Kinetic investigation of the reduction of pinacolone by borane catalyzed by oxazaborolidines in THF. Hydride shift as rate determining step
Holger Jockela, Reinhard Schmidt*a, Helge Jopea and Hans-Günther Schmalza
aInstitut für Physikalische und Theoretische Chemie, J. W. Goethe-Universität, Marie-Curie-Str. 11, D60439, Frankfurt/M, Germany
bAnalytic Jena GmbH, Konrad-Zuse-Str. 1, D07745, Jena, Germany
cInstitut für Organische Chemie, Universität zu Köln, Greinstr. 4, D50939, Köln, Germany
First published on UnassignedUnassigned23rd December 1999
Abstract
The kinetics of the reduction of the ketone pinacolone (P) by borane (B) catalyzed by the oxazaborolidines (OAB) (S)-1-methyl-3,3-diphenylperhydropyrrolo[1,2-c][1,3,2]oxazaborolidine and (S)-2,4-dimethyl-3,3-diphenyl-1,3,2-oxazaborolidine have been studied in THF. The findings are discussed in comparison with the kinetic results obtained previously for the same reaction using two different OABs as catalysts (C). The reaction order in the ketone P changes in the series of OABs from first-order to zero-order. This surprising behaviour excludes the addition of P to the binary complex C–B forming the ternary complex P–C–B as the rate determining step. However, the kinetic data are consistently explained if the intramolecular hydride shift in P–C–B with subsequent rearrangement leading to the complex of monoalkoxyborane and catalyst is assumed to be rate determining. The rate equations of the catalytic reactions of the four OABs, the corresponding ee values, and apparent activation energies are given.
Introduction
The development in organic chemistry during the last decades has led to various methods for asymmetric synthesis.1–5 One of them is the reduction of prochiral ketones catalyzed by oxazaborolidines (OAB).1,6 This reaction is widely used due to its very high enantioselectivity and excellent ee. Itsuno et al. started working with OABs in 1981 and used them to introduce defined chirality to alcohols.7–13 A little later Corey et al. developed a conclusive mechanism for the OAB catalyzed reaction.1,14,15 Since this time there have even been attempts to immobilize OABs on polymeric supports.16–20 Borane sources other than BH3–THF have also been examined.20–27 The principal intermediates of the reaction sequence are known from experimental![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 6,14,15,28–33 as well as theoretical investigations.6,28,34–41 However, the rate determining step of the catalytic reaction is still unknown. The addition of the ketone to the borane–catalyst complex forming a ternary complex or the subsequent hydride shift are assumed to determine the rate of the catalytic process.1,25,38
6,14,15,28–33 as well as theoretical investigations.6,28,34–41 However, the rate determining step of the catalytic reaction is still unknown. The addition of the ketone to the borane–catalyst complex forming a ternary complex or the subsequent hydride shift are assumed to determine the rate of the catalytic process.1,25,38Few kinetic studies investigating the dependence of the reaction rate on the concentrations of catalyst and reagents, which could give further insight into the mechanism of the catalyzed reaction, have been published, although such information is also of particular interest for finding the optimum reaction conditions. Recently, we published the first detailed kinetic study on the reduction of a ketone by borane catalyzed by oxazaborolidines in THF.42 The direct, uncatalyzed reduction, which competes with the catalytic cycle leading to a decrease in enantioselectivity, was also investigated.43 Pinacolone was used as prochiral ketone, (S)-1-phenyl-3,3-diethylperhydropyrrolo[1,2-c][1,3,2]oxazaborolidine 1a and (S)-1,3,3-triphenylperhydropyrrolo[1,2-c][1,3,2]oxazaborolidine 1b as catalysts, see Scheme 1. It was shown by the analysis of the kinetic data that the reaction of ketone P and binary complex catalyst–borane leading to the complex catalyst–monoalkoxyborane is rate determining with both OABs. Actually this reaction consists of the reaction steps ketone addition, hydride shift, and rearrangement. Based on the kinetic data, however, it was not possible to distinguish which of the single steps is rate determining. In order to increase our knowledge about the OAB catalyzed reduction of ketones, we extended the kinetic investigations and report in the present paper our results with (S)-1-methyl-3,3-diphenylperhydropyrrolo[1,2-c][1,3,2]oxazaborolidine 1c and (S)-4-methyl-2,5,5-triphenyl-1,3,2-oxazaborolidine 1d, see Scheme 1.
 | ||
| Scheme 1 | ||
Results and discussion
Rates of reaction
Borane (B) exists in THF (T) as a 1∶1 BH3–THF complex (= B–T).44–49 In our previous investigations we observed that the direct borane reduction of pinacolone (P) as well as the catalyzed reduction occurred much slower if we used freshly prepared, pure B–T instead of commercial B–T as reducing agent.43 It was shown that a contaminant, the stabilizer NaBH4 of the commercial reagent, leads to a change in the reaction kinetics and to much faster reactions. Therefore, we performed all our further experiments with pure B–T.50,51Fig. 1 shows representative kinetic traces of the overall reaction of P with B–T in the presence of catalyst (C) OAB 1c at 20 °C.![Kinetic traces of the reduction of P by borane in THF catalyzed by OAB 1c at 20 °C. [C]o = 0.0025 mol dm−3; [B–T]o = 0.405 (upper trace), 0.115 mol dm−3 (lower trace).](/image/article/2000/P2/a906935f/a906935f-f1.gif) | ||
| Fig. 1 Kinetic traces of the reduction of P by borane in THF catalyzed by OAB 1c at 20 °C. [C]o = 0.0025 mol dm−3; [B–T]o = 0.405 (upper trace), 0.115 mol dm−3 (lower trace). | ||
The time required for 50% conversion of the ketone in the presence of the overall concentration [C]o = 0.0025 mol dm−3 of OAB catalyst 1c amounts to τ1/2 = 11 s at [B/T]o = 0.405 mol dm−3 and to 29 s at [B/T]o = 0.115 mol dm−3. Without catalyst the corresponding conversions of P require about 460 and 1600 s. Thus, a strong acceleration of the reaction is caused already at this low concentration of OAB 1c. A closer look at Fig. 1 demonstrates that both kinetic curves distinctly deviate from first-order behavior. The reaction rates are almost constant in the beginning and decrease distinctly only after about 50% conversion of P. In this respect we notice a significant difference from OABs 1a and 1b, for which the reaction was in a good approximation first-order in the ketone P. With OAB 1c, however, the reaction is between zero-order and first-order in P. An even stronger shift in the direction of zero-order kinetics is observed with OAB 1d. As is demonstrated by the kinetic traces of Fig. 2, the reaction rates decrease only slightly during almost the complete course of reaction until about 95% of P is consumed.
