Enantiodifferentiating photocyclodimerization of cyclohexa-1,3-diene sensitized by chiral arenecarboxylates
Sadayuki Asaokaa, Motohiro Ooia, Peiyun Jianga, Takehiko Wadaa and Yoshihisa Inoue*ab
aDepartment of Molecular Chemistry and Venture Business Laboratory, Faculty of Engineering, Osaka University, 2-1 Yamada-oka, Suita, Osaka, 565-0871, Japan
bInoue Photochirogenesis Project, ERATO, JST, 4-6-3 Kamishinden, Toyonaka, 565-0085, Japan
First published on UnassignedUnassigned23rd December 1999
Abstract
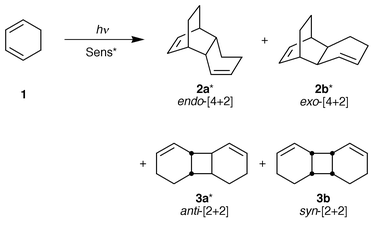
The enantiodifferentiating photosensitized cyclodimerization of cyclohexa-1,3-diene 1 was performed over a range of temperatures in the presence of chiral arene(poly)carboxylates, giving endo- and exo-[4 + 2] cyclodimers (2a, 2b) and anti- and syn-[2 + 2] cyclodimers (3a, 3b). Among the three chiral cyclodimers (2a, 2b, 3a), only 2b was obtained as an optically active species with an enantiomeric excess (ee) of up to 8.2%. The detailed reaction mechanism and the origin of the enantiodifferentiation have been elucidated, and the crucial role played by the ‘microenvironmental polarity’ around the chromophore in determining the photoreactivity and the ee of the product is also discussed.
Enantiodifferentiating photosensitization, which necessitates only a catalytic amount of chiral sensitizer, is one of the most promising methodologies for inducing chirality into prochiral substrates through the electronically excited state.1 Since the first report on the asymmetric photosensitization of trans-1,2-diphenylcyclopropane by Hammond and Cole,2 a great deal of effort has been devoted to the study of enantiodifferentiating photosensitized isomerizations, but the reported enantiomeric excesses (ee’s) have rarely exceeded 10%, until recently.2–12,20h We have demonstrated that the enantiodifferentiating geometrical photoisomerization of (Z
![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) )-cyclooctene, sensitized by chiral benzenepolycarboxylates gives the optically active (E)-isomer in exceptionally high ee’s of up to 64% at −89 °C, and displays the interesting property of product chirality inversion, induced by temperature and pressure changes.5b,h,k
)-cyclooctene, sensitized by chiral benzenepolycarboxylates gives the optically active (E)-isomer in exceptionally high ee’s of up to 64% at −89 °C, and displays the interesting property of product chirality inversion, induced by temperature and pressure changes.5b,h,kIn contrast to the unimolecular enantiodifferentiating photoisomerizations, only a few attempts have been reported on bimolecular enantiodifferentiating reactions. The enantiodifferentiating [2 + 2] photocyclodimerizations of aryl vinyl ether and 4-methoxystyrene in acetonitrile were examined in the presence of some chiral naphthalenecarboxylates to give the corresponding cyclodimers in good chemical yields, but no enantiodifferentiation occurred (ee < 1%).13 Kim and Schuster reported that the [4 + 2] photocycloaddition of trans-β-methylstyrene with cyclohexa-1,3-diene sensitized by (−)-1,1′-bis(2,4-dicyanonaphthalene), gave the cyclodimer with 15% ee at −65 °C.14f
Recently we reported that the enantiodifferentiating photoaddition of alcohols to 1,1-diphenylalkenes sensitized by chiral naphthalene(di)carboxylates gives the anti-Markovnikov adduct.15 In this bimolecular asymmetric photosensitization, we observed an unusual temperature effect on the enantioselectivity of the product. It was found that the product chirality was inverted by temperature at the critical point (T0), which enabled us to obtain both of the enantiomeric products simply by changing the irradiation temperature, also allowing higher ee’s to be obtained at higher temperatures beyond T0.15 We have also found that the chemical and optical yields of the product are critically controlled by the ‘microenvironmental polarity’ around the sensitizer chromophore, showing that the introduction of saccharide substituent(s) to the sensitizer works as a new effective strategy for overcoming the trade-off between the chemical and optical yields in such photoaddition reactions involving a radical ion intermediate. By combining the unusual temperature effect and the enhanced microenvironmental polarity by introducing saccharide substituent(s) to the sensitizer, we obtained the optimized ee of 33%.15
Photocycloaddition initiated by energy or electron transfer is one of the most widely investigated photochemical reactions.16 The photocycloadditions of 1,3-dienes to arenes have been used in the syntheses of various types of novel cyclic compounds.14e,17–20 The photocyclodimerization of cyclohexa-1,3-diene (1) which gives isomeric [4 + 2] and [2 + 2] cyclodimers (2 and 3) (Scheme 1) has also been investigated under a variety of conditions, for which several reaction mechanisms involving different intermediates have been proposed, depending on the mode of excitation.14b,20–22 Here, we report the result of our study of the enantiodifferentiating photocyclodimerization of 1 sensitized by chiral arene(poly)carboxylates. The use of chiral sensitizers with saccharide and non-saccharide substituents has enabled us to obtain definitive evidence for the cyclodimerization mechanism. Furthermore, it has allowed exploration into the enhancement of the microenvironmental polarity to increase the chemical yield without decreasing the ee of the product, by preventing the dissociation of the photochemically generated radical ion pair.
