large lateral dipole.3–5 Lateral fluoro substituents have long been used to generate a lateral dipole because their small size and high electronegativity serve to minimise any reduction in liquid crystal phase stability and to minimise viscosity.1,6The ortho-difluoroterphenyls (I and II) make excellent host materials for ferroelectric mixtures; they have low melting points, wide smectic C phase temperature ranges and a low viscosity, they also have a high lateral dipole due to the two fluoro substituents that are fixed on the same side of the molecule.1 In order to enhance the lateral dipole, recent work has concentrated on locating a third fluoro substituent next to the existing two fluoro substituents so as to create a 2,2′-difluoro substitution pattern within a trifluoroterphenyl structure (III and IV). It is known from many different sources that the 2,2′-difluoro substituents in a biphenyl moiety tend to attract each other to their van der Waals minimum so providing a high lateral dipole.7–9 Another important molecular structural feature that considerably increases the lateral dipole is the special relationship between an ether oxygen of the terminal alkoxy chain and the adjacent fluoro substituent in a phenyl ring; it is thought that in this case there is a mesomeric effect from the oxygen which enhances the polarity of the fluoro substituent.1,10

Recently, we have reported on a series of trifluoroterphenyls of types III and IV where at least one of the terminal chains is alkoxy, thus combining the useful structural combinations mentioned above.6 These materials showed particularly high lateral dipole moments and enhanced the dielectric biaxiality of ferroelectric mixtures. However, the viscosity of the alkoxy-alkyl-difluoroterphenyls is considerably higher than for the dialkyl analogues.1,10 Hence, the aim of this work was to synthesise a series of analogous trifluoroterphenyls (III and IV), but with two alkyl terminal chains in order to minimise viscosity. Melting points of dialkyl compounds are nearly always lower than for the analogous alkoxy-alkyl systems due to the reduced polarity.1,11 Provided melting points are low, the dialkyl compounds of type III will certainly exhibit the smectic C phase (the outer-edge fluoro substituent enhances lamellar attractions and the lateral polarity from the fluoro substituents promotes molecular tilting),1,11 albeit to lower temperature than for the alkoxy-alkyl systems where the ether oxygen enhances polarisability and lateral dipole. The dialkyl compounds of type IV will have particularly low viscosity because the fluoro substituents are away from the outer edge of the core, but the lack of an outer-edge fluoro substituent combined with more interannular twisting means that they will most certainly not exhibit smectic phases and hence be nematogens.1,11 Despite the obvious expected disadvantages in terms of mesophase morphology and transition temperatures, the expected low melting points and low viscosity will ensure some considerable potential as additives to enhance the dielectric biaxiality of ferroelectric mixtures for τVmin applications. Those materials that are nematogenic will possibly be useful for vertically aligned nematic (VAN) devices![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 12–14 where a negative dielectric anisotropy and a low viscosity are required.
12–14 where a negative dielectric anisotropy and a low viscosity are required.
A wide range of homologues of types III and IV of the dialkyltrifluoroterphenyls has been synthesised in order to (a) fully evaluate the effect of a third lateral fluoro substituent on melting points, transition temperatures and mesophase morphology, and (b) find the optimum combination of terminal chain lengths that generates a material with as low a melting point as possible and that exhibits a smectic C phase over as wide a temperature range as possible.
Synthesis of materials
Palladium-catalysed cross-coupling reactions are virtually essential in the efficient synthesis of novel liquid crystals designed to meet the very exacting requirements for future advanced technology. Palladium-catalysed cross-coupling reactions are very efficient and general in the synthesis of multi-aryl materials because they allow the individual construction of aromatic rings of the desired substitution pattern; they thus facilitate the synthesis of materials that are virtually impossible to generate through other methods. Those cross-coupling reactions involving boronic acids offer particular advantages in terms of tolerance towards other functional groups, selectivity and provide high yields of easily-purified multi-aryl materials.1,6,15–18The requirement for fluoro substituents in these novel liquid crystals (and many reported previously) actually facilitates the synthesis. The electron-withdrawing effect of the two fluoro substituents renders adjacent protons acidic, and such sites were exploited sequentially by a strong base at low temperature and then functionalised, firstly to introduce the desired alkyl chain, and finally to provide substituted difluorophenylboronic acids 5a–c (Scheme 1). Subsequent palladium-catalysed cross-coupling with the substituted aryl bromide 6 (itself prepared through a selective cross-coupling) provided 7a as an example of a dialkyl liquid crystalline trifluoroterphenyl. However, this particular cross-coupling involving a combination of boronic acid and bromide is disadvantaged by the slight steric hindrance due to the fluoro substituents and the tendency of ortho-fluoroarylboronic acids (e.g., 5a–c) to undergo hydrodeboronation.
 | ||
| Scheme 1 1a ... (i) n-BuLi, THF; (ii) R′′CH2CHO, THF; (iii) NH4Cl, H2O; 1b ... P2O5, pentane or PTSA, toluene; 1c ... H2, Pd/C, ethanol–THF; 1d ... (i) n-BuLi, THF; (ii) (MeO)3B, THF; (iii) 10% HCl; 1e ... Pd(PPh3)4, 2 M Na2CO3, DME. | ||
Scheme 2 shows the alternative approach of a selective coupling reaction involving the alkyldifluorophenylboronic acids 5a–c and the commercially available 4-bromo-2-fluoro-1-iodobenzene (8) to provide a range of bromo-substituted alkyl-trifluorobiphenyls. The final coupling reactions are now particularly efficient because the boronic acids 10a–c are not prone to hydrodeboronation and there is less steric hindrance. Overall, this approach is better than that shown in Scheme 1 because of the commercial availability of compound 8 and the activation of the iodo leaving group due to the ortho fluoro substituent which facilitates good selectivity in the first coupling reaction.
