Preparations, X-ray crystal structure determinations, and base strength measurements of substituted tritylamines
Received (in Cambridge, UK) 19th August 1999, Accepted 25th October 1999
First published on UnassignedUnassigned23rd December 1999
Abstract
A range of tritylamines TrNRR′, and 4-methoxy-, 4,4′-dimethoxy-, and 4,4′,4′′-trimethoxy-substituted analogues, have been prepared from (substituted) trityl chloride, bromide, or tetrafluoroborate with ammonia or with amines HNRR′ where R and R′ are hydrogen, alkyl, or aryl. Crystal structures of 4,4′,4′′-trimethoxytritylamine, N-tritylglycine methyl ester, tritylammonium chloride, and N-tritylglycine have been determined. The central C–N bond of tritylamine is not significantly affected by the introduction of p-methoxy substituents into the trityl group, or by N-alkylation, but is lengthened upon protonation of the amino group. Some degree of planarisation of the three C–C bonds to the central carbon of the trityl group is also associated with this C–N bond lengthening. N-Tritylglycine is shown to be a zwitter-ion in the crystalline state and has pI = 6.4 in aqueous acetonitrile. Base strengths of a range of tritylamines have been measured in aqueous acetonitrile. The pKBH+ values (pKa values of the corresponding tritylammonium ions), including ones for N-tritylglycine methyl ester and a range of N-tritylanilines, are remarkable for their similarity at pKBH+ = ca. 9, i.e. characteristic of values for simple alkylamines. It is proposed that the (substituted) trityl group sterically inhibits solvation of the protonated tritylglycine ester cation selectively, and prevents significant resonance interaction between the arene ring and the amino group in the anilines.
Introduction
Various substituted forms of the trityl group continue to be used as site-selective protecting groups in the synthesis of amines and alcohols.1 The parent trityl group is most easily introduced but the most difficult to remove requiring extended reaction times under strongly acidic conditions. Increasing the number of p-methoxy substituents appreciably increases the ease of removal of the protecting group. We have already reported kinetics results for the deprotection of several substituted N-trityl,N-alkylamines,2 and a detailed mechanistic investigation of the deamination of 4,4′-dimethoxytritylamine under controlled acidic conditions.3,4 We report here preparations, crystal structure determinations, and base strength measurements of compounds in our ongoing mechanistic investigation in this area. The X-ray structure determinations were required both for confirmation of structures, especially of N-tritylglycine in view of an earlier report that this compound exists as the non-zwitter-ion form,5 and also to investigate whether there is a Bürgi–Dunitz effect![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 6 upon the central carbon–nitrogen bond length when the nitrogen is protonated and as p-methoxy substituents are introduced into the trityl group. The base strengths were required first to facilitate the isolation of N-tritylglycine which required knowledge of its isoelectric point, and also to investigate whether different base strengths are a contributing cause of the extraordinary rate ratios between substituted tritylamines and their N-alkyl analogues in deamination reactions.2
6 upon the central carbon–nitrogen bond length when the nitrogen is protonated and as p-methoxy substituents are introduced into the trityl group. The base strengths were required first to facilitate the isolation of N-tritylglycine which required knowledge of its isoelectric point, and also to investigate whether different base strengths are a contributing cause of the extraordinary rate ratios between substituted tritylamines and their N-alkyl analogues in deamination reactions.2Results and discussion
Preparations
The majority of compounds reported here and shown in Fig. 1 were prepared by conventional methods described in the experimental section and require little comment. In general, the substituted trityl tetrafluoroborates (which had to be prepared) are the most convenient tritylating reagents,7 but very satisfactory yields may be obtained for some compounds using the corresponding trityl halides (some of which are commercially available). In all cases, spectroscopic and analytical data support the structures assigned; when compounds had been reported earlier (but without spectroscopic data which we now report), presently obtained samples were shown to be identical with one exception (see later). |
| | Fig. 1 Structures of compounds.
| |
The preparations of the parent and p-methoxy-substituted tritylamines (1a–4a) were unexceptional, and it was possible to make the hydrochloride (1b) from 1a in aqueous acetone. It proved to be quite stable, and was recrystallised and fully characterized. The 4-methoxy-analogue (2b) had to be made in non-aqueous solution and, although isolated and characterized, was unstable. Attempts at isolation and purification of the di- and tri-p-methoxy-substituted tritylammonium analogues failed. Our preparation of N-tritylglycine methyl ester (1c) from trityl chloride followed a literature method8 and served as a basis for the preparation of the mono- and di-p-methoxy-analogues, 2c and 3c; the tri-p-methoxy-compound (4c) was made by the same method but from the tetrafluoroborate salt.7N-Tritylglycine (1d) had been made previously by acidification of the product of alkaline hydrolysis of N-tritylglycine methyl ester (1c).8 By this method, we prepared a crystalline sample for X-ray crystallographic investigation. We were unable, however, to adapt the method successfully for the preparation of the 4-methoxy-analogue (2d) or to prepare (2d) by another literature method in aqueous solution.9 An alternative procedure for 2d was successful in which the carboxylate of glycine was first protected by the trimethylsilyl group;10 4-methoxytritylation of the amino residue followed by desilylation of the carboxylate under very mildly acidic conditions (5% aqueous citric acid) gave a low but adequate yield of 2d. We were unable to prepare the di- and tri-p-methoxy-substituted tritylglycines, all attempts leading to the formation of the correspondingly substituted trityl alcohols. We are also unable to reconcile the high melting point of our sample of 2d purified by recrystallisation from methanol (mp 210–212 °C) with the low melting point of a sample made differently and reportedly recrystallised from ether–petroleum ether (mp 76–78 °C).9
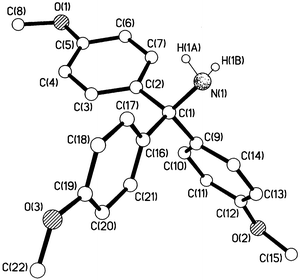 |
| | Fig. 2 Crystal structure of 4,4′,4′′-trimethoxytritylamine, 4a.