 | ||
| Scheme 1 | ||
Results and discussion
Photocyclodimerization of cyclohexa-1,3-diene (1)
The [4 + 2] and [2 + 2] cyclodimerizations of 1 have been investigated under a wide variety of thermal and photochemical conditions.14b,20–25 The thermal dimerization requires a long reaction time and affords the endo- and exo-Diels–Alder adducts 2a and 2b in poor yields, with an endo∶exo ratio of ca. 4∶1.21a,25 However, photochemical reactions of 1 lead to the formation of both [4 + 2] and [2 + 2] cyclodimers. Direct irradiation of neat 1 at 254 nm produces the exo-[4 + 2] adduct 2b and the anti- and syn-[2 + 2] adducts 3a and 3b in 1∶4.4∶2.3 ratio together with other dimers,22 whereas photosensitization with a triplet sensitizer such as phenanthrene and benzophenone gives the same products 2b, 3a and 3b but in higher combined yields. The relative product ratio (2b∶3a∶3b = ca. 1∶3∶1) is appreciably different from that obtained in the direct excitation and is independent of the triplet energy and structure of the sensitizer employed.20h In contrast, the photoinduced electron-transfer reaction of 1 leads to the endo-adduct 2a in improved yield and selectivity.20e,h We performed the electron-transfer and triplet-sensitized photocyclodimerization of 1, using 1-cyanonaphthalene (1-CN) and benzophenone (BP). As can be seen from Table 1 (runs 1 and 2), the photoinduced electron-transfer with 1-CN gave 2a as the major product along with much smaller amounts of 2b, 3a and 3b in a ratio of 27.8∶4.2∶3.0∶1.0. The triplet sensitization with BP gave only 2b, 3a and 3b in a ratio of 0.8∶3.0∶0.8, which is in good agreement with the results reported by Mattay et al.20ha
| % yield (% eeb) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Sensitizer | Solvent | Temperature/°C | Irradiation time/h | Conversion (%) | 2a | 2b | 3a | 3b |
| a [1] = 100 mmol dm−3; [Sens*] = 5 mM unless noted otherwise.b Enantiomeric excess determined by chiral GC.c Not determined.d [1] = 50 mmol dm−3.e [1] = 20 mmol dm−3.f [1] = 10 mmol dm−3. | |||||||||
| 1 | 1-CN | Acetonitrile | 25 | 6 | 61 | 21.3 (−0.1) | 3.2 (−0.3) | 2.3 (−0.3) | 0.8 |
| 2 | BP | Acetonitrile | 25 | 6 | >99 | 0 | 8.7 (+0.2) | 30.9 (+1.0) | 8.4 |
| 3 | 4a | Pentane | 25 | 2 | 44 | 0 | 2.3 (+2.5) | 6.0 (−0.9) | 1.9 |
| 4 | −43 | 4 | 49 | 0 | 1.3 (+2.8) | 3.4 (−1.5) | 0.8 | ||
| 5 | Toluene | 25 | 2 | 56 | 0 | 1.7 (−0.6) | 4.3 (−1.0) | 1.7 | |
| 6 | −41 | 4 | 22 | 0 | 1.1 (+0.3) | 2.6 (−0.7) | 0.6 | ||
| 7 | 5a | Pentane | 25 | 2 | 16 | 0 | 1.6 (+0.7) | 3.6 (−0.8) | 1.4 |
| 8 | −41 | 4 | 17 | 0 | 2.0 (−0.1) | 5.2 (−1.8) | 1.1 | ||
| 9 | Toluene | 25 | 2 | 23 | 0 | 1.0 (+2.5) | 2.5 (−1.1) | 0.4 | |
| 10 | −41 | 4 | 13 | 0 | 1.1 (+4.0) | 2.6 (−1.1) | 0.6 | ||
| 11 | 6a | Pentane | 25 | 2 | 40 | 0 | 1.7 (−0.8) | 4.5 (−2.0) | 1.4 |
| 12 | −41 | 4 | 41 | 0 | 0.6 (−0.3) | 1.7 (−1.8) | 0.4 | ||
| 13 | Toluene | 25 | 2 | 21 | 0 | 1.3 (−0.1) | 3.4 (−2.5) | 1.1 | |
| 14 | −41 | 4 | c | 0 | 1.2 (+0.7) | 3.1 (−1.5) | 0.8 | ||
| 15 | 6b | Toluene | 25 | 2 | 30 | 0 | 5.1 (−0.5) | 13.2 (−0.6) | 4.6 |
| 16 | −41 | 4 | 24 | 0 | 2.8 (−0.2) | 7.4 (−0.6) | 1.5 | ||
| 17 | 6c | Toluene | 25 | 2 | 34 | 0 | 1.7 (+0.2) | 4.3 (+0.7) | 1.5 |
| 18 | −41 | 4 | 35 | 0 | 1.0 (−0.2) | 2.5 (+0.8) | 0.6 | ||
| 19 | 7a | Pentane | 25 | 2 | 10 | 0 | 0.2 (+1.0) | 0.6 (−1.6) | 0.2 |
| 20 | −34 | 4 | 49 | 0 | 0.1(c) | 0.3 (−0.9) | 0.1 | ||
| 21 | Toluene | 25 | 2 | 11 | 0 | <0.1(c) | 0.8 (+0.3) | <0.1 | |
| 22 | −41 | 4 | 10 | 0 | 0.6 (+0.5) | 1.5 (0.0) | <0.1 | ||
| 23 | 7b | Toluene | 25 | 2 | 36 | 0 | 0.6 (−0.8) | 1.7 (+0.6) | 0.6 |
| 24 | −41 | 4 | 29 | 0 | 0.8 (−0.1) | 2.2 (+0.4) | 0.5 | ||
| 25 | 7c | Toluene | 25 | 2 | 23 | 0 | 1.8 (−0.1) | 4.8 (−0.2) | 1.7 |
| 26 | −41 | 4 | 24 | 0 | 1.8 (0.0) | 4.7 (−0.1) | 1.2 | ||
| 27 | 8a | Pentane | 25 | 2 | 92 | 0 | 17.9 (0.0) | 47.5 (−1.9) | 15.9 |
| 28 | −43 | 4 | 36 | 0 | 1.6 (+0.8) | 4.0 (−1.4) | 1.0 | ||
| 29 | Toluene | 27 | 2 | 76 | 0 | 15.6 (+0.1) | 41.4 (−0.9) | 14.0 | |
| 30 | −41 | 4 | 59 | 0 | 3.8 (+0.9) | 10.0 (+0.1) | 2.4 | ||
| 31 | Ether | 25 | 2 | 16 | 0 | 2.1 (+1.4) | 5.4 (−0.3) | 1.8 | |
| 32 | −41 | 4 | 13 | 0 | 1.5 (+0.4) | 3.8 (−0.2) | 0.9 | ||
| 33 | Acetonitrile | 25 | 2 | 78 | 26.4 (−0.1) | 5.0 (+0.2) | 7.8 (−1.1) | 1.9 | |
| 34 | 8b | Toluene | 25 | 2 | 65 | 0 | 4.4 (+1.4) | 11.2 (−1.4) | 4.2 |
| 35 | −41 | 4 | 48 | 0 | 3.5 (+3.0) | 8.8 (−1.5) | 2.2 | ||
| 36 | 8c | Pentane | 25 | 2 | 51 | 0 | 7.3 (+0.4) | 18.1 (−1.3) | 5.4 |