 | ||
| Scheme 2 2a ... Pd(PPh3)4, 2 M Na2CO3, DME. | ||
As part of a long and on-going investigation into the scope and limitations of palladium-catalysed cross-coupling reactions in the synthesis of multi-aryl materials, two arylboronic esters (11a and 11b) were prepared from their parent arylboronic acids (10a and 10b). Arylboronic acids are very difficult to purify and characterise because of their tendency to exist as mixtures of the boronic acid and the trimeric anhydride, whereas the analogous esters can be purified and characterised easily. Another reported advantage of the boronic esters is their reduced tendency towards hydrodeboronation19 in palladium-catalysed cross-coupling reactions when used in anhydrous conditions, a point not relevant here because boronic acids 10a and 10b are not prone to hydrodeboronation. Boronic esters can also be used with the same aqueous base conditions as boronic acids, in which case hydrolysis to the boronic acid occurs. For the purposes of cross-coupling reactions there is usually no point in preparing the boronic esters, except where hydrodeboronation is a problem, as the boronic acids are perfectly satisfactory, a point shown by the close similarity (around 80%) of yields (before recrystallisation) of materials from Schemes 2 and 3.
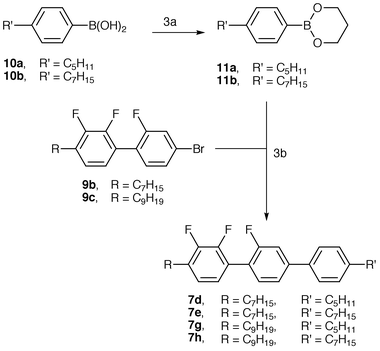 | ||
| Scheme 3 3a ... Propane-1,3-diol, hexane; 3b ... Pd(PPh3)4, K3PO4, DMF. | ||
Scheme 4 shows the synthesis of those trifluoroterphenyls (17a–i) with the fluoro substituents in the centre of the molecule. Once again palladium-catalysed cross-coupling reactions are essential for such syntheses. The exploitation of an acidic proton in compound 1 at low temperature generated the required arylboronic acid (12) which was then involved in a palladium-catalysed cross-coupling reaction to yield difluorobiphenyls 14a–c. The other acidic proton was exploited to generate difluorobiarylboronic acids 15a–c which were then coupled to the appropriate fluoro-substituted aryl bromides to generate the required liquid crystals in reasonably good yields.
 | ||
| Scheme 4 4a ... (i) n-BuLi, THF; (ii) (MeO)3B, THF; (iii) 10% HCl; 4b ... Pd(PPh3)4, 2 M Na2CO3, DME. | ||
Discussion of mesophase morphology and transition temperatures
Table 1 shows the transition temperatures of a series of nine homologous dialkyl trifluoroterphenyls with the fluoro substituents at the outermost positions of the core and a typical alkoxy-alkyl analogue (18)6 for comparison with compound 7f. In terms of expectation, the outer-edge position tends to fill space at the site of the terminal chain which enhances lateral inter-molecular attractions and hence offers scope for the generation of smectic phases, in particular the tilted smectic C phase due to the strong lateral dipole. However, two of the fluoro substituents are at an inter-annular position and the one in the centre ring will cause additional broadening of the molecules, hence tending to depress smectic phase stability and leave nematogens. With three lateral fluoro substituents the polarity is high and hence the usual influence of a lateral fluoro substituent in reducing melting point may not be expected to hold. However, the melting points of the compounds in Table 1 are remarkably low considering the location of a fluoro substituent at an outer-edge position; compound 7b in particular exhibits a smectic C phase at room temperature and does not crystallise down to −100 °C. Such low melting points are of obvious benefit in compounds where the thermal stabilities of the smectic C phase are moderate at best.
| |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | Transition temperatures/°C | ||||||||||
| No. | R | R′ | Cryst | SC | SA | N | Iso | ||||
| 7a | C5H11 | C5H11 | · | 31.8 | · | (20.3) | — | — | · | 81.5 | · |
| 7b | C5H11 | C7H15 | · | ? | · | 37.2 | · | 51.7 | · | 75.0 | · |
| 7c | C5H11 | C9H19 | · | 30.8 | · | 49.2 | · | 78.6 | · | 83.1 | · |
| 7d | C7H15 | C5H11 | · | 22.7 | · | (8.2) | — | — | · | 75.5 | · |
| 7e | C7H15 | C7H15 | · | 38.1 | · | 50.8 | · | 64.6 | · | 80.8 | · |
| 7f | C7H15 | C9H19 | · | 28.0 | · | 52.0 | · | 78.5 | · | 81.0 | · |
| 7g | C9H19 | C5H11 | · | 36.1 | · | — | — | — | · | 77.5 | · |
| 7h | C9H19 | C7H15 | · | 37.0 | · | 51.3 | · | 67.4 | · | 79.6 | · |
| 7i | C9H19 | C9H19 | · | 45.6 | · | 54.7 | · | 79.8 | · | 80.8 | · |
| 186 | C6H13O | C9H19 | · | 47.9 | · | 91.5 | — | — | · | 109.6 | · |

The lack of an ether oxygen is particularly detrimental towards the smectic C phase stability of compounds 7a–i where in most cases the smectic phase stability continues way beyond the tilted smectic C through the exhibition of a smectic A phase. Interestingly, where the smectic phase stability is weak (e.g., the short-chain homologues, 7a and 7d) it is the smectic C phase and not the smectic A phase that is exhibited; in fact 7g is nematogenic. Where the terminal chains are long (particularly R′) the smectic A phase is particularly dominant; e.g., compare compounds 7a–c where the smectic A phase thermal stability increases more dramatically than that of the smectic C phase. The alkoxy-alkyl analogues did not generally generate a smectic A phase and the whole smectic tendency was exhibited as the tilted smectic C phase because of the additional polarity from the ether oxygen. It is particularly interesting to compare compound 7f with the analogous alkoxy-alkyl system (18);6 for 7f the nematic phase stability has been reduced by around 30 °C, but the smectic phase stability has been reduced by only 13 °C. However, the smectic C phase stability has been reduced by 40 °C, and combined with a reduction in melting point of only 20 °C results in a narrower smectic C temperature range. Generally, the very low melting points and reasonable smectic C phase thermal stabilities of compounds 7a–i makes them ideally suited as components in ferroelectric mixtures.