![Kinetic traces of the reduction of P by borane in THF catalyzed by OAB 1d at 20 °C. [C]o = 0.0050 mol dm−3; [B–T]o = 0.455 mol dm−3.](/image/article/2000/P2/a906935f/a906935f-f2.gif) | ||
| Fig. 2 Kinetic traces of the reduction of P by borane in THF catalyzed by OAB 1d at 20 °C. [C]o = 0.0050 mol dm−3; [B–T]o = 0.455 mol dm−3. | ||
Because of the approximate zero-order character of the reaction, the time required for 50% conversion of P changes strongly with the initial concentration [P]i. At [C]o = 0.0050 mol dm−3 of OAB 1d and [B/T]o = 0.455 mol dm−3τ1/2 amounts to 60 s for [P]i = 0.134 mol dm−3 and to only 7.5 s for [P]i = 0.016 mol dm−3 at 20 °C. Without catalyst the corresponding conversion of P is achieved after about 410 s. Again, we observe a strong acceleration of the reaction, which is, however, weaker than in the case of OAB 1c.
The lack of first-order kinetics prevents the determination of rate constants. Instead, reaction rates r = −d[P]/dt have been determined as slopes of linear least squares fits to the kinetic traces, describing the consumption of P over about 25% conversion of P with OAB 1c and 50% conversion of P with OAB 1d. The direct reduction of P by B–T, competing with the actual catalytic process, is considered by calculation of the corresponding rate rd = kdf[P]i[B–T]o with kd = 0.0037 dm3 mol−1 s−1 (20 °C).43 The factors f = 0.9 (OAB 1c) and f = 0.75 (OAB 1d) indicate that the mean concentration of P in the linear range of the reaction rate is lower than [P]i. Thus, we obtain the rate of the catalytic reaction as rc = r − rd. The importance of the correction by rd generally increases with increasing [P]i, [B–T]o and decreasing [C]o. The ratio rd/r ranges only from 0.01 to 0.08 for OAB 1c, but becomes important with 0.01 ≤ rd/r ≤ 0.4 for OAB 1d.
Kinetic scheme
It was previously shown by us that the following three reactions R1 to R3 with corresponding rate constants k1 to k3 (20 °C) are sufficient for a simulation of the experimental kinetic curves of the direct borane reduction by numerical integration.42| B–T + P → M–T k1 = 0.0037 dm3 mol−1 s−1 | (R1) |
| M–T + P → D–T k2 = 0.003 dm3 mol−1 s−1 | (R2) |
| 2 M–T → D–T + B–T k3 = 0.2 dm3 mol−1 s−1 | (R3) |
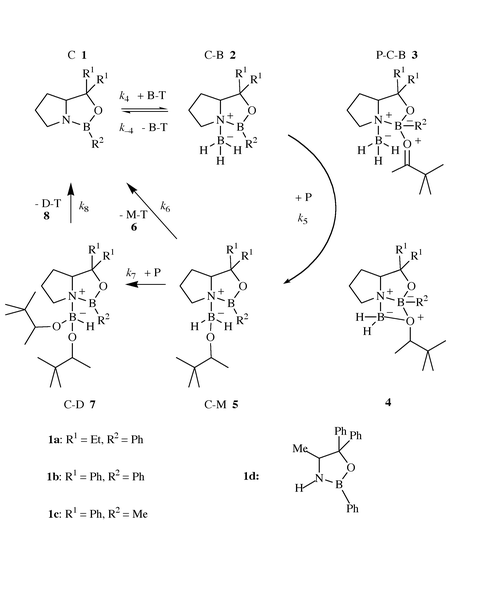
M–T represents the monoalkoxyborane–THF complex derived from P (M = mono(3,3-dimethyl-2-butoxy)borane). D–T is the corresponding dialkoxyborane complex. Dialkoxyboranes are the only reaction products formed in the direct borane reduction of ketones.28,52 The distribution reaction to trialkoxyborane is too slow and must therefore not be considered.43,53 Reaction R1 describes the main part of the overall direct reduction of P. Scheme 1 presents a simplified catalytic cycle for the reduction, which considers the results of experimental and theoretical studies and which proved to be sufficient to interpret the results with OABs 1a and 1b.