| 37 | Toluene | 25 | 2 | 64 | 0.5(c) | 4.5 (−5.3) | 10.8 (0.0) | 4.1 | |
| 38 | −41 | 4 | 17 | 0.9(c) | 3.2 (−2.2) | 7.1 (−0.6) | 2.1 | ||
| 39 | Ether | 25 | 2 | c | 0.3(c) | 2.8 (−4.6) | 6.3 (−0.3) | 2.0 | |
| 40 | −41 | 4 | c | 0.5(c) | 1.3 (−6.4) | 2.7 (−0.4) | 0.6 | ||
| 41 | 8d | Toluene | 25 | 2 | 21 | 0.4(c) | 5.3 (−2.8) | 11.9 (−0.8) | 4.8 |
| 42 | −41 | 4 | 38 | 1.1 (+0.8) | 3.0 (−2.0) | 6.7 (−0.4) | 2.0 | ||
| 43 | Ether | 25 | 2 | c | 0.3(c) | 3.1 (−2.9) | 7.7 (0.0) | 2.4 | |
| 44 | −41 | 4 | c | 0.6(c) | 2.5 (−3.6) | 5.9 (−0.2) | 1.3 | ||
| 45 | 8e | Toluene | 25 | 2 | 52 | 0.5(c) | 3.8 (−7.6) | 8.1 (−0.3) | 3.1 |
| 46 | −41 | 4 | 49 | 0.6(c) | 1.4 (−0.2) | 3.2 (−0.4) | 0.5 | ||
| 47 | Ether | 25 | 2 | c | 0.4(c) | 2.3 (−4.1) | 4.7 (+0.3) | 1.4 | |
| 48 | −41 | 4 | c | 0.5(c) | 1.1 (−2.5) | 2.3 (−0.7) | 0.6 | ||
| 49 | 8f | Toluene | 25 | 2 | 31 | 0.6(c) | 5.0 (−5.1) | 10.8 (−0.3) | 4.2 |
| 50 | 25d | 2 | 42 | 1.3(c) | 8.8 (−2.8) | 15.5 (−0.7) | 5.8 | ||
| 51 | 25e | 2 | 78 | 4.8(c) | 17.9 (+0.2) | 25.1 (−0.7) | 6.6 | ||
| 52 | 25f | 2 | 96 | 10.5(c) | 22.5 (+0.4) | 26.2 (−0.3) | 3.7 | ||
| 53 | −41 | 4 | 24 | 0.8(c) | 4.1 (−2.4) | 9.7 (−0.8) | 1.2 | ||
| 54 | Ether | 25 | 2 | c | 0.3(c) | 2.0 (−6.7) | 4.6 (+0.3) | 1.3 | |
| 55 | −41 | 4 | c | 0.3(c) | 0.8 (−8.2) | 1.8 (−1.3) | 0.4 | ||
| 56 | Acetonitrile | 25 | 2 | 54 | 21.1 (0.0) | 1.9 (+0.4) | 1.4 (+0.7) | 0.4 | |
| 57 | −41 | 4 | 36 | 20.0 (−0.1) | 2.5 (+0.1) | 4.1 (−0.1) | 1.1 | ||
| 58 | 9a | Pentane | 27 | 2 | 71 | 0 | 7.6 (−0.2) | 19.9 (−1.2) | 6.1 |
| 59 | −43 | 4 | 54 | 0 | 7.1 (+0.1) | 18.1 (−0.1) | 4.3 | ||
| 60 | Toluene | 27 | 2 | 95 | 0 | 21.4 (+0.4) | 56.8 (−1.4) | 20.4 | |
| 61 | −41 | 4 | >99 | 0 | 19.6 (−0.4) | 53.6 (−0.3) | 13.0 | ||
| 62 | 9b | Toluene | 25 | 2 | >99 | 0 | 9.7 (+0.7) | 26.2 (+1.0) | 9.7 |
| 63 | −41 | 4 | 95 | 0 | 11.3 (+0.4) | 30.8 (+0.2) | 7.5 | ||
| 64 | 9c | Toluene | 25 | 2 | >99 | 0 | 8.8 (+1.3) | 22.3 (−1.3) | 7.1 |
| 65 | −41 | 4 | >99 | 0 | 8.2 (+0.2) | 21.2 (−0.3) | 5.1 | ||
| 66 | 10a | Pentane | 25 | 2 | c | 0 | 0.6 (+1.9) | 1.4 (−1.0) | 0.4 |
| 67 | −39 | 3 | 21 | 0 | 0.3 (+1.2) | 0.8 (−0.7) | 0.2 | ||
| 68 | Toluene | 25 | 2 | 23 | 0 | 2.0 (+1.6) | 5.0 (+1.1) | 1.8 | |
| 69 | −41 | 3 | 52 | 0 | 1.3 (+0.1) | 3.3 (−1.0) | 0.9 | ||
| 70 | 10b | Toluene | 25 | 2 | 23 | 0 | 2.5 (+0.8) | 6.6 (−0.3) | 2.5 |
| 71 | −41 | 4 | 36 | 0 | 1.5 (+0.9) | 3.8 (−0.5) | 1.0 | ||
| 72 | 10c | Toluene | 25 | 2 | c | 0 | 2.1 (−0.2) | 5.4 (+0.6) | 2.0 |
| 73 | −41 | 4 | 39 | 0 | 1.9 (+0.3) | 5.0 (−1.4) | 1.2 | ||
| 74 | 11a | Pentane | 25 | 2 | 12 | 0 | 0.4 (−2.3) | 1.0 (−1.3) | 0.3 |
| 75 | −41 | 4 | 18 | 0 | 0.2 (c) | 0.5 (−1.2) | 0.1 | ||
| 76 | Toluene | 28 | 2 | 16 | 0 | 2.8 (+0.9) | 7.0 (−0.8) | 2.6 | |
| 77 | −41 | 4 | 52 | 0 | 1.5 (−0.2) | 3.5 (−0.3) | 1.0 | ||
| 78 | 11b | Toluene | 25 | 2 | 30 | 0 | 1.3 (−0.1) | 3.2 (−0.2) | 1.0 |
| 79 | −41 | 4 | 62 | 0 | 1.0 (−1.4) | 2.8 (+0.7) | 0.7 | ||
| 80 | 11c | Toluene | 25 | 2 | 26 | 0 | 2.4 (+0.2) | 6.3 (0.0) | 2.3 |
| 81 | −41 | 4 | 61 | 0 | 2.3 (−1.5) | 6.0 (+0.7) | 1.7 | ||
| 82 | 12a | Toluene | 25 | 2 | 53 | 0 | 1.7 (+0.4) | 4.3 (−0.8) | 1.6 |
| 83 | −41 | 4 | 55 | 0 | 1.6 (+0.2) | 4.2 (−1.0) | 1.1 | ||
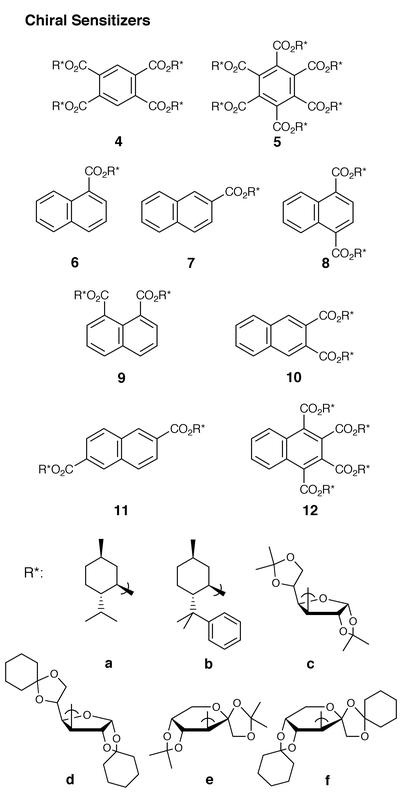
In the present study, we have employed a variety of optically active (poly)alkyl benzene- and naphthalene(poly)carboxylates (4–12) as chiral sensitizers for the enantiodifferentiating photocycloaddition of cyclohexa-1,3-diene 1, as illustrated in Chart 1. Although arene(poly)carboxylates have not frequently been used as sensitizers in photoinduced electron transfer reactions of aromatic alkenes,26,27 they are prominent and effective chiral sensitizers15b for the enantiodifferentiating photoaddition, which will allow us to examine a wide variety of chiral auxiliaries introduced into the vicinity of the chromophore.
 | ||
| Chart 1 | ||
In performing optically and chemically efficient enantiodifferentiation in a photoreaction that involves an electron transfer process and radical ionic species, one of the most important factors is the choice of solvent. In general, the use of a polar solvent is an essential condition for high chemical yield, which however often accompanies a decreased optical yield of photoproduct as a result of the intervention of free or solvent-separated radical ion pairs between the chiral sensitizer and substrate.5i,14f,15 We have therefore employed nonpolar or less polar solvents in the present enantiodifferentiating photocyclodimerization of 1.