Table 2 shows some difluoroterphenyls,1 and it is most interesting to look at the influence of the additional fluoro substituent in the centre ring on liquid crystal behaviour (compare compounds 7a, 7b and 7d with 19a, 19b and 19c respectively). Melting points have been reduced by around 40 °C, which is quite surprising as previous views on fluoro substitution indicate that one fluoro, and possibly two, may reduce melting point significantly, but the high polarity from three fluoro substituents is usually expected to cause very high melting points. It is however, useful that melting points are so low since the all-important smectic C phase stability has been reduced by around 90 °C, although the trifluoroterphenyls are certainly far more influenced by chain lengths. Longer terminal chains (especially R′) are essential even for moderate smectic C phase stability. Nematic phase stability is less affected by the third inner-core fluoro substituent and is reduced by around 60 °C, showing the steric effect of the increased molecular breadth.
1,8
| |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | Transition temperatures/°C | ||||||||||
| No. | R | R′ | Cryst | SC | SA | N | Iso | ||||
| 19a | C5H11 | C5H11 | · | 81.0 | · | 115.5 | · | 131.5 | · | 142.0 | · |
| 19b | C5H11 | C7H15 | · | 65.5 | · | 118.5 | · | 135.0 | · | 137.0 | · |
| 19c | C7H15 | C5H11 | · | 56.0 | · | 105.5 | · | 131.0 | · | 136.0 | · |
 | |||||||||||

A comparison of compound 7a with difluoroterphenyl 208 shows the influence of the outer-edge fluoro substituent. Compound 20 supercools to −100 °C without the generation of a smectic C phase, yet the trifluoroterphenyl 7a exhibits a monotropic smectic C phase at 20.3 °C despite the presence of an additional fluoro substituent. The additional fluoro substituent at the outer-edge position does not broaden the molecule, but its polar nature aids that lamellar attraction of the molecules dramatically enhancing smectic phase stability, but has little effect on the nematic phase stability which is similar for both compounds.
Compounds 17a–i (Table 3) were prepared because the pattern of three fluoro substituents in the centre ring was expected to generate very low melting points, almost certainly lower than for the isomeric compounds shown in Table 1; hence it is very surprising that the melting points of compounds 17a–i are considerably higher. It was always expected that compounds 17a–i would have low liquid crystal phase stability, and probably not exhibit the smectic C phase, and this has been proved. However, the fluoro-substitution pattern shown by compounds 17a–i is ideal for low viscosity and so the compounds would make ideal additives for ferroelectric mixtures. Since compounds 17a–i are nematogens of high negative dielectric anisotropy they would also be useful as components in mixtures for vertically aligned nematic devices. Unlike those trifluoroterphenyls shown in Table 1, the nematic phase stabilities are remarkably independent of terminal chain length; this is quite unusual since longer terminal chains are expected to have reduced nematic phase stability.
| |||||||
|---|---|---|---|---|---|---|---|
| Compound | Transition temperatures/°C | ||||||
| No. | R | R′ | Cryst | N | Iso | ||
| 17a | C5H11 | C5H11 | · | 62.4 | · | 71.1 | · |
| 17b | C5H11 | C7H15 | · | 44.2 | · | 68.9 | · |
| 17c | C5H11 | C9H19 | · | 50.5 | · | 69.1 | · |
| 17d | C7H15 | C5H11 | · | 41.2 | · | 69.9 | · |
| 17e | C7H15 | C7H15 | · | 56.4 | · | 69.5 | · |
| 17f | C7H15 | C9H19 | · | 48.1 | · | 70.0 | · |
| 17g | C9H19 | C5H11 | · | 50.4 | · | 69.2 | · |
| 17h | C9H19 | C7H15 | · | 51.2 | · | 69.5 | · |
| 17i | C9H19 | C9H19 | · | 61.0 | · | 70.7 | · |
The nematic phase stabilities of the difluoroterphenyls (21a–d, Table 4)1 do vary with chain length in the expected manner, and the third fluoro substituent has reduced the values by around 40 to 50 °C. Taking account of substantial supercooling of compounds 17a–i (Table 3), the third fluoro substituent has reduced the smectic C phase stability by at least 100 °C.