The reversible addition of borane to the catalyst C 1 forms the complex C–B 2 in reaction R4. It has been shown by NMR spectroscopy of solutions of C–B in THF15 and by single-crystal X-ray spectroscopy of solid C–B28,33 that BH3 is coordinated to the nitrogen atom of the OAB ring in C–B. Since the lone pair electrons of the nitrogen can no longer be shared with the boron atom of the heterocycle, the acidity of the boron atom of the OAB ring is distinctly increased in C–B relative to C.1 The ketone P couples with the sterically better accessible free electron pair of the oxygen to the electrophilic ring-boron of C–B to form the ternary complex P–C–B 3.1,32,34 It is generally accepted that hydride transfer occurs from the NBH3 unit of P–C–B to the carbonyl carbon via a six-membered cyclic transition state.1,14,15,31,40 The hereby formed intermediate 4 rearranges to yield the monoalkoxyborane adduct C–M of the oxazaborolidine catalyst. Since we observed only very small spectral changes in the reaction spectra of the catalytic cycle, we assume for the complex C–M in THF at room temperature structure 5, which is very similar to the structure 2 of complex C–B.42 C–M reacts with THF and dissociates into C and M–T 6 in reaction R6 or adds and reduces a second ketone molecule and yields the dialkoxyborane adduct C–D 7 in the reaction sequence R7, which is very similar to the first reaction sequence R5 of the catalytic cycle. Finally, C–D reacts with T and dissociates into C and D–T 8. Thus, in addition to the direct borane reduction reactions R4 to R8 had to be considered for a quantitative description of the kinetics.42
| C + B–T ⇌ C–B + T K4 = k4/k−4 | (R4) |
| C–B + P → C–Mk5 | (R5) |
| C–M + T → C + M–Tk6 | (R6) |
| C–M + P → C–Dk7 | (R7) |
| C–D + T → C + D–Tk8 | (R8) |
Besides k1, k2, and k3 the rate constants k4 and k−4 and thus the equilibrium constant K4 of the borane addition of the catalyst had been determined for OAB 1b.42 Based on these known constants it was actually possible to resolve the experimental kinetics by numerical integration, since experimental kinetic traces had been recorded in dependence of [B/T]o, [C]o and [P]i.42 It was shown by the analysis of the numerical integration data that reaction R5 is rate determining in the catalytic process. Actually, R5 is a reaction sequence which is composed of several steps, the addition of P to C–B 2 yielding P–C–B 3, and the hydride shift to 4 with subsequent rearrangement of 4 forming C–M 5. Eqn. (1) was derived for the rate rc of the catalyzed reaction, which was found to be first-order in P with OABs 1a and 1b.
| rc = k5(K4/[T])[B–T]o[P][C]o/{1 + (K4/[T])[B–T]o} | (1) |
Already Figs. 1 and 2 reveal that the catalytic reaction is not first-order in P with OABs 1c and 1d. Thus, we notice distinct changes in the reaction kinetics of the OAB catalyzed reduction of P by B–T. We have no reason to assume important changes in the kinetic scheme of the catalytic cycle, if we compare OABs 1a and 1b with 1c and 1d. However, the graduated shift from first-order kinetics to zero-order kinetics in P demonstrates that the addition of P to C–B forming the ternary complex P–C–B cannot be the rate determining step. Thus, the intramolecular hydride shift in the ternary complex P–C–B with subsequent rearrangement forming C–M must be rate determining. Therefore, it is necessary to split reaction R5 into reaction R5a, the reversible addition of P to C–B, and reaction R5b, the rate determining hydride shift with subsequent rearrangement producing C–M.
| C–B + P ⇌ P–C–BK5a | (R5a) |
| P–C–B → C–Mk5b | (R5b) |
If C–M reacts much faster than it is formed, then a stationary state is established, in which reaction R5b is rate determining. Then, the rate k5b[P–C–B] becomes equal to the rate rc of the OAB catalyzed consumption of P. Since we put together hydride shift and rearrangement in one reaction R5b, the result of the formal kinetic treatment would be the same if the last step, the rearrangement, but not the hydride shift were rate determining which, however, appears to be less realistic. P–C–B is formed in the two coupled equilibrium reactions R4 and R5a. As long as the conditions [P] ≫ [C]o, [B–T]o ≫ [C]o and [B–T]o ≪ [T] = 12.3 mol dm−3 hold true, the equilibrium concentration [P–C–B] and the rate rc of the catalytic reaction are described by eqns. (2) and (3), respectively.
| [P–C–B] = (K4/[T])[B–T]oK5a[P][C]o/{1 + (K4/[T])[B–T]o(1 + K5a[P])} | (2) |
| rc = k5b(K4/[T])[B–T]oK5a[P][C]o/{1 + (K4/[T])[B–T]o(1 + K5a[P])} | (3) |
Eqn. (3) simplifies with k5 = k5bK5a to Eqn. (1), if K5a[P] ≪ 1. Then, the catalyzed reaction becomes first-order in P, as was actually found with OABs 1a and 1b. Eqn. (1) is also derived if the addition of P to C–B is the rate determining step. Thus, we realize that we cannot distinguish on the basis of the kinetics which is first-order in P and follows Eqn. (1), whether the addition of P to C–B or the subsequent hydride shift is the rate determining step. However, since we observe a shift in direction to zero-order kinetics in P with OABs 1c and 1d, the addition of P to C–B cannot be rate determining. Instead, the hydride shift with subsequent rearrangement has to be assumed to be rate determining. Eqn. (3) but not Eqn. (1) can describe a kinetics which is zero-order in P.
The spectrophotometric determination of K4, k4 and k−4 failed for the OABs 1c and 1d, since the spectral changes following the addition of borane are too fast and/or too small to be measured with sufficient accuracy with our stopped-flow apparatus. Furthermore, the kinetic scheme becomes more complex with the replacement of reaction R5 by the reactions R5a and R5b. Thus, it is not surprising that the kinetics of the reduction of P catalyzed by OABs 1c and 1d cannot be resolved by numerical integration. Therefore, we will discuss the kinetic data in the following paragraphs on the basis of eqn. (3).