| |
Our preparations of N-tritylpyrrolidines (1e–4e) and N-tritylalkylamines (1f–4f) were unexceptional, some compounds having been reported previously.11 Similarly, preparations of known12,13 and new N-tritylarylamines (1g–3g) were straightforward and are sufficiently described in the experimental section.
Crystal structures
A number of crystal structures of trityl and substituted tritylamines have been reported.14,15 Consideration of Fig. 3 and the data in Table 1 shows that replacement of one of the hydrogens on nitrogen of tritylamine by an uncharged (though polar) CH2CO2Me group has no significant effect upon the structure of the rest of the molecule. This finding is in accord with previous results.15 The central C–N bond lengths of compounds with 12 reported partial structures Ph3C–NRR′ (R and R′ = alkyl) in the Cambridge Structural Database15 cover a relatively narrow range, the average being 1.495 Å (SD = 0.008). For 29 compounds with partial structures Ph3C–NHR, the corresponding range is also quite narrow, the average now being 1.482 Å (SD = 0.012). From the present results, we additionally see that introduction of three methoxy substituents into the trityl has no significant effect upon the structure of the rest of the molecule. In particular, the central C(1)–N bond length remains virtually the same, and the sums of the three C–C(1)–C bond angles are the same.
Table 1 Selected bond lengths and angles for compounds 1a, 1b·Me2CO, 1c, 1d and 4a
| | 1a14 | 1b·Me2CO | 1c | 1d | 4aa |
|---|
| Two molecules in the crystallographic asymmetric unit; both values are given. |
|---|
| C(1)–N(1) | 1.481(3) | 1.519(2) | 1.484(2) | 1.551(7) | 1.484(2) |
| | | | | | 1.481(2) |
| C(1)–Cipso(Ph) | | | | | |
| 1 | 1.539(3) | 1.540(2) | 1.542(2) | 1.534(5) | 1.540(3) |
| | | | | | 1.541(2) |
| 2 | 1.541(3) | 1.538(2) | 1.540(2) | 1.550(5) | 1.537(2) |
| | | | | | 1.542(2) |
| 3 | 1.541(3) | 1.532(2) | 1.543(2) | 1.522(5) | 1.539(2) |
| | | | | | 1.538(2) |
| Sum of angles | 331.87 | 334.76 | 331.34 | 333.60 | 330.56 |
| C–C(1)–C | | | | | 329.70 |
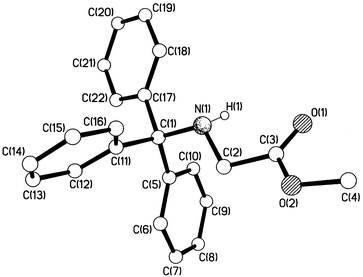 |
| | Fig. 3 Crystal structure of N-tritylglycine methyl ester, 1c.
| |
We know from kinetics results that the deamination reactions occur exclusively from the protonated form of the (substituted) tritylamine,2–4i.e. the (substituted) tritylammonium ion, and heterolysis of the C–N bond of the free base in solution does not occur. Consideration of Fig. 4 and the data in Table 1 shows that the C–N bond becomes longer (from 1.486 to 1.519 Å) when tritylamine is protonated. Elongation of the C(1)–N bond upon N-protonation has been observed previously for other amines16 including an increase from 1.489 to 1.523 Å upon protonation of N-tritylazetidine.17 The C–N bond lengthening between 1a and 1b is accompanied by some degree of planarisation of the C(1)–C bonds, i.e. the sum of the three central C–C(1)–C bond angles increases from 331.9 to 334.8° between 1a and 1b. From N-tritylazetidine to its conjugate acid, the degree of planarisation at C(1) is somewhat smaller (from 330.9 to 332.6°).17 Corresponding effects are observed between N-tritylglycine methyl ester and N-tritylglycine (which in Fig. 5 is now shown to be a zwitter-ion in the crystalline state in contrast to what was reported5 previously). Here, the C–N bond is lengthened from 1.484 to 1.551 Å and the C–C(1)–C bond angle sums are 331.3 and 333.6°, respectively. For tritylamines, therefore, we have clear structural evidence that protonation of the nitrogen, but not introduction of three p-methoxy substituents, begins progression along the deamination reaction coordinate.