1
| |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | Transition temperatures/°C | ||||||||||||||||||||||
| No. | R | R′ | Cryst | SC | SA | N | Iso | ||||||||||||||||
| 21a | C5H11 | C5H11 | · | 60.0 | — | — | — | — | · | 120.0 | · | ||||||||||||
| 21b | C5H11 | C7H15 | · | 36.5 | · | (24.0) | — | — | · | 111.5 | · | ||||||||||||
| 21c | C5H11 | C9H19 | · | 42.5 | · | 66.0 | — | — | · | 110.0 | · | ||||||||||||
| 21d | C7H15 | C9H19 | · | 49.0 | · | 77.0 | · | 93.0 | · | 108.5 | · | ||||||||||||
Conclusions
A wide range of materials have been synthesised with multiple fluoro substituents in strategic lateral positions in order to generate a large lateral dipole to provide a high dielectric biaxiality. Some interesting synthetic methods have been used including the development of selective palladium-catalysed cross-coupling reactions and selective low temperature lithiations to generate vital fluoro-substituted intermediates. It has been shown that highly polar materials with multiple lateral fluoro substituents can have low melting points and generate a reasonably high smectic C phase stability, which makes them suitable components in ferroelectric mixtures. These fluoro-substituted materials are of low viscosity and confer a high dielectric biaxiality on ferroelectric mixtures and a forthcoming paper by our collaborators at DERA (Malvern) will discuss the whole range of physical properties of the materials and their effect in ferroelectric mixtures (e.g., dielectric biaxiality, viscosity and switching times). Additionally, those materials that are nematogenic will possibly be useful for vertically aligned nematic (VAN) devices where a negative dielectric anisotropy and a low viscosity are required.Experimental
Confirmation of the structures of intermediates and products was obtained by 1H and 13C NMR spectroscopy (JEOL JNM-GX270 spectrometer), infrared spectroscopy (Perkin-Elmer 457 grating spectrophotometer) and mass spectrometry (Finnigan-MAT 1020 GC/MS spectrometer). Elemental analysis (Fisons EA1108 CHN) data were obtained for each final compound prepared (7a–i and 17a–i). The progress of reactions was frequently monitored using a Chrompack 9001 capillary gas chromatograph fitted with a CP-SIL 5 CB 10 m × 0.25 mm, 0.12 μm column (Cat. No. 7700). Transition temperatures were measured using a Mettler FP5 hot-stage and control unit in conjunction with an Olympus BH2 polarising microscope and these were confirmed using differential scanning calorimetry (Perkin-Elmer DSC-7 and IBM data station). The purities of intermediates were checked by GLC analysis (see above) and the purity of each final compound (7a–i and 17a–i) was checked by HPLC analysis (Merck-Hitachi with Merck RP 18 column, Cat. No. 16 051) and was found to be >99.9% pure in each case.The preparation of intermediates (2a–c, 3a–c, 4a–c, 5a–c),1 (6, 10a–c)8 and 16a–c11 have been reported previously. Tetrakis(triphenylphosphine)palladium(0) was prepared according to the literature procedure.20 Compounds 1 and 8 were purchased from Aldrich.
2,2′,3-Trifluoro-4,4′′-dipentylterphenyl 7a
Quantities: compound 5a (0.97 g, 4.25 mmol); compound 6 (1.14 g, 3.54 mmol). The experimental procedure was as described in a previous publication.6 The crude product was purified by column chromatography (silica gel; hexane) to give a colourless solid which was recrystallised from ethanol–ethyl acetate (5∶1) to yield colourless crystals. Yield 0.49 g (43%); transitions (°C) Cryst 31.8 (SC 20.3) N 81.5 Iso; δH (270 MHz; CDCl3) 0.91 (3H, t), 0.92 (3H, t), 1.27–1.45 (8H, m), 1.66 (4H, 2 × quint), 2.66 (2H, t), 2.71 (2H, t), 7.01 (1H, ddd), 7.09 (1H, ddd), 7.28 (2H, d), 7.35–7.48 (3H, m), 7.53 (2H, d); MS m/z 424 (M+); found C, 79.17%; H, 7.35%; C28H31F3 requires: C, 79.21%; H, 7.36%.4′-Bromo-2,2′,3-trifluoro-4-pentylbiphenyl 9a
Quantities: compound 5a (10.53 g, 0.046 mol); compound 8 (12.65 g, 0.042 mol). The experimental procedure was as described in a previous publication.6 The crude product was purified by column chromatography (silica gel; hexane) and Kugelrohr distillation (160 °C at 0.1 mmHg) to give a colourless oil. Yield 8.72 g (58%); δH (270 MHz; CDCl3) 0.91 (3H, t), 1.28–1.45 (4H, m), 1.65 (2H, quint), 2.69 (2H, t), 6.95–7.05 (2H, m), 7.25 (1H, dd), 7.32–7.40 (2H, m); MS m/z 358 (M+), 356 (M+).4′-Bromo-2,2′,3-trifluoro-4-heptylbiphenyl 9b