Kinetics of the reduction of P catalyzed by OAB 1c
Eqn. (3) describes the variation of rc with the overall concentrations of C and B–T as well as the concentration of P. Fig. 3 illustrates the dependence of rc on [C]o for constant [B–T]o and [P]i = 0.11 mol dm−3.![Rates of the catalyzed reaction as a function of the overall catalyst concentration, OAB 1c. [P]i = 0.11 mol dm−3; [B–T]o = 0.62, 0.405, 0.23, 0.115 mol dm−3 (from top to bottom).](/image/article/2000/P2/a906935f/a906935f-f3.gif) | ||
| Fig. 3 Rates of the catalyzed reaction as a function of the overall catalyst concentration, OAB 1c. [P]i = 0.11 mol dm−3; [B–T]o = 0.62, 0.405, 0.23, 0.115 mol dm−3 (from top to bottom). | ||
The experiments performed with [B–T]o = 0.405 and 0.23 mol dm−3 and varying catalyst concentrations demonstrate that actually a linear correlation exists, i.e., the reaction is first-order in catalyst. Thus, it is reasonable to analyze in the further discussion rates of the catalyzed reaction, which are normalized to the catalyst concentration: k′ = rc/[C]o. In Fig. 3 the rc values determined for only one single catalyst concentration at [B–T]o = 0.62 and 0.115 mol dm−3 are also given. The slopes of the interconnecting lines and both linear least squares fits correspond to normalized rates at constant pinacolone concentration k′P, which we indicate by the subscript P. The values of k′P increase with increasing [B–T]o. Fig. 4 illustrates the dependence of k′P on [B–T]o for [P]i = 0.114 and [P]i = 0.046 mol dm−3.
![Normalized rates of the catalyzed reaction as a function of the overall borane concentration, OAB 1c. [P]i = 0.114 (upper curve), 0.046 mol dm−3 (lower curve).](/image/article/2000/P2/a906935f/a906935f-f4.gif) | ||
| Fig. 4 Normalized rates of the catalyzed reaction as a function of the overall borane concentration, OAB 1c. [P]i = 0.114 (upper curve), 0.046 mol dm−3 (lower curve). | ||
Despite the scatter of the data it is obvious that the dependence of k′P on [B–T]o weakens distinctly with increasing [B–T]o. Considering that [P] is constant, we rearrange eqn. (3) and derive eqn. (4), which gives an analytical expression for the dependence of k′P on [B–T]o at constant [P]i.
| k′P = f1[B–T]o/(1 + f2[B–T]o) | (4) |
| f1 = k5bK5a(K4/[T])[P] | (4a) |
| f2 = K4/[T] + K5a(K4/[T])[P] | (4b) |
The curves of Fig. 4 describing the experimental data have been calculated with the constants f1 = 10 M−1 s−1 and f2 = 5.0 M−1 at [P]i = 0.114 mol dm−3 and f1 = 6.0 M−1 s−1 and f2 = 3.5 M−1 at [P]i = 0.046 mol dm−3. Eqn. (4a) demands direct proportionality of f1 with [P], which seems to be fulfilled. The corresponding slope is k5bK5a(K4/[T]) = 90 M−2 s−1. If we calculate the straight line connecting the two data points f2;[P]i according to eqn. (4b) we obtain as slope K5a(K4/[T]) = 36 M−2 and as intercept (K4/[T]) = 1.8 M−1. From these results we finally get k5b = 2.5 s−1, K5a = 20 M−1 and K4 = 23.
The analysis of the normalized rates k′B, determined at constant B–T concentration, with respect to their dependence on the concentration of pinacolone allows a test of the consistency of evaluation. Taking [B–T]o as constant, we derive eqn. (5) as an analytical expression for the dependence of k′B on [P]i at constant [B–T]o.
| k′B = g1g2[P]i/(1 + g2[P]i) | (5) |
| g1 = k5b | (5a) |
| g2 = (K4/[T])[B–T]oK5a/{1 + (K4/[T])[B–T]o} | (5b) |
Thus, only parameter g2 should vary with [B–T]o. Fig. 5 shows the dependence of k′B on [P]i for three different constant borane concentrations.
![Normalized rates of the catalyzed reaction as a function of the initial pinacolone concentration, OAB 1c. [B–T]o = 0.62, 0.23, 0.115 mol dm−3 (from top to bottom).](/image/article/2000/P2/a906935f/a906935f-f5.gif) | ||
| Fig. 5 Normalized rates of the catalyzed reaction as a function of the initial pinacolone concentration, OAB 1c. [B–T]o = 0.62, 0.23, 0.115 mol dm−3 (from top to bottom). | ||
The lines drawn in Fig. 5 roughly describe the experimental data. They have been calculated by eqn. (5) with g1 = 2.5 s−1 and g2 = 11.0 M−1 ([B–T]o = 0.62 mol dm−3), g2 = 6.0 M−1 (0.23) and g2 = 3.4 M−1 (0.115). Using eqns. (5a) and (5b) and the above derived values of k5b, K5a and K4/[T] we calculate g1 = 2.5 s−1, g2 = 10.5 M−1 ([B–T]o = 0.62 mol dm−3), g2 = 5.8 M−1 (0.23) and g2 = 3.4 M−1 (0.115). Hence, it is actually possible to describe both the dependence of k′P on [B–T]o and the dependence of k′B on [P]i with the common set of constants of k5b = 2.5 s−1, K5a = 20 M−1 and K4 = 23 for OAB 1c. Thus, the evaluation appears to be consistent.
It has to be noted that the value of K4 obtained from the kinetic analysis of the catalyzed reduction of P is by one order of magnitude smaller than the value K4 = 260, which was determined by Mathre et al. studying the concentration dependent changes of the NMR spectrum of the 1∶1 complex of OAB 1c and BH3 in THF-d8 at 25 °C.28 The reason for that discrepancy is not fully understood. We tried to determine the equilibrium constant K4 spectrophotometrically. However, the spectral changes following the addition of borane to OAB 1c are too fast to be measured time-resolved with our stopped-flow apparatus. We observed slow spectral changes developing during the time scale of several minutes. The corresponding slow reaction, however, cannot be of relevance for the catalyzed reaction, which occurs on a faster time scale with conversions Δ[P] ≥ 20[C]o, indicating borane–catalyst turnover numbers of at least 10 during one or two minutes, see Fig. 1. Since the time span between sample preparation and NMR measurement was probably distinctly longer than some seconds, the relevance of the corresponding value of K4 for the kinetics of the catalyzed reaction has to be questioned.