Arenecarboxylate sensitizers
In search of the most effective arenecarboxylate sensitizers for the photocyclodimerization of 1, benzenepolycarboxylates (4 and 5) and naphthalene(poly)carboxylates (6–12) with several chiral auxiliaries (a–f) (shown in Chart 1) were examined. Using optically active arenecarboxylates (5 mmol dm−3), the photosensitized cyclodimerization of 1 (100 mmol dm−3) was performed in either pentane, toluene, diethyl ether or acetonitrile at −41 and + 25 °C to give 2a, 2b, 3a and/or 3b. Of these four cyclodimers, 2a, 2b and 3a are chiral, as indicated by an asterisk in Scheme 1. The chemical yields and the enantiomeric excesses (ee’s), as determined by chiral stationary-phase gas chromatography, are summarized in Table 1. Since the enantiomers of the cyclodimers could not be isolated on a preparative scale, the sign of the reported ee value is just a tentative one representing the order of elution from the Supelco β-Dex 120 and 325 columns, and therefore may not coincide with the direction of the optical rotation of the product. Thus, a positive value means the predominant formation of the first-eluted enantiomer.The photocyclodimerizations sensitized by chiral arene(poly)carboxylates possessing (−)-menthyl and (−)-8-phenylmenthyl auxiliaries were performed in pentane and toluene at 25 and −41 °C. Polymenthyl benzenepolycarboxylates which were used as singlet energy-transfer sensitizers for photoisomerization of cycloalkenes5 were examined first. As can be seen from Table 1, the singlet sensitization with benzenetetracarboxylate 4a (runs 3–6) and benzenehexacarboxylate 5a (runs 7–10) gave cyclodimers 2b, 3a and 3b in low chemical yields but never produced the endo-dimer 2a. Irrespective of the solvent and sensitizer used, effectively the same product ratio was obtained at 25 °C, i.e.2b∶3a∶3b = 1.2∶3.0∶1.0, which is slightly different from that observed for triplet sensitization with BP, i.e.2b∶3a∶3b = 0.8∶3.0∶0.8. However, the product distribution was affected by the irradiation temperature, with the ratio of 3b decreasing at lower temperatures, while the 2b∶3a ratio stayed constant. The ee of 2b was generally low (<2.5%) at 25 °C but was appreciably enhanced to 4.0% in toluene at −41 °C, upon sensitization with 5a. Conversely, low ee’s (<2%) were obtained for 3a at 25 °C and were not improved even at −41 °C.
We further examined chiral naphthalene(poly)carboxylates, which are often used in photoinduced electron-transfer reactions.15 Photosensitized cyclodimerization of 1 using naphthalenecarboxylates 6a and 7a (runs 11-14 and 19–22, respectively) gave 2b, 3a and 3b in low chemical yields (2b and 3b in <2%, 3a in <5%), these yields were slightly enhanced in toluene, however no 2a was formed in either pentane or toluene. The product ratios 2b∶3a∶3b were 1.2∶3.0∶1.0 and 1.2∶3.0∶0.7 at 25 and −41 °C respectively, which are exactly the same as those obtained in the benzenepolycarboxylate sensitizations described above. This agreement suggests that the photocyclodimerizations of 1 sensitized by benzenecarboxylates 4a and 5a, and by naphthalenecarboxylates 6a and 7a proceed through a common intermediate such as a singlet biradical. Unfortunately, the photosensitizations with the naphthalenemonocarboxylates 6a–c and 7a–c gave practically racemic 2b and 3a in both pentane and toluene even at the low temperature.
Chemical yields were greatly improved upon sensitization with naphthalene-1,4- and 1,8-dicarboxylates 8a and 9a (runs 27–30 and 58–61), up to 14–20% for 2b and 3b and 41–57% for 3a. But sensitization with naphthalene-2,3- and 2,6-dicarboxylates 10a and 11a (runs 66-69 and 74–77, respectively) was ineffective in enhancing the chemical yields, resulting in low ee’s (< 2.5%) in all cases. In general, the use of toluene as solvent slightly enhanced the chemical yields but did not improve the products’ ee. Judging from the facts that the product ratios obtained upon sensitization with 8a–11a agree with those obtained with the benzenepolycarboxylates 4a and 5a, and that the endo-adduct 2a was not formed under these conditions, we deduce that the photosensitization with the naphthalenedicarboxylates in non-polar solvents proceeds through the singlet energy-transfer mechanism involving a singlet biradical or other common intermediate, as is the case with the benzenepolycarboxylates. The ee’s were not enhanced by using the (−)-8-phenylmenthyl naphthalenedicarboxylates 8b–11b (runs 34–35, 62–63, 70–71 and 78–79, respectively). Neither chemical yield nor ee’s were improved upon by using the highly substituted tetramenthyl naphthalenetetracarboxylate 12a in toluene (runs 82–83). Based on these results we may conclude that the product ratio is independent of the energy and structure of sensitizers in non-polar solvents, and also that the simple singlet energy-transfer sensitization is ineffective in inducing chirality in the cyclodimers.
In order to elucidate the origin of the sensitizer-dependent chemical yields, we calculated the Rehm–Weller free energy change (ΔGet)28 from the oxidation potential of 1 (Eox = 1.15 V),20c the reduction potentials (Ered) and fluorescence 0–0 bands (λ0–0) of sensitizers 4a–12a. The relevant data are listed in Table 2. The observed differences in photoreactivity are well accounted for in terms of the calculated ΔGet values. Apart from the highly hindered naphthalene-1,2,3,4-tetracarboxylate 12a,29 the naphthalene-1,4- and 1,8-dicarboxylates 8a and 9a gave the most negative ΔGet values among the sensitizers examined, which is the primary reason for the high chemical yields obtained upon sensitization with 8 and 9. As the singlet energies of naphthalene(poly)carboxylates 6a–12a are significantly lower than those of benzenepolycarboxylates 4a and 5a, the simple singlet energy transfer mechanism cannot rationalize the photoreactivity, consequently we may conclude that the photocyclodimerization of 1 sensitized by naphthalene(poly)carboxylates (at least with 8 and 9) proceeds through the electron transfer mechanism which involves an exciplex with high charge-transfer character or a contact ion pair even in the nonpolar solvents.