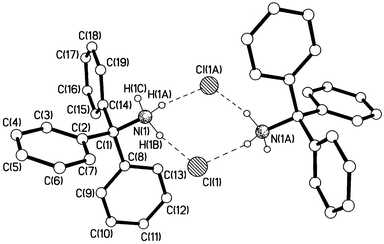 |
| | Fig. 4 Crystal structure of tritylammonium chloride, 1b.
| |
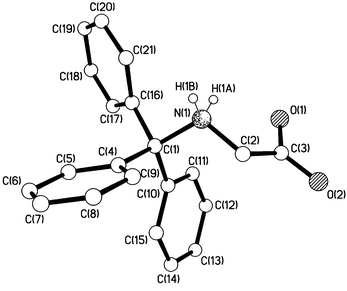 |
| | Fig. 5 Crystal structure of N-tritylglycine, 1d.
| |
Base strengths
The organic bases were titrated against standard perchloric or hydrochloric acid with acetonitrile as cosolvent to overcome solubility difficulties in water alone. Results in Table 2 for compounds 3g ii and 3g iii, however, are seen to be only slightly affected by the solvent composition over the range we have used. Sequential p-methoxy substituents in the trityl residue and wide-ranging N-alkyl substituents have only small effects upon the base strength of tritylamine; all are strong bases in aqueous solution and comparable with much simpler alkylamines.18 We may now conclude, therefore, that the dramatic deamination rate increases seen upon N-alkylation of tritylamines are not due to changes in base strength.2
Table 2 pKBH+ values of tritylamines (pKa values of tritylammonium ions) in aqueous acetonitrile at 21 °C and ionic strength = 0.1 mol dm−3 (NaClO4)a
| Compound | pKBH+ | %CH3CN |
|---|
| pKBH+ = 9.0 was determined for benzylamine under our experimental conditions compared with literature values of 9.29 and 9.35 in water at 20 and 25 °C, respectively.30 22 °C, ionic strength = 1.0 mol dm−3. The same result had been obtained in 1% acetonitrile in water.2 Compound kindly supplied by Dr A. P. Henderson of Newcastle University. |
|---|
| 1a | 9.2 | 22b |
| 1c | 10.1 | 22b |
| 1d | 9.1 | 68 |
| | 9.5 | 25 |
| | 10.0 | 22b |
| 2a | 9.3 | 10b |
| 2d | 9.3 | 20% DMSOb |
| 2f | 10.5 | 38 |
| 3c | 9.7 | 21 |
| 3e | 8.7 | 21 |
| 3g(i) | 8.9 | 39 |
| 3g(ii) | 8.8 | 17 |
| | 8.9 | 42 |
| | 9.0 | 83 |
| 3g(iii) | 9.2 | 17 |
| | 9.7 | 42 |
| | 9.6 | 83 |
| 3g(iv) | 9.5 | 53 |
| 3g(v) | 9.3 | 87 |
| 4c | 9.4c | 23b |
| 4e | 8.2 | 21 |
| 4f(ii)d | 9.7 | 21b |
| 4f(iii)d | 9.4 | 21b |
Results for 3c and 4c show that substituted N-trityl groups have substantial but very similar base-strengthening effects upon glycine methyl ester (pKBH+ = 6.84–7.59 depending upon the experimental conditions).19 It is difficult to ascribe this effect (ca. 2–2.5 pK units) to anything other than the size of the (substituted) trityl groups, and in particular to a steric effect upon the relative solvation of protonated and unprotonated forms of the base.
For ammonium ions with pKa > ca. 6, ΔG![[circle, cut, short horiz bar]](https://www.rsc.org/images/entities/char_e0eb.gif) 298 K for eqn. (1) is substantially positive and, unlike the dissociation of carboxylic acids, typically dominated by large positive ΔH terms.18 The reduced degree of solvation as R′ is changed from H to (substituted) trityl will be greater for the protonated base than for the base, which will render ΔH for eqn. (1) less positive and ΔS more negative (or less positive). Since this structural change is base-strengthening overall for glycine methyl ester (ΔG for eqn. (1) becomes more positive), we conclude that the change in −TΔS for the reaction of eqn. (1) is greater than the change in ΔH. In other words, the effect of the N-trityl group upon the base strength of glycine methyl ester is principally a sterically induced entropy of solvation effect.
298 K for eqn. (1) is substantially positive and, unlike the dissociation of carboxylic acids, typically dominated by large positive ΔH terms.18 The reduced degree of solvation as R′ is changed from H to (substituted) trityl will be greater for the protonated base than for the base, which will render ΔH for eqn. (1) less positive and ΔS more negative (or less positive). Since this structural change is base-strengthening overall for glycine methyl ester