Kinetics of the reduction of P catalyzed by OAB 1d
Both kinetic traces of Fig. 2 indicate that the reaction catalyzed by OAB 1d is almost zero-order in pinacolone. This conclusion is supported by the quantitative evaluation. Fig. 6 plots rates rc determined at constant [B–T]o = 0.23 mol dm−3 but different catalyst concentrations of [C]o = 0.0010, 0.0025 and 0.0050 mol dm−3versus [P]i.![Rates of the catalyzed reaction as a function of the initial pinacolone concentration, OAB 1d. [B–T]o = 0.23 mol dm−3; [C]o = 0.0050, 0.0025, 0.0010 mol dm−3 (from top to bottom).](/image/article/2000/P2/a906935f/a906935f-f6.gif) | ||
| Fig. 6 Rates of the catalyzed reaction as a function of the initial pinacolone concentration, OAB 1d. [B–T]o = 0.23 mol dm−3; [C]o = 0.0050, 0.0025, 0.0010 mol dm−3 (from top to bottom). | ||
No significant dependence of rc on [P]i can be seen in the pinacolone concentration range investigated. According to eqn. (3), rc becomes independent of [P] only if K5a[P] ≫ 1 andK5a[P](K4/[T])[B–T]o ≫ 1 hold true. Then, eqn. (3) simplifies to rc ≈ k5b[C]o and rc should also be independent of [B–T]o. Because of the constancy of the rc data of Fig. 5 it is reasonable to calculate average values, covering the whole investigated pinacolone concentration range. Fig. 7 shows the plot of the average values of rcversus [C]o at constant [B–T]o = 0.23 mol dm−3.
![Rate of the catalyzed reaction as a function of the overall catalyst concentration, OAB 1d. Values of rc are averages of the rc data in the 0.015 ≤ [P]i ≤ 0.13 mol dm−3 concentration range of Fig. 6; [B–T]o = 0.23 mol dm−3.](/image/article/2000/P2/a906935f/a906935f-f7.gif) | ||
| Fig. 7 Rate of the catalyzed reaction as a function of the overall catalyst concentration, OAB 1d. Values of rc are averages of the rc data in the 0.015 ≤ [P]i ≤ 0.13 mol dm−3 concentration range of Fig. 6; [B–T]o = 0.23 mol dm−3. | ||
A strong linear correlation is obtained in agreement with the expectations based on eqn. (3). k5b = 0.174 s−1 results as slope. Further experiments have been carried out at [B–T]o = 0.445 mol dm−3 and [C]o = 0.0050 mol dm−3. The rate rc was again found to be independent of [P]i between 0.015 and 0.13 mol dm−3. The mean value of k′ = rc/[C]o = 0.205 s−1 yields a further estimate of k5b. Both results differ only in the mutual limits of experimental error. Thus, we obtain as average k5b = 0.2 s−1 for OAB 1d.
Comparison of the catalysts
The catalyzed reaction is first-order in P for OABs 1a and 1b. The corresponding rate follows Eqn. (1), which allows no distinction as to whether the addition of P to C–B forming P–C–B (R5a) or the hydride shift with subsequent rearrangement (R5b) is the rate determining step. However, the change from first- to zero-order kinetics in P observed with OABs 1c and 1d definitely indicates for these catalysts reaction R5b to be rate determining. Thus, for the first time it is shown that the hydride shift with subsequent rearrangement determines the rate of the catalytic process. Since we see no reason why the chemistry developing at the OAB catalyst should change from OAB 1a to 1d, we conclude that the reaction R5b is rate determining for all OABs investigated by us and use eqn. (3) as a common base for the comparison of the catalysts.For OABs 1a and 1bK5a[P]i ≪ 1 and hence K5a ≤ 2 M−1 must hold true, since only in this case the catalyzed reaction becomes first-order in P in the investigated concentration range. For OAB 1cK5a = 20 M−1 was obtained and for OAB 1dK5a[P]i ≫ 1 corresponding to K5a ≥ 100 M−1 results. It is the question, how this strong variation of the equilibrium constant of the addition of P to C–B can be understood. The excellent effectiveness of the enantioselectively catalyzed reduction of prochiral ketones by OABs is based on the almost perfect application of the lock and key principle, which implies that for each OAB–borane complex (lock) the highest effectiveness is only obtained with an optimum fitting substrate (key). Besides electronic and structural properties of the OAB–borane complex C–B the structure of the substrate P has to be taken into account. The magnitude of the value of K5a seems to be determined by steric restrictions hindering the access of the oxygen atom of the bulky pinacolone molecule to the ring boron atom in the case of the rather rigid bicyclic OABs 1a, 1b and 1c. According to AM1 calculations,54 the bond between the ring boron atom and its substituent stands almost perpendicular to the plane of the oxazaborolidine ring. Therefore, the access of P to C–B is rather restricted for the OABs 1a and 1b, which have a phenyl substituent bound to the ring boron atom. The restriction is smaller for OAB 1c, since the phenyl substituent is replaced by a methyl substituent at the ring boron, leading to a larger value of K5a. Because of the missing structure stabilizing effect of the fused second ring, the flexibility of the monocyclic OAB 1d is increased, possibly allowing for the larger value of K5a due to the better accessibility of the ketone P to the ring boron atom, despite its phenyl substituent.