Quantities: compound 5b (11.01 g, 0.043 mol); compound 8 (11.74 g, 0.039 mol). The experimental procedure was as described for the preparation of compound 9a to give a colourless oil. Yield 6.97 g (46%); δH (270 MHz; CDCl3) 0.89 (3H, t), 1.20–1.44 (8H, m), 1.64 (2H, quint), 2.69 (2H, t), 6.95–7.05 (2H, m), 7.25 (1H, dd), 7.32–7.40 (2H, m); MS m/z 386 (M+), 384 (M+).4′-Bromo-2,2′,3-trifluoro-4-nonylbiphenyl 9c
Quantities: compound 5c (11.36 g, 0.040 mol); compound 8 (10.93 g, 0.036 mol). The experimental procedure was as described for the preparation of compound 9a to give a colourless oil. Yield 6.22 g (31%); δH (270 MHz; CDCl3) 0.88 (3H, t), 1.18–1.44 (12H, m), 1.64 (2H, quint), 2.69 (2H, t), 6.95–7.05 (2H, m), 7.24 (1H, dd), 7.32–7.39 (2H, m); MS m/z 414 (M+), 412 (M+).2,2′,3-Trifluoro-4′′-heptyl-4-pentylterphenyl 7b
Quantities: compound 10b (1.75 g, 7.97 mmol); compound 9a (2.37 g, 6.64 mmol). The experimental procedure was as described for the preparation of compound 7a. The crude product was purified by column chromatography (silica gel; hexane) to afford a colourless liquid which was further purified by Kugelrohr distillation (175 °C at 0.15 mmHg) in order to remove volatiles. The resultant smectic liquid crystal phase was again purified by column chromatography (silica gel; hexane) to afford a colourless smectic liquid crystal. Yield 1.54 g (54%); transitions (°C) Cryst ? SC 37.2 SA 51.7 N 75.0 Iso; δH (270 MHz; CDCl3) 0.89 (3H, t), 0.92 (3H, t), 1.20–1.45 (12H, m), 1.66 (4H, 2 × quint), 2.65 (2H, t), 2.71 (2H, t), 7.01 (1H, ddd), 7.09 (1H, ddd), 7.27 (2H, d), 7.34–7.47 (3H, m), 7.53 (2H, d); MS m/z 452 (M+); found C, 79.58%; H, 7.75%; C30H35F3 requires: C, 79.61%; H, 7.79%.2,2′,3-Trifluoro-4′′-nonyl-4-pentylterphenyl 7c
Quantities: compound 10c (1.86 g, 7.50 mmol); compound 9a (2.23 g, 6.25 mmol). The experimental procedure was as described for the preparation of compound 7a. The crude product was purified by column chromatography (silica gel; hexane) to give a colourless solid which was recrystallised from ethanol–ethyl acetate (4∶1) to yield colourless crystals. Yield 1.80 g (60%); transitions (°C) Cryst 30.8 SC 49.2 SA 78.6 N 83.1 Iso; δH (270 MHz; CDCl3) 0.88 (3H, t), 0.92 (3H, t), 1.19–1.45 (16H, m), 1.65 (4H, 2 × quint), 2.65 (2H, t), 2.70 (2H, t), 7.00 (1H, ddd), 7.09 (1H, ddd), 7.27 (2H, d), 7.34–7.47 (3H, m), 7.53 (2H, d); MS m/z 480 (M+); found C, 79.91%; H, 8.16%; C32H39F3 requires: C, 79.96%; H, 8.18%.2,2′,3-Trifluoro-4-heptyl-4′′-nonylterphenyl 7f
Quantities: compound 10c (1.23 g, 4.96 mmol); compound 9b (1.60 g, 4.16 mmol). The experimental procedure was as described for the preparation of compound 7a. The crude product was purified by column chromatography (silica gel; hexane) to give a colourless solid which was recrystallised from ethanol to yield colourless crystals. Yield 1.23 g (58%); transitions (°C) Cryst 28.0 SC 52.0 SA 78.5 N 81.0 Iso; δH (270 MHz; CDCl3) 0.89 (6H, 2 × t), 1.15–1.45 (20H, m), 1.65 (4H, 2 × quint), 2.65 (2H, t), 2.70 (2H, t), 7.01 (1H, ddd), 7.09 (1H, ddd), 7.27 (2H, d), 7.34–7.49 (3H, m), 7.53 (2H, d); MS m/z 508 (M+); found C, 80.22%; H, 8.48%; C34H43F3 requires: C, 80.28%; H, 8.52%.2,2′,3-Trifluoro-4,4′′-dinonylterphenyl 7i
Quantities: compound 10c (1.39 g, 5.59 mmol); compound 9c (1.93 g, 4.66 mmol). The experimental procedure was as described for the preparation of compound 7a. The crude product was purified by column chromatography (silica gel; hexane) to give a colourless solid which was recrystallised from ethanol–ethyl acetate (5∶1) to yield colourless crystals. Yield 1.84 g (74%); transitions (°C) Cryst 45.6 SC 54.7 SA 79.8 N 80.8 Iso; δH (270 MHz; CDCl3) 0.88 (6H, 2 × t), 1.19–1.45 (24H, m), 1.65 (4H, 2 × quint), 2.65 (2H, t), 2.70 (2H, t), 7.00 (1H, ddd), 7.09 (1H, ddd), 7.27 (2H, d), 7.35–7.48 (3H, m), 7.53 (2H, d); MS m/z 536 (M+); found C, 80.54%; H, 8.83%; C36H47F3 requires: C, 80.56%; H, 8.83%.1-(4-Pentylphenyl)-2,6,1-dioxaborinane 11a