| Sensitizer | Ereda/V | λ0–0b/nm | ΔGetc/kJ mol−1 |
|---|---|---|---|
| a Reduction potentials estimated from the half-wave potentials measured using a platinum electrode, relative to the Ag/AgCl electrode using 0.1 mol dm−3 tetrabutylammonium perchlorate as the electrolyte in acetonitrile. b Fluorescence maxima of highest energy emission in frozen EPA (diethyl ether∶isopentane:ethanol = 5∶5∶2) glass at 77 K. c Based on Weller equation: ΔGet = 23.06 (Eox(D+/D) − Ered(A/A–)) −
ΔG0–0 − wp; oxidation potential of 1 (Eox) estimated as 0.028 V before the peak potential (Ep = 1.33 V14a); Coulombic attraction term (wp) taken to be −5.4 kJ mol−1. d Not determined due to low solubility of 4a and 5a in acetonitrile. | |||
| 4a | d | 315 | — |
| 5a | d | 309 | — |
| 6a | −2.30 | 334 | −5.2 |
| 7a | −2.39 | 339 | 8.8 |
| 8a | −1.84 | 371 | −13.9 |
| 9a | −2.22 | 334 | −12.9 |
| 10a | −2.30 | 341 | 2.2 |
| 11a | −2.02 | 357 | −9.1 |
| 12a | −1.89 | 345 | −33.7 |
Effect of saccharide auxiliary
In our recent study,15b we demonstrated that the use of protected saccharides as chiral auxiliaries of the photosensitizer can enhance both the chemical and optical yields in the enantiodifferentiating photoaddition of alcohols to 1,1-diphenylalkene, through the increased ‘microenvironmental polarity’ around the sensitizer chromophore. In this context it is interesting to examine the effects of saccharide derivatives (c–f). The photocyclodimerizations of 1 sensitized by naphthalene(di)carboxylates 6c–11c, which possess diacetone glucose (DAG) auxiliaries were first examined in pentane and in toluene (runs 17–18, 25–26, 36–38, 64–65, 72–73 and 80–81 respectively). Unexpectedly, the DAG ester chiral sensitizers 6c, 7c, 9c, 10c and 11c did not particularly improve the chemical or optical yields, and the product ratios obtained were very similar to those for the menthyl esters 6a–11a. However, the photosensitization with naphthalene-1,4-dicarboxylates with saccharide auxiliaries 8c–f (runs 36–57) showed distinctly different behaviour in toluene. The endo-adduct 2a which is derived from the radical cation intermediate usually generated in polar solvent under the electron transfer conditions was obtained in low but appreciable yield, along with the slightly enhanced formation of 2b (the average product ratio of 2b∶3a∶3b is 1.3∶3.0∶1.1 and 1.3∶3.0∶0.7 at 25 and −41 °C, respectively). The ee of 2b was increased to 7.6% upon sensitization with 8e in toluene at 25 °C (run 45), whereas the 3a obtained was practically racemic in any solvent and at any temperature examined. Unfortunately in most cases the ee of 2a could not be determined by chiral GC as a result of low chemical yields, though 2a obtained from toluene at −41 °C (run 42) was racemic.In order to investigate the influence of solvent polarity, photosensitization by 8c–f was performed in diethyl ether (runs 39–40, 43–44, 47–48, 54–55) and in acetonitrile (runs 56–57). In diethyl ether, the yield of 2a relative to 3a was slightly enhanced for all saccharide sensitizers and the highest ee (8.2%) was obtained for 2b upon sensitization with 8f in ether at −41 °C (run 55), although the ee of 3a was not improved in polar solvents. In contrast, the photosensitization with menthyl ester 8a in ether gave no endo-adduct 2a, and the resulting product ratio is comparable to that obtained in non-polar solvent (runs 31 and 32). Hence, the formation of 2a and the altered product ratios obtained upon sensitizations with saccharide esters are attributable to the enhanced microenvironmental polarity around the sensitizer chromophore. Under such conditions, the charge-transfer interaction is encouraged by the enhanced microenvironmental polarity, and the dissociation of the resulting radical ion pair is discouraged by the low bulk polarity. The combined effects keep the stereochemical interaction between chiral sensitizer and the substrate more intimate, resulting in increased ee’s. Judging from the fact that the highest ee was obtained in ether, the enhancement of the microenvironmental polarity is not canceled by ether’s lower bulk polarity. In acetonitrile the effect of the saccharide auxiliaries seems to disappear completely, as the photosensitization with both the methyl ester 8a and the saccharide ester 8f gave the electron-transfer product 2a as the main product, and all of the chiral products obtained were racemic.
Effect of substrate concentration
The product ratio of the electron-transfer dimerization of 1 is known to be sensitive to the reaction conditions, e.g. solvent,19,20c,f,30 concentration of 1,20c,e,f,24 wavelength20c,22 and sensitizer.20c Upon sensitization with 1,4-dicyanonaphthalene or chloranil in acetonitrile the product ratio depends critically on the concentration of the substrate 1, with the endo-isomer 2a more favoured at lower concentrations. This has been accounted for in terms of the involvement of differently solvated radical ion pairs. At high concentration of 1, the polarized exciplex or contact ion pair (CIP), which is formed after (partial) electron transfer from 1 to excited sensitizer, is efficiently quenched by a second molecule of 1 to afford exo-adduct 2b. At low concentration of 1, the CIP dissociates to solvent-separated ion pairs (SSIP), which in turn give endo-adduct 2a.20c,e In the present study, we observed considerable concentration effects on the product distribution upon photosensitization with 8f in diethyl ether (runs 49–52). Thus, on decreasing the concentration of 1 from 100 to 10 mmol dm−3, the endo/exo ratio (2a/2b) increased from 0.12 to 0.47. Since these results coincide exactly with the reported observations,20c,e,24 it can be concluded that the electron-transfer mechanism operates in this reaction, and that the mechanism involves differently solvated radical-ion pairs. On the other hand, the ee of 2b was reduced with decreasing concentration of 1, and eventually no enantiodifferentiation was observed at concentrations less than 20 mmol dm−3 (runs 51 and 52), where the endo-isomer 2a is favoured. It has been shown that a contact ion pair or exciplex with high charge-transfer character shows different selectivities in the photoinduced electron-transfer dimerization of 1,19,20b,c,d compared to a solvent-separated ion pair or free radical cation. The endo-dimer 2a is formed from the solvent-separated ion pair or free radical cation. Since the insufficiently cationic 1 in the contact ion pair or exciplex is not efficiently trapped by ground-state 1 at low concentrations, it thus tends to form a solvent-separated ion pair for which effective enantiodifferentiation is not expected to occur.Quenching of sensitizer fluorescence
In order to elucidate the excited state and mechanism involved in the photosensitized cyclodimerization, fluorescence quenching experiments were performed with the menthyl esters 6a–12a in aerated pentane and acetonitrile. The fluorescence of sensitizers was efficiently quenched upon the addition of 1 up to 100 mmol dm−3. Representative quenching behaviour of 8a in pentane is shown in Fig. 1. Even at high concentrations of 1 no emission attributable to exciplex or triplex intermediates was observed for any sensitizer.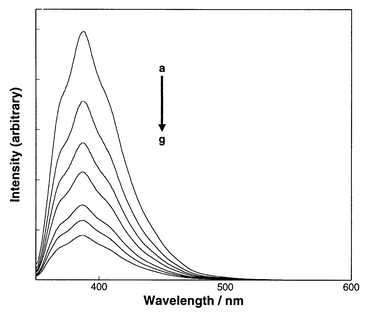 | ||
| Fig. 1 Quenching of fluorescence of 8a (1 mmol dm−3 in pentane), excited at 340 nm, by 1 at various concentrations: (a) 0, (b) 5, (c) 10, (d) 16, (e) 26, (f) 37, and (g) 52 mmol dm−3.
| ||
Using the conventional Stern–Volmer treatment of the quenching data [eqn. (1)], the relative fluorescence intensity (IF0/IF) was plotted as a function of the concentration of 1 added, and an excellent straight line was obtained for each sensitizer, as exemplified in Fig. 2. From the Stern–Volmer constant (kQτ0) obtained from the slope of the plot and the fluorescence lifetime (τ0) determined independently by using the single photon counting technique, the quenching rate constant (kQ) for each sensitizer was calculated. The results are summarized in Table 3.