The equilibrium constant of the borane addition to the catalyst was recently spectrophotometrically determined by us as K4 = 220 for OAB 1b.42 This value is one order of magnitude larger than the value of K4 = 23 presently obtained for OAB 1c. Because of the smaller steric hindrance to the addition of borane to the nitrogen atom in OAB 1c reasons other than steric ones must be responsible for the smaller value of K4. Possibly, the basicity of the nitrogen atom is decreased in 1c compared with 1b due to the replacement of the phenyl substituent at the ring boron atom by the methyl substituent, which is the weaker electron donor.
The rate constant of the hydride shift amounts to k5b = 2.5 s−1 for OAB 1c and 0.2 s−1 for OAB 1d. This difference is the main reason why 1c is the better catalyst. Electronic as well as steric factors may influence the graduation in k5b. A detailed interpretation of this interesting effect could eventually be given on the basis of high level theoretical calculations.
The comparison of the catalytic effect of the OABs depends on the experimental conditions of the reactions, as the reaction kinetics change in the series of catalysts. For OABs 1a and 1b second-order rate constants of catalysis kc have recently been calculated for constant [B–T]o from the pseudo-first-order rate constants kp1 and kd of the overall and of the direct borane reduction, respectively, by kc = (kp1 − kd)/[C]o, whereby kc depends on [B–T]o.42 Values of kc = 0.23 dm3 mol−1 s−1 (OAB 1a, [B–T]o = 0.26 mol dm−3, 0.08 ≤ [P]i ≤ 0.12 mol dm−3) and kc = 9 dm3 mol−1 s−1 (OAB 1b, [B–T]o = 0.23 mol dm−3, 0.08 ≤ [P]i ≤ 0.14 mol dm−3) resulted at 20 °C. These data can be used to calculate the normalized rate k′ of the catalytic reaction as k′ = kc[P]i. We obtain at [B–T]o = 0.26 mol dm−3k′ = 0.23[P]i s−1 for OAB 1a and k′ = 9[P]i s−1 for OAB 1b. For OAB 1ck′ = 16[P]i/(1 + 6.4[P]i) s−1 and for OAB 1d finally k′ = 0.20 s−1 results in the 0.015 ≤ [P]i ≤ 0.13 mol dm−3 range at [B–T]o = 0.26 mol dm−3. Thus, the catalytic activity of the OABs decreases in the series 1c ≈ 1b > 1d ≫ 1a.
We performed as complementary to the kinetic experiments ee determinations at 20 °C under the experimental conditions [C]o = 0.0050, [P]i = 0.10, and [B–T]o = 0.23 mol dm−3. The following ee values (%) have been obtained: 61 (OAB 1a), 96 (OAB 1b), 97 (OAB 1c), and 85 (OAB 1d). The fact that the graduation of ee values follows the same sequence as the graduation of catalytic activity strongly indicates a close relationship between the two quantities. However, it should be noted that these graduations hold true for pinacolone. The investigation of other ketones will probably result in different graduations.
Temperature dependence
The kinetic investigation revealed that the rate of the catalytic reactions depends in a complex manner on equilibrium and rate constants. Therefore, temperature dependent measurements only lead to apparent activation energies, which may even depend on [P] and [B–T]o. Since they cannot be directly related with one defined reaction step, these values are of more practical interest but less suited for a discussion of a particular transition state.Measurements of the overall reaction rates r have been performed with OABs 1c and 1d at intervals of 5 °C from 10 to 30 °C. The temperature dependent rate of the direct borane reduction of P was calculated using the previously determined rate constant kd = 0.0037 dm3 mol−1 s−1 (20 °C) and the corresponding activation energy of Ea = 54.3 kJ mol−1. The rate of the catalytic reaction was obtained for each temperature as rc = r − rd. From Arrhenius type plots of ln (rc) versus 1/T apparent activation energies “Ea” have been obtained, which are collected together with calculated values of k′ in Table 1. As expected, with all catalysts the apparent activation energies are smaller than the activation energy of the direct reduction.
Conclusions
The reaction order in the ketone P changes in the series of OABs from first-order to zero-order. This kinetic result demonstrates for the first time that it is not the addition of P to the binary complex C–B but the hydride shift in the ternary complex P–C–B with subsequent rearrangement which is the rate determining step in the OAB catalyzed reduction of pinacolone.Experimental
Techniques
Melting points were measured with a Büchi 510 apparatus and are uncorrected. FT-infrared spectra were recorded with a Nicolet Magna FT-IR spectrometer. 1H NMR and 13C NMR spectra were recorded on a Bruker AM 270 spectrometer. Chemical shifts are reported in ppm and were referenced to the residual CHCl3 resonances (7.26 ppm/77.00 ppm). Elemental analyses were obtained with a Perkin-Elmer CHNO/S-Analysator 2400 II or a Heraeus CHN-Rapid. Optical rotations were measured at 20 °C in CHCl3 on a Perkin-Elmer 241 polarimeter. The reactions were followed spectrophotometrically using a stopped flow apparatus and a computerized Hewlett-Packard HP8452A diode array spectrophotometer as described earlier.42,43 The handling of the materials and the preparation of the solutions was done under Ar to avoid contamination by water. If not otherwise stated, all kinetic experiments were carried out at 20 °C.Determination of enantiomeric excess