A mixture of compound 10a (16.90 g, 0.088 mol) and propane-1,3-diol (12.90 g, 0.170 mol) in hexane (150 ml) was stirred at room temperature for 16 h. The organic layer was decanted off from the aqueous propane-1,3-diol, dried (MgSO4) and the solvent was removed in vacuo to yield a colourless oil. Yield 16.60 g (81%); δH (270 MHz; CDCl3) 0.85 (3H, t), 1.25–1.40 (4H, m), 1.60 (2H, quint), 2.03 (2H, quint), 2.59 (2H, t), 4.14 (4H, 2 × t), 7.15 (2H, d), 7.67 (2H, d); MS m/z 232 (M+).1-(4-Heptylphenyl)-2,6,1-dioxaborinane 11b
Quantities: compound 10b (17.38 g, 0.079 mol). The experimental procedure was as described for the preparation of compound 11a to yield a colourless oil. Yield 25.85 g (99%); δH (270 MHz; CDCl3) 0.87 (3H, t), 1.17–1.41 (8H, m), 1.60 (2H, quint), 2.03 (2H, quint), 2.60 (2H, t), 4.14 (4H, 2 × t), 7.16 (2H, d), 7.67 (2H, d); MS m/z 260 (M+).2,2′,3-Trifluoro-4-heptyl-4′′-pentylterphenyl 7d
A stirred mixture of compound 11a (0.83 g, 3.58 mmol), compound 9b (1.15 g, 2.99 mmol), anhydrous potassium phosphate (0.95 g, 4.49 mmol) and tetrakis(triphenylphosphine)palladium(0) (0.17 g, 0.15 mmol) in dry DMF (30 ml) under dry nitrogen was heated under reflux for 16 h (GLC analysis revealed a complete reaction) and the cooled mixture was poured into water. The product was extracted into ether, the ethereal extract was washed with brine, dried (MgSO4), and the solvent was removed in vacuo. The crude product was purified by column chromatography (silica gel; hexane) to afford a colourless liquid which was further purified by Kugelrohr distillation (175 °C at 0.15 mmHg) in order to remove volatiles. The resultant nematic liquid crystal phase was again purified by column chromatography (silica gel; hexane) to afford a colourless nematic liquid crystal. Yield 1.11 g (82%); transitions (°C) Cryst 22.7 (SC 8.2) N 75.5 Iso; δH (270 MHz; CDCl3) 0.89 (3H, t), 0.91 (3H, t), 1.21–1.45 (12H, m), 1.66 (4H, 2 × quint), 2.65 (2H, t), 2.70 (2H, t), 7.00 (1H, ddd), 7.09 (1H, ddd), 7.27 (2H, d), 7.35–7.48 (3H, m), 7.53 (2H, d); MS m/z 452 (M+); found C, 79.60%; H, 7.76%; C30H35F3 requires: C, 79.61%; H, 7.79%.2,2′,3-Trifluoro-4,4′′-diheptylterphenyl 7e
Quantities: compound 11b (1.30 g, 3.38 mmol); compound 9b (1.57 g, 5.21 mmol). The experimental procedure was as described for the preparation of compound 7d. The crude product was purified by column chromatography (silica gel; hexane) to give a colourless solid which was recrystallised from ethanol–ethyl acetate (5∶1) to yield colourless crystals. Yield 1.01 g (62%); transitions (°C) Cryst 38.1 SC 50.8 SA 64.6 N 80.8 Iso; δH (270 MHz; CDCl3) 0.89 (6H, 2 × t), 1.20–1.45 (16H, m), 1.65 (4H, 2 × quint), 2.65 (2H, t), 2.70 (2H, t), 7.00 (1H, ddd), 7.09 (1H, ddd), 7.27 (2H, d), 7.35–7.48 (3H, m), 7.53 (2H, d); MS m/z 480 (M+); found C, 79.94%; H, 8.17%; C32H39F3 requires: C, 79.96%; H, 8.18%.2,2′,3-Trifluoro-4-nonyl-4′′-pentylterphenyl 7g
Quantities: compound 11a (1.15 g, 4.94 mmol); compound 9c (1.70 g, 4.12 mmol). The experimental procedure was as described for the preparation of compound 7d. The crude product was purified by column chromatography (silica gel; hexane) to give a colourless solid which was recrystallised from ethanol–ethyl acetate (3∶1) to yield colourless crystals. Yield 1.17 g (59%); transitions (°C) Cryst 36.1 N 77.5 Iso; δH (270 MHz; CDCl3) 0.89 (3H, t), 0.91 (3H, t), 1.20–1.45 (16H, m), 1.66 (4H, 2 × quint), 2.65 (2H, t), 2.70 (2H, t), 7.00 (1H, ddd), 7.09 (1H, ddd), 7.27 (2H, d), 7.34–7.48 (3H, m), 7.53 (2H, d); MS m/z 480 (M+); found C, 79.92%; H, 8.15%; C32H39F3 requires: C, 79.96%; H, 8.18%.2,2′,3-Trifluoro-4′′-heptyl-4-nonylterphenyl 7h
Quantities: compound 11b (1.51 g, 5.81 mmol); compound 9c (2.00 g, 4.84 mmol). The experimental procedure was as described for the preparation of compound 7d. The crude product was purified by column chromatography (silica gel; hexane) to give a colourless solid which was recrystallised from ethanol–ethyl acetate (5∶1) to yield colourless crystals. Yield 1.55 g (63%); transitions (°C) Cryst 37.0 SC 51.3 SA 67.4 N 79.6 Iso; δH (270 MHz; CDCl3) 0.88 (6H, 2 × t), 1.19–1.45 (20H, m), 1.65 (4H, 2 × quint), 2.65 (2H, t), 2.70 (2H, t), 7.00 (1H, ddd), 7.08 (1H, ddd), 7.27 (2H, d), 7.34–7.48 (3H, m), 7.53 (2H, d); MS m/z 508 (M+); found C, 80.22%; H, 8.49%; C34H43F3 requires: C, 80.28%; H, 8.52%.2,2′,3′-Trifluoro-4,4′′-dipentylterphenyl 17a