| IF0/IF = 1 + kQτ0 [Q] | (1) |
a
| Sensitizer | Solvent | kQτ/mol−1dm3 | τb/ns | kQ/1010 mol−1 dm3 s−1 |
|---|---|---|---|---|
| a Measured with 0.01 mmol dm−3 aerated solution of sensitizers at 25 °C.b Fluorescence lifetime of sensitizers in aerated solution at 25 °C. | ||||
| 6a | Pentane | 21 | 0.78 | 2.7 |
| 7a | Pentane | 87 | 8.0 | 1.1 |
| 8a | Pentane | 88 | 3.6 | 2.4 |
| Acetonitrile | 92 | 8.2 | 1.1 | |
| 9a | Pentane | 21 | 1.5 | 1.4 |
| 10a | Pentane | 78 | 6.6 | 1.2 |
| 11a | Pentane | 121 | 9.9 | 1.2 |
| 12a | Pentane | 30 | 2.9 | 1.1 |
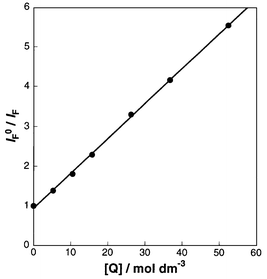 | ||
| Fig. 2 Stern–Volmer plot for fluorescence quenching of 8a by 1 in pentane. | ||
The quenching of sensitizer singlet by 1 proceeds very efficiently at rates of 1.1–2.7 × 1010 mol−1 dm3 s−1, which are almost comparable to the diffusion controlled rate in pentane (kdiff = 4.4 × 1010 mol−1 dm3 s−1)31 and acetonitrile (kdiff = 2.9 × 1010 mol−1 dm3 s−1).31 Although no exciplex emission was observed, it is inferred that the quenching leads to an exciplex intermediate with high charge-transfer character, or directly to a contact radical ion pair in nonpolar solution. If the ΔGet value is not sufficiently negative to develop a positive charge on 1, the subsequent attack of the second 1 (forming a dimer biradical) should be decelerated, which should account for the low chemical yields obtained upon sensitization with 6a, 7a, 10a and 11a. Contrary to this, the much higher chemical yields obtained upon sensitization with 8a and 9a are attributable to the highly negative ΔGet values for 8a and 9a and the accompanying development of positive charge on 1, which accelerates the subsequent attack of 1.
Mechanism
On the basis of the mechanism reported previously,14b,19,20 we propose a modified mechanism illustrated in Scheme 2, which is compatible with the previous and present results. In view of the relatively low concentrations of 1 (10–100 mmol dm−3) employed in this study, we may exclude the serious contribution of the triplex intermediate,14a–d,30 intervention of which has been proposed at much higher concentrations of 0.2–2 mol dm−3 in non-polar or less polar solvents.14a–d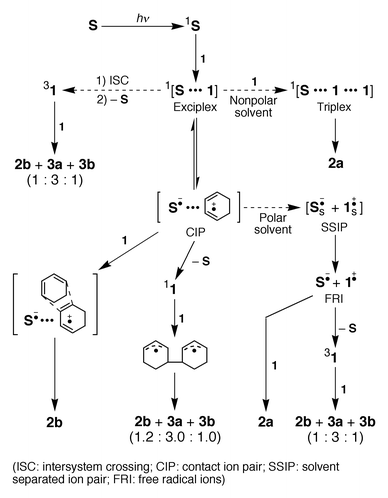 | ||
| Scheme 2 | ||
Although the enantiodifferentiating photocyclodimerization of 1 sensitized by various chiral sensitizers can potentially give four isomeric cyclodimers 2a, 2b, 3a and 3b as described above, significant ee’s were obtained exclusively for the exo-[4 + 2]-cyclodimer 2b upon sensitization with saccharide esters 8c–f in pentane or ether. This means that in addition to the biradical and radical ionic routes illustrated in Scheme 2, there is an independent cyclodimerization pathway that involves either the exciplex or contact ion pair of 1 with chiral sensitizer and affords preferentially 2b. In the case of saccharide esters 8c–f, the highly negative ΔGet values and the enhanced microenvironmental polarity around the chromophore may stabilize such an exciplex or contact ion pair intermediate in nonpolar solvents, allowing the transfer of chiral information from the sensitizer to the cyclodimer. Because the product ratios obtained in nonpolar solvents do not greatly deviate from the average value (2b∶3a∶3b = 1.15∶ 3.00∶1.02 and 1.17∶3.00∶0.74 at 25 and −41 °C, respectively) for most of the naphthalene(di)carboxylate sensitizers, except for the saccharide esters 8c–f. Then, we may estimate the ‘net’ ee of 2b produced through this ‘independent’ exciplex route by assuming that 2b, 3a and 3b formed through the singlet biradical intermediate are racemic (or inherently achiral), and their ratio is fixed at 1.15∶3.00∶1.02, irrespective of the sensitizer and solvent used. Also that 2b is produced exclusively through either the exciplex or singlet biradical mechanism, as demonstrated in the literature.20,32 In the case of the photosensitization by 8e in toluene at 25 °C, 19% of 2b is estimated to be formed via the exciplex, with a ‘net’ ee of 40%. In the case of the photosensitization by 8f in ether at 25 and −41 °C, 10 and 12% of 2b is similarly estimated to be formed via the exciplex, and the ‘net’ ee’s are 65% and 70%, respectively.
Finally, we would like to emphasize that although the overall ee’s are not very high (<8%) in the present case as a result of the contamination from the racemic product of the other route, the introduction of polar saccharide moieties into the sensitizer can raise the ee of the product through the enhancement of the microenvironmental polarity around the sensitizer chromophore.
Experimental
General
Melting points were measured with a YANACO MP-300 apparatus and are uncorrected. 1H NMR spectra were obtained on a JEOL GX-400 or GSX-270 spectrometer in [2H2]-chloroform (CDCl3). Infrared spectra were obtained on a JASCO FT/IR-230 instrument. Electronic absorption and fluorescence spectra were recorded on JASCO V-550 and FP-777 instruments, respectively. Optical rotations were determined at 589 nm in a thermostatted conventional 10 cm cell, using a JASCO DIP-1000 polarimeter.Fluorescence lifetimes were measured with 1 × 10−5 mol dm−3 solution of sensitizers in aerated pentane or toluene by means of the time-correlated single-photon-counting method on a Horiba NAES-1100 instrument equipped with a pulsed H2 light source. The radiation from the lamp was made monochromatic by a 10-cm monochromator, and the emission from sample solution was detected through a Toshiba UV-33, 35 or 37 filter.
Enantiomeric excesses of 2a, 2b and 3a were determined by gas chromatography over a 30 m chiral capillary column (SUPELCO β-Dex325 and/or 120) at 100 °C, using a Shimadzu GC-14B instrument connected to a Shimadzu C-R6A integrator. Calibrations with racemic 2a, 2b and 3a indicated that the GC analysis gave a systematic error of ±0.8% ee.
Materials
Pentane used as solvent was stirred over concentrated sulfuric acid until the acid layer no longer turned yellow, washed with water, neutralized with aqueous sodium hydrogen carbonate, dried over sodium sulfate, and then fractionally distilled. Toluene was fractionally distilled from melting sodium. Diethyl ether was refluxed with potassium hydroxide and then fractionally distilled from sodium. Spectrograde acetonitrile (Dojin) was used without further purification. Cyclohexa-1,3-diene 1 (Aldrich) was purified by fractional distillation, followed by column chromatography on activated aluminum oxide (ICN Biomedicals).Optically active alcohols used in the preparation of the sensitizers were commercially available: (−)-menthol from TCI; (−)-8-phenylmenthol from Aldrich.