The procedure for the determination of ee values under the conditions of our stopped-flow experiments was the same for all catalysts. Solution 1 (50 ml) contained [C]o = 0.010 and [B–T]o = 0.455 mol dm−3 and solution 2 (50 ml) [P] = 0.20 mol dm−3. 1 ml of each solution was mixed in the stopped flow apparatus at 20 °C. The mixing was repeated every 60 s until the entire solutions had reacted. The reaction mixture (100 ml) was quenched by 10 ml of water and refluxed for 1 h to destroy the unreacted borane and to transform the dialkoxyboranes to the product alcohol 3,3-dimethylbutan-2-ol (pinacolyl alcohol). Finally the solution was distilled. The fraction at ≥72 °C contained pinacolyl alcohol and THF as checked by 1H NMR spectroscopy. The THF was removed from 40 ml of this fraction in a rotary evaporator. The residue was separated by using a column of packed Kieselgel F60 and hexane–ethyl acetate (ratio 3∶1). The progress was controlled by gas chromatography (conditions: column Lipodex E of Machery + Nagel/Düren, injector block 100 °C, flame ionisation injector block 200 °C, oven 50 °C at isothermal cond., pressure of H2 carrier gas 45 kPa, approx. split ratio 60∶1). 0.5 g of the isolated pinacolyl alcohol (4.9 mmol) were dissolved in 10 ml of dry CH2Cl2. Then 2.5 ml of dry pyridine and 2.55 g acetic anhydride (25 mmol, 5 eq.) were added. The solution was stirred at room temperature for 5 h, forming pinacolyl acetate. Then 5 ml of 2 M HCl were added and the solution was washed several times with ether. The combined ether phases were washed with 1 M NaOH and brine. After drying over K2CO3 the ether was removed in vacuo. The residue was separated from pyridine by chromatography (column of packed Kieselgel F60, solvent hexane–ethyl acetate, ratio 3∶1). The pinacolyl acetate passes the column first (Rf = 1), pyridine is slower (Rf = 0.5). After separation, the solvent hexane–ethyl acetate is removed at 40 °C at 200 mbar. The yellow volatile liquid residue is purified pinacolyl acetate (yield ca. 270 mg), characterized by 1H NMR spectroscopy. The ester was separated into its enantiomers by using the above described GC method. Based on the integrals over the peaks of the R (retention time 6.5–6.69 min) and the S enantiomer (retention time 7.15–7.5 min), we found that in every case mainly the R configuration of pinacolyl alcohol was formed.Preparations
(S)-1-Methyl-3,3-diphenylperhydropyrrolo[1,2-c][1,3,2]oxazaborolidine 1c.. Mp 73–75 °C (74–86 °C);15 [α]589 = −211.5°, c = 1.58 (CHCl3, 20 °C); 1H NMR (CDCl3, 270 MHz): δ = 7.65–7.1 (m, 10H), 4.35 (dd, J = 5.6 and 9.8 Hz, 1H), 3.44–3.32 (m, 1H), 3.1–2.95 (m, 1H), 1.9–1.55 (m, 3H), 0.9–0.75 (m, 1H), 0.4 (s, 3H); 13C NMR (CDCl3, 270 MHz): δ = 147.8, 143.9, 128.2, 128.1, 127.7, 127.1, 126.5, 126.3, 126.2, 125.8, 87.8, 72.6, 42.9, 30.2, 26.4.
(S)-4-Methyl-2,5,5-triphenyl-1,3,2-oxazaborolidine 1d.. [α]589 = −182°, c = 0.54 (CHCl3, 25 °C); 1H NMR (CDCl3, 400 MHz): δ = 7.92 (m, 2H), 7.75 (m, 2H), 7.56–7.61 (m, 11H), 4.66 (q, 1H), 3.9 (s, br., 1H), 0.99 (d, 3H); 13C NMR (CDCl3, 100 MHz): δ = 147, 143, 133.9, 130.4, 129, 128.4, 128.2, 128, 127.8, 127.6, 127.1, 126.7, 126.4, 126.1, 125.9, 125.5, 125.4, 89.9, 59.3, 22.5.
Acknowledgements
Generous financial support by the Bundesministerium für Bildung, Wissenschaft, Forschung und Technologie, grant nos. 03D0029B3 and 03D0029A0, and by the Adolf Messer Stiftung is gratefully acknowledged. We also thank the Fonds der Chemischen Industrie for support.References
- E. J. Corey and C. J. Helal, Angew. Chem., 1998, 110, 2093; Angew. Chem., Int. Ed. Engl., 1998, 37, 1986. Search PubMed.
- D. Valentine and J. W. Scott, Synthesis, 1978, 329 CAS.
- V. K. Singh, Synthesis, 1992, 605 CrossRef.
- L. Deloux and M. Srebnik, Chem. Rev., 1993, 93, 763 CrossRef CAS.
- Y. H. Kim, D. H. Park, I. S. Byun, I. K. Yoon and C. S. Park, J. Org. Chem., 1993, 58, 4511 CrossRef CAS.
- S. Wallbaum and J. Martens, Tetrahedron: Asymmetry, 1992, 3, 1475 CrossRef CAS.
- S. Itsuno, Y. Sakurai, K. Ito, A. Hirao and S. Nakahama, Bull. Chem. Soc. Jpn., 1987, 60, 395 CAS.
- A. Hirao, S. Itsuno, S. Nakahama and N. Yamazaki, J. Chem. Soc., Chem. Commun., 1981, 315 RSC.
- S. Itsuno, K. Ito, A. Hirao and S. Nakahama, J. Chem. Soc., Chem. Commun., 1983, 469 RSC.
- S. Itsuno, M. Nakano, K. Miyazaki, H. Masuda and K. Ito, J. Chem. Soc., Perkin Trans. 1, 1985, 2039 RSC.
- S. Itsuno, Y. Sakurai, K. Shimizu and K. Ito, J. Chem. Soc., Perkin Trans. 1, 1990, 1859 RSC.
- S. Itsuno, A. Hirao, S. Nakahama and N. Yamazaki, J. Chem. Soc., Perkin Trans. 1, 1983, 1673 RSC.
- S. Itsuno, K. Ito, A. Hirao and S. Nakahama, J. Org. Chem., 1984, 49, 555 CrossRef CAS.
- E. J. Corey, R. K. Bakshi and S. Shibata, J. Am. Chem. Soc., 1987, 109, 5551 CrossRef CAS.
- E. J. Corey, R. K. Bakshi and S. Shibata, J. Am. Chem. Soc., 1987, 109, 7925 CrossRef CAS.