Quantities: compound 15a (2.37 g, 7.79 mmol); compound 16a (1.73 g, 7.08 mmol). The experimental procedure was as described for the preparation of compound 7a. The crude product was purified by column chromatography (silica gel; hexane) to give a colourless solid which was recrystallised from ethanol to yield colourless crystals. Yield 1.42 g (47%); transitions (°C) Cryst 62.4 N 71.1 Iso; δH (270 MHz; CDCl3) 0.91 (6H, 2 × t), 1.21–1.45 (8H, m), 1.67 (4H, 2 × quint), 2.66 (4H, 2 × t), 7.02 (1H, dd), 7.06 (1H, ddd), 7.13–7.36 (5H, m), 7.50 (2H, d); MS m/z 424 (M+); found C, 79.19%; H, 7.33%; C28H31F3 requires: C, 79.21%; H, 7.36%.2,2′,3′-Trifluoro-4′′-heptyl-4-pentylterphenyl 17b
Quantities: compound 15b (2.65 g, 7.97 mmol); compound 16a (1.63 g, 6.64 mmol). The experimental procedure was as described for the preparation of compound 7a. The crude product was purified by column chromatography (silica gel; hexane) to give a colourless solid which was recrystallised from ethanol–ethyl acetate (5∶1) to yield colourless crystals. Yield 1.48 g (49%); transitions (°C) Cryst 44.2 N 68.9 Iso; δH (270 MHz; CDCl3) 0.89 (3H, t), 0.92 (3H, t), 1.21–1.45 (12H, m), 1.66 (4H, 2 × quint), 2.66 (4H, 2 × t), 7.02 (1H, dd), 7.06 (1H, ddd), 7.14–7.36 (5H, m), 7.50 (2H, d); MS m/z 452 (M+); found C, 79.57%; H, 7.75%; C30H35F3 requires: C, 79.61%; H, 7.79%.2,2′,3′-Trifluoro-4′′-nonyl-4-pentylterphenyl 17c
Quantities: compound 15c (2.70 g, 7.50 mmol); compound 16a (1.53 g, 6.25 mmol). The experimental procedure was as described for the preparation of compound 7a. The crude product was purified by column chromatography (silica gel; hexane) to give a colourless solid which was recrystallised from ethanol–ethyl acetate (5∶1) to yield colourless crystals. Yield 1.45 g (48%); transitions (°C) Cryst 50.5 N 69.1 Iso; δH (270 MHz; CDCl3) 0.88 (3H, t), 0.92 (3H, t), 1.20–1.45 (16H, m), 1.66 (4H, 2 × quint), 2.66 (4H, 2 × t), 7.02 (1H, dd), 7.06 (1H, ddd), 7.13–7.37 (5H, m), 7.50 (2H, d); MS m/z 480 (M+); found C, 79.90%; H, 8.14%; C32H39F3 requires: C, 79.96%; H, 8.18%.2,2′,3′-Trifluoro-4-heptyl-4′′-pentylterphenyl 17d
Quantities: compound 15a (2.42 g, 7.97 mmol); compound 16b (1.81 g, 6.64 mmol). The experimental procedure was as described for the preparation of compound 7a. The crude product was purified by column chromatography (silica gel; hexane) to give a colourless solid which was recrystallised from ethanol to yield colourless crystals. Yield 1.16 g (39%); transitions (°C) Cryst 41.2 N 69.9 Iso; δH (270 MHz; CDCl3) 0.89 (3H, t), 0.91 (3H, t), 1.21–1.44 (12H, m), 1.66 (4H, 2 × quint), 2.65 (4H, 2 × t), 7.02 (1H, dd), 7.06 (1H, ddd), 7.14–7.36 (5H, m), 7.50 (2H, d); MS m/z 452 (M+); found C, 79.59%; H, 7.76%; C30H35F3 requires: C, 79.61%; H, 7.79%.2,2′,3′-Trifluoro-4,4′′-diheptylterphenyl 17e
Quantities: compound 15b (2.49 g, 7.50 mmol); compound 16b (1.71 g, 6.25 mmol). The experimental procedure was as described for the preparation of compound 7b. The crude product was purified by column chromatography (silica gel; hexane) to give a colourless solid which was recrystallised from ethanol to yield colourless crystals. Yield 1.66 g (55%); transitions (°C) Cryst 56.4 N 69.5 Iso; δH (270 MHz; CDCl3) 0.89 (6H, 2 × t), 1.21–1.44 (16H, m), 1.66 (4H, 2 × quint), 2.66 (4H, 2 × t), 7.02 (1H, dd), 7.06 (1H, ddd), 7.14–7.36 (5H, m), 7.50 (2H, d); MS m/z 480 (M+); found C, 79.94%; H, 8.15%; C32H39F3 requires: C, 79.96%; H, 8.18%.2,2′,3′-Trifluoro-4-heptyl-4′′-nonylterphenyl 17f
Quantities: compound 15c (2.55 g, 7.09 mmol); compound 16b (1.61 g, 5.91 mmol). The experimental procedure was as described for the preparation of compound 7a. The crude product was purified by column chromatography (silica gel; hexane) to give a colourless solid which was recrystallised from ethanol to yield colourless crystals. Yield 1.63 g (54%); transitions (°C) Cryst 48.1 N 70.0 Iso; δH (270 MHz; CDCl3) 0.90 (3H, t), 0.91 (3H, t), 1.22–1.45 (20H, m), 1.67 (4H, 2 × quint), 2.67 (4H, 2 × t), 7.03 (1H, dd), 7.07 (1H, ddd), 7.15–7.37 (5H, m), 7.52 (2H, d); MS m/z 508 (M+); found C, 80.22%; H, 8.48%; C34H43F3 requires: C, 80.28%; H, 8.52%.2,2′,3′-Trifluoro-4-nonyl-4′′-pentylterphenyl 17g