Sugar derivatives were prepared from D-glucose and D-fructose according to the procedures reported by Kartha et al.33 and Kang et al.,34 respectively.15b 1,2∶5,6-Di-O-cyclohexylidene-α-D-glucofuranose and 1,2∶4,5-di-O-cyclohexylidene-β-D-fructopyranose were prepared in a similar manner. 1,2∶5,6-Di-O-cyclohexylidene-α-D-glucofuranose: [α]D31 +3.51° (c 2.15, CHCl3) (lit.35 [α]D31 +1.65° (c 2.10, CHCl3)); mp 139–140 °C; δH(CDCl3) 1.24–1.87 (m, 20H), 2.60 (d, J = 2.9 Hz, 1H), 3.96 (dd, J = 2.9, 5.9 Hz, 1H), 4.06 (dd, J = 2.4, 4.9 Hz, 1H), 4.14–4.18 (m, 1H), 4.33–4.34 (m, 1H), 4.52 (d, J = 3.4 Hz, 1H), 5.95 (d, J = 3.4 Hz, 1H). 1,2∶4,5-Di-O-cyclohexylidene-β-D-fructopyranose: [α]D25 −108.3° (c 0.52, CHCl3); mp 130–131 °C; δH(CDCl3) 1.59–1.79 (m, 20H), 3.64 (dd, J = 6.8, 8.3 Hz, 1H), 3.96 (d, J = 8.8 Hz, 1H), 4.01–4.05 (m, 2H), 4.10 (t, J = 2.4 Hz, 2H), 4.12–4.22 (m, 2H).
1-Cyanonaphthalene (TCI) and benzophenone (Wako) used as achiral sensitizers were purified by recrystallization from methanol. Optically active benzenepolycarboxylates employed as chiral sensitizers were prepared as reported previously.36 Chiral naphthalene(di)carboxylates were prepared from the corresponding alcohols and acid chlorides, which were prepared from the corresponding carboxylic acids or anhydrides.15b While most of the carboxylic acids and anhydrides were commercially available: naphthalene-1-, -2- and -1,4-(di)carboxylic acid from Wako, naphthalene-1,8- and -2,3-dicarboxylic anhydride from TCI, naphthalene-2,6-dicarboxylic acid dipotassium salt from Aldrich, naphthalene-1,2,3,4-tetracarboxylic acid was obtained by the hydrolysis of the tetramethyl ester, which was prepared according to the procedures reported by Cadogan et al.37
Photolysis
All irradiations were performed in a temperature-controlled water (25 °C), methanol–propan-2-ol (−40 °C) bath. The light sources employed were a conventional 300 W high-pressure mercury lamp for irradiations at 25 °C and an equivalent lamp fitted with a transparent Pyrex vacuum sleeve designed for low-temperature irradiation (Eikosha). A solution (4 cm3), containing cyclohexa-1,3-diene 1 (100 mmol dm−3), optically active sensitizer 4–12 (5 mmol dm−3), and n-dodecane (5 mmol dm−3) added as an internal standard, was irradiated at >300 nm under an argon atmosphere in a Pyrex tube (1 cm i.d.) placed near the lamp surface or in an annular Pyrex vessel surrounding the lamp, the whole system being immersed in the cooling bath.Acknowledgements
We would like to thank Dr Guy Hembury for assistance in the preparation of this manuscript.References
- For reviews, see H. Rau, Chem. Rev., 1983, 83, 535 Search PubMed; (a) Y. Inoue, Chem. Rev., 1992, 92, 471; (b) S. R. L. Everitt and Y. Inoue , in Organic Molecular Photochemistry, V. Ramamurthy and K. Schanze, Eds.; Marcel Dekker, New York, 1999;p. 71. Search PubMed.
- G. S. Hammond and R. S. Cole, J. Am. Chem. Soc., 1965, 87, 3256 CrossRef CAS.
- C. Ouannès, R. Beugelmans and G. Roussi, J. Am. Chem. Soc., 1973, 95, 8472 CrossRef CAS.
- G. Balavoine, S. Jugè and H. B. Kagan, Tetrahedron Lett., 1973, 4159 CrossRef CAS.
- (a) Y. Inoue, Y. Kunitomi, S. Takamuku and H. Sakurai, J. Chem. Soc., Chem. Commun., 1978, 1024 RSC; (b) Y. Inoue, S. Takamuku, Y. Kunitomi and H. Sakurai, J. Chem. Soc., Perkin Trans. 2, 1980, 1672 RSC; (c) S. Goto, S. Takamuku, H. Sakurai, Y. Inoue and T. Hakushi, ibid., 1980, 1678 Search PubMed; (d) Y. Inoue, T. Yokoyama, N. Yamasaki and A. Tai, Nature, 1989, 341, 225 CrossRef CAS; (e) Y. Inoue, T. Yokoyama, N. Yamasaki and A. Tai, J. Am. Chem. Soc., 1989, 111, 6480 CrossRef CAS; (f) Y. Inoue, H. Shimoyama, N. Yamasaki and A. Tai, Chem. Lett., 1991, 593; (g) Y. Inoue, N. Yamasaki, T. Yokoyama and A. Tai, J. Org. Chem., 1992, 57, 1332 CrossRef CAS; (h) Y. Inoue, N. Yamasaki, T. Yokoyama and A. Tai, J. Org. Chem., 1993, 58, 1011 CrossRef CAS; (i) Y. Inoue, N. Yamasaki, H. Shimoyama and A. Tai, J. Org. Chem., 1993, 58, 1785 CrossRef CAS; (j) Y. Inoue, H. Tsuneishi, T. Hakushi and A. Tai, J. Am. Chem. Soc., 1997, 119, 472 CrossRef CAS; (k) Y. Inoue, E. Matsushima and T. Wada, J. Am. Chem. Soc., 1998, 120, 10687 CrossRef CAS.
- (a) O. Piva, F. Henin, J. Muzart and J.-P. Pete, Tetrahedron Lett., 1986, 27, 3001 CrossRef CAS; (b) O. Piva, R. Mortezaei, F. Henin, J. Muzart and J.-P. Pete, J. Am. Chem. Soc., 1990, 112, 9263 CrossRef CAS.
- (a) H. Rau and M. Hormann, J. Photochem., 1981, 16, 231 Search PubMed; (b) H. Rau and R. Ratz, Angew. Chem., 1983, 95, 552 CAS; Angew. Chem., Int. Ed. Engl., 1983, 22, 550. Search PubMed; (c) H. Rau and F. Totter, J. Photochem. Photobiol. A: Chem., 1992, 63, 337 Search PubMed.
- (a) M. Demuth, P. R. Raghavan, C. Carter, K. Nakano and K. Schaffner, Helv. Chim. Acta, 1980, 63, 2434 CrossRef CAS; (b) M. Demuth, A. Palomer, H.-D. Sluma, A. K. Dey, C. Kruger and Y.-H. Tsay, Angew. Chem., 1986, 98, 1093 CAS; Angew. Chem., Int. Ed. Engl., 1986, 25, 117. Search PubMed.
- N. Hoshi, Y. Furukawa, H. Hagiwara, H. Uda and K. Sato, Chem. Lett., 1980, 47 CAS.
- (a) K. Ohkubo, T. Hamada, T. Inaoka and H. Ishida, Inorg. Chem., 1989, 28, 2021 CrossRef CAS; (b) K. Ohkubo, H. Ishida, T. Hamada and T. Inaoka, Chem. Lett., 1989, 1545 CAS.