- S. Itsuno, T. Wakasugi, K. Ito, A. Hirao and S. Nakahama, Bull. Chem. Soc. Jpn., 1985, 58, 1669 CAS.
- S. Itsuno, K. Ito, A. Hirao and S. Nakahama, J. Chem. Soc., Perkin Trans. 1, 1984, 2887 RSC.
- S. Itsuno, M. Nakano, K. Ito, A. Hirao, M. Owa, N. Kanda and S. Nakahama, J. Chem. Soc., Perkin Trans. 1, 1985, 2615 RSC.
- C. Cazé, N. El Moualij, P. Hodge, C. J. Lock and J. Ma, J. Chem. Soc., Perkin Trans. 1, 1995, 345 RSC.
- C. Franot, G. B. Stone, P. Engeli, C. Spöndlin and E. Waldvogel, Tetrahedron: Asymmetry, 1995, 6, 2755 CrossRef CAS.
- G. B. Stone, Tetrahedron: Asymmetry, 1994, 5, 465 CrossRef CAS.
- A. M. Salunkhe and E. R. Burkhardt, Tetrahedron Lett., 1997, 38, 1523 CrossRef CAS.
- E. J. Corey and J. O. Link, J. Am. Chem. Soc., 1992, 114, 1906 CrossRef CAS.
- E. J. Corey and R. K. Bakshi, Tetrahedron Lett., 1990, 31, 611 CrossRef CAS.
- E. J. Corey, J. O. Link and R. K. Bakshi, Tetrahedron Lett., 1992, 33, 7107 CrossRef CAS.
- E. J. Corey and J. O. Link, Tetrahedron Lett., 1992, 33, 3431 CrossRef.
- E. J. Corey and C. J. Helal, Tetrahedron Lett., 1995, 36, 9153 CrossRef CAS.
- D. J. Mathre, A. S. Thompson, A. W. Douglas, K. Hoogsteen, J. D. Carroll, E. G. Corley and E. J. J. Grabowski, J. Org. Chem., 1993, 58, 2880 CrossRef CAS.
- A. W. Douglas, D. M. Tschaen, R. A. Reamer and Y.-J. Shi, Tetrahedron: Asymmetry, 1996, 7, 1303 CrossRef CAS.
- J. G. Quallich, J. F. Blake and T. M. Woodall, J. Am. Chem. Soc., 1994, 116, 8516 CrossRef.
- G. J. Quallich and T. M. Woodall, Tetrahedron Lett., 1993, 34, 4145 CrossRef CAS.
- E. J. Corey, X.-M. Cheng, K. A. Cimprich and S. Sarshar, Tetrahedron Lett., 1991, 32, 6835 CrossRef CAS.
- E. J. Corey, M. Azimioara and S. Sarshar, Tetrahedron Lett., 1992, 33, 3429 CrossRef CAS.
- V. Nevalainen, Tetrahedron: Asymmetry, 1991, 2, 429 CrossRef CAS.
- V. Nevalainen, Tetrahedron: Asymmetry, 1993, 4, 1597 CrossRef CAS.
- V. Nevalainen, Tetrahedron: Asymmetry, 1994, 5, 289 CrossRef CAS.
- V. Nevalainen, Tetrahedron: Asymmetry, 1996, 7, 2655 CrossRef CAS.
- L. P. Linney, C. R. Self and I. H. Williams, J. Chem. Soc., Chem. Commun., 1994, 1651 RSC.
- L. P. Linney, C. R. Self and I. H. Williams, Tetrahedron: Asymmetry, 1994, 5, 813 CrossRef CAS.
- D. Liotta and D. K. Jones, Adv. Mol. Modelling, 1995, 3, 1 Search PubMed.
- D. K. Jones, D. C. Liotta, I. Shinkai and D. J. Mathre, J. Org. Chem., 1993, 58, 799 CrossRef CAS.
- R. Schmidt, H. Jockel, H.-G. Schmalz and H. Jope, J. Chem. Soc., Perkin Trans. 2, 1997, 2725 RSC.
- H. Jockel and R. Schmidt, J. Chem. Soc., Perkin Trans. 2, 1997, 2719 RSC.
- C. F. Lane, Chem. Rev., 1976, 76, 773 CrossRef CAS.
- J. R. Elliot, W. L. Roth, G. F. Roedel and E. M. Boldebuck, J. Am. Chem. Soc., 1952, 74, 5211 CrossRef.
- C. Than and H. Morimoto, J. Org. Chem., 1995, 60, 7503 CrossRef CAS.
- C. Camacho, G. Uribe and R. Contreras, Synthesis, 1982, 1027 CrossRef CAS.
- A. T. Whatley and R. N. Pease, J. Am. Chem. Soc., 1954, 76, 835 CrossRef CAS.
- B. Rice, J. A. Livasy and G. W. Schaeffer, J. Am. Chem. Soc., 1955, 77, 2750 CrossRef CAS.
- H. C. Brown , Hydroboration, W. A. Benjamin Inc., New York, 1962, p. 18. Search PubMed.
- G. F. Freegard and L. H. Long, Chem. Ind., 1963, 471 Search PubMed.
- H. C. Brown, H. I. Schlesinger and A. B. Burg, J. Am. Chem. Soc., 1939, 61, 673 CrossRef CAS.
- J. D. Pasto and B. Lepeska, J. Am. Chem. Soc., 1976, 98, 1091 CrossRef.
- HyperChem software package release 4.5, 1995, Hypercube, Inc., Waterloo, Ontario. Search PubMed.
- D. J. Mathre, T. K. Jones, L. C. Xavier, T. J. Blacklock, R. A. Reamer, J. J. Mohan, E. T. T. Jones, K. Hoogsteen, M. W. Baum and E. J. J. Grabowski, J. Org. Chem., 1991, 56, 751 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2000 |