Quantities: compound 15a (1.82 g, 6.00 mmol); compound 16c (1.57 g, 5.21 mmol). The experimental procedure was as described for the preparation of compound 7a. The crude product was purified by column chromatography (silica gel; hexane) to give a colourless solid which was recrystallised from ethanol–ethyl acetate (5∶1) to yield colourless crystals. Yield 1.19 g (48%); transitions (°C) Cryst 50.4 N 69.2 Iso; δH (270 MHz; CDCl3) 0.89 (3H, t), 0.92 (3H, t), 1.20–1.45 (16H, m), 1.66 (4H, 2 × quint), 2.66 (4H, 2 × t), 7.02 (1H, dd), 7.05 (1H, ddd), 7.13–7.36 (5H, m), 7.51 (2H, d); MS m/z 480 (M+); found C, 79.93%; H, 8.14%; C32H39F3 requires: C, 79.96%; H, 8.18%.2,2′,3′-Trifluoro-4′′-heptyl-4-nonylterphenyl 17h
Quantities: compound 15b (2.35 g, 7.09 mmol); compound 16c (1.78 g, 5.91 mmol). The experimental procedure was as described for the preparation of compound 7a. The crude product was purified by column chromatography (silica gel; hexane) to give a colourless solid which was recrystallised from ethanol–ethyl acetate (5∶1) to yield colourless crystals. Yield 2.18 g (73%); transitions (°C) Cryst 51.2 N 69.5 Iso; δH (270 MHz; CDCl3) 0.89 (6H, 2 × t), 1.21–1.44 (20H, m), 1.66 (4H, 2 × quint), 2.66 (4H, 2 × t), 7.02 (1H, dd), 7.06 (1H, ddd), 7.14–7.36 (5H, m), 7.50 (2H, d); MS m/z 508 (M+); found C, 80.28%; H, 8.50%; C34H43F3 requires: C, 80.28%; H, 8.52%.2,2′,3′-Trifluoro-4,4′′-dinonylterphenyl 17i
Quantities: compound 15c (2.42 g, 6.72 mmol); compound 16c (1.69 g, 5.60 mmol). The experimental procedure was as described for the preparation of compound 7a. The crude product was purified by column chromatography (silica gel; hexane) to give a colourless solid which was recrystallised from ethanol to yield colourless crystals. Yield 1.90 g (63%); transitions (°C) Cryst 61.0 N 70.7 Iso; δH (270 MHz; CDCl3) 0.89 (6H, 2 × t), 1.19–1.44 (24H, m), 1.66 (4H, 2 × quint), 2.66 (4H, 2 × t), 7.02 (1H, dd), 7.05 (1H, ddd), 7.13–7.36 (5H, m), 7.50 (2H, d); MS m/z 536 (M+); found C, 80.52%; H, 8.79%; C36H47F3 requires: C, 80.56%; H, 8.83%.Acknowledgements
The work reported here is published by permission of the Director, HMSO, and was supported by the Ministry of Defence. We express our thanks to Dr D. F. Ewing, Mrs B. Worthington, Mr R. Knight, Mr A. D. Roberts and Mr A. T. Rendell for various spectroscopic measurements.References
- G. W. Gray, M. Hird, D. Lacey and K. J. Toyne, J. Chem. Soc., Perkin Trans. 2, 1989, 2041 RSC.
- C. Escher, Kontakte (Darmstadt), 1986, 3 Search PubMed.
- J. C. Jones, M. J. Towler and J. R. Hughes, Displays, 1993, 14, 86 CrossRef CAS.
- A. J. Slaney, V. Minter and J. C. Jones, Ferroelectrics, 1996, 178, 65 Search PubMed.
- U. Finkenzeller, A. E. Pausch, E. Poetsch and J. Suermann, Kontakte (Darmstadt), 1993, 3 Search PubMed.
- M. E. Glendenning, J. W. Goodby, M. Hird and K. J. Toyne, J. Chem. Soc., Perkin Trans. 2, 1999, 481 RSC.
- C. Vauchier, F. Vinet and N. Maiser, Liq. Cryst., 1989, 5, 141 CAS.
- M. Hird, K. J. Toyne, G. W. Gray, D. G. McDonnell and I. C. Sage, Liq. Cryst., 1995, 18, 1 CAS.
- A. C. Littlejohn and J. W. Smith, J. Chem. Soc., 1954, 2552 RSC.
- G. W. Gray, M. Hird, D. Lacey and K. J. Toyne, Mol. Cryst. Liq. Cryst., 1990, 191, 1 Search PubMed.
- G. W. Gray, M. Hird and K. J. Toyne, Mol. Cryst. Liq. Cryst., 1991, 204, 43 Search PubMed.
- T. Takahashi, S. Saito and T. Akahane, Jpn. J. Appl. Phys., 1997, 36, 3531 CrossRef CAS.
- H. Vithana, D. Johnson, P. Bos, R. Herke, Y. K. Fung and S. Jamal, Jpn. J. Appl. Phys., 1996, 35, 2222 CrossRef CAS.
- P. Kirsch, M. Heckmeier and K. Tarumi, Liq. Cryst., 1999, 26, 449 CrossRef CAS.
- M. Hird, G. W. Gray and K. J. Toyne, Mol. Cryst. Liq. Cryst., 1991, 206, 187 Search PubMed.
- N. Miyaura, T. Yanagi and A. Suzuki, Synth. Commun., 1981, 11, 513 CrossRef CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS.
- M. Hird, R. A. Lewis, K. J. Toyne, J. J. West and M. K. Wilson, J. Chem. Soc., Perkin Trans. 1, 1998, 3479 RSC.
- T. Watanabe, N. Miyaura and A. Suzuki, Synlett, 1992, 207 CrossRef.
- D. R. Coulson, Inorg. Synth., 1972, 13, 121.
| This journal is © The Royal Society of Chemistry 2000 |