- T. Hikita, K. Tamaru, A. Yamagishi and T. Iwamoto, Inorg. Chem., 1989, 28, 2221 CrossRef CAS.
- K. Okada, H. Sakai, M. Oda, A. Yoshimura and T. Ohno, Tetrahedron Lett., 1989, 30, 1091 CrossRef CAS.
- Y. Inoue, T. Okano, N. Yamasaki and A. Tai, J. Photochem. Photobiol. A: Chem., 1992, 66, 61 Search PubMed.
- (a) N. J. Peacock and G. B. Schuster, J. Am. Chem. Soc., 1983, 105, 3632 CrossRef CAS; (b) G. C. Calhoun and G. B. Schuster, J. Am. Chem. Soc., 1984, 106, 6870 CrossRef CAS; (c) G. C. Calhoun and G. B. Schuster, J. Am. Chem. Soc., 1986, 108, 8021 CrossRef CAS; (d) D. Hartsough and G. B. Schuster, J. Org. Chem., 1989, 54, 3 CrossRef CAS; (e) N. Aubulut, D. Hartsough, J.-I. Kim and G. B. Schuster, J. Org. Chem., 1989, 54, 2549 CrossRef; (f) J.-I. Kim and G. B. Schuster, J. Am. Chem. Soc., 1990, 112, 9635 CrossRef CAS.
- (a) Y. Inoue, T. Okano and N. Yamasaki and A. Tai, J. Chem. Soc., Chem. Commun., 1993, 718 RSC; (b) S. Asaoka, T. Kitazawa, T. Wada and Y. Inoue, J. Am. Chem. Soc., 1999, 121, 8486 CrossRef CAS.
- For review, see F. Müller and J. Mattay, Chem. Rev., 1993, 93, 99 Search PubMed.
- T.-y. Wang, J.-d. Ni, J. Masnovi and N. C. Yang, Tetrahedron Lett., 1982, 23, 1231 CrossRef CAS.
- (a) W. K. Smothers and J. Saltiel, J. Am. Chem. Soc., 1983, 105, 2794 CrossRef CAS; (b) J. Saltiel, R. Dabestani, D. F. Sears Jr., W. M. McGowan and E. F. Hilinski, J. Am. Chem. Soc., 1995, 117, 9129 CrossRef CAS.
- J. Mlcoch and E. Steckhan, Angew. Chem., Int. Ed. Engl., 1985, 24, 412 CrossRef; Tetrahedron Lett., 1987, 28, 1081. Search PubMed.
- (a) J. Mattay, J. Gersdorf and J. Mertes, J. Chem. Soc., Chem. Commun., 1985, 1088 RSC; (b) J. Mattay, Angew. Chem., Int. Ed. Engl., 1987, 26, 825 CrossRef; (c) J. Mattay, G. Trampe and J. Runsink, Chem. Ber., 1988, 121, 1991 Search PubMed; (d) J. Mattay, Nachr. Chem., Tech. Lab., 1988, 36, 376 Search PubMed; (e) J. Mattay, M. Vondenhof and R. Denig, Chem. Ber., 1989, 122, 951 Search PubMed; (f) W. S. Chung, N. J. Turro, J. Martes and J. Mattay, J. Org. Chem., 1989, 54, 4881 CrossRef CAS; (g) J. Mattay, Synthesis, 1989, 233 CrossRef CAS; (h) J. Mattay and M. Vondenhof, Chem. Ber., 1990, 123, 2457; Tetrahedron Lett., 1989, 30, 1091. Search PubMed; (i) J. Mattay and M. Vondenhof, Top. Curr. Chem., 1991, 159, 219 Search PubMed.
- (a) D. Valentine, N. J. Turro and G. S. Hammond, J. Am. Chem. Soc., 1964, 86, 5202 CrossRef CAS; (b) T. L. Penner, D. G. Whitten and G. S. Hammond, J. Am. Chem. Soc., 1970, 92, 2861 CrossRef CAS.
- G. O. Schenck, S. P. Mannsfeld, G. Schomburg and C. H. Krauch, Z. Naturforsch., Teil B, 1964, 19, 18 Search PubMed.
- (a) N. L. Bauld, D. J. Bellville, B. Harirchian, K. T. Lorenz, R. A. Pabon Jr., D. W. Reynolds, D. D. Wirth, H. S. Chiou and B. K. Marsh, Acc. Chem. Res., 1987, 20, 371 CrossRef CAS; (b) D. J. Bellville, D. D. Wirth and N. L. Bauld, J. Am. Chem. Soc., 1981, 103, 718 CrossRef CAS; (c) D. W. Raynolds, K. T. Lorenz, H. S. Chiou, D. J. Bellville, R. A. Padon and N. L. Bauld, J. Am. Chem. Soc., 1987, 109, 4960 CrossRef; (d) K. T. Lorenz and N. L. Bauld, J. Am. Chem. Soc., 1987, 109, 1157 CrossRef CAS.
- R. Schutte and G. R. Freeman, J. Am. Chem. Soc., 1969, 91, 3715 CrossRef CAS.
- K. Alder and G. Stein, Justus Liebigs Ann. Chem., 1932, 496, 197 Search PubMed; Angew. Chem., 1933, 50, 514. Search PubMed.
- K. Mizuno, I. Nakanishi, N. Ichinose and Y. Otsuji, Chem. Lett., 1989, 1095 CAS.
- For reviews, see S. L. Mattes and S. Farid , Organic Photochemistry, A. Padwa, Marcel Dekker, New York, NY, 1983; 6, Ch. 4; Search PubMed (a) M. Chanon and L. Eberson , Photoinduced Electron Transfer, M. A. Fox and M. Chanon, Elsevier, Amsterdam, 1988, Part A, Ch. 1.11; Search PubMedF. D. Lewis , ibid., Part C, Ch. 4.1. Search PubMed.
- D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259 Search PubMed.
- The severe steric hindrance of 12a caused by four menthoxycarbonyl groups would prevent the intimate electron-transfer interaction between the substrate and the sensitizer, resulting in the poor chemical yields..
- C. R. Jones, B. J. Allman, A. Mooring and B. Spahic, J. Am. Chem. Soc., 1983, 105, 652 CrossRef CAS.
- S. L. Murov , Handbook of Photochemistry; Marcel Dekker, New York, 1973, p. 207. Search PubMed.
- If there is another path for the formation of exo-adduct 2b, the contribution of the direct formation path from the exciplex is reduced and the ee should be a higher value. Thus, the evaluated value is the lowest limit..
- K. P. R. Kartha, Tetrahedron Lett., 1986, 27, 3415 CrossRef CAS.
- J. Kang, G. J. Lim, S. K. Yoon and M. Y. Kim, J. Org. Chem., 1995, 60, 564 CrossRef CAS.
- R. C. Hockett, R. E. Miller and A. Scattergood, J. Am. Chem. Soc., 1949, 71, 3072 CrossRef CAS.
- N. Yamasaki, Y. Inoue, T. Yokoyama, A. Tai, A. Ishida and S. Takamuku, J. Am. Chem. Soc., 1991, 113, 1933 CrossRef CAS.
- B. D. Baigrie, J. Brennan, J. I. G. Cadogan, J. Cook and J. T. Sharp, J. Chem. Soc., Perkin Trans. 1, 1975, 1060 RSC.
| This journal is © The Royal Society of Chemistry 2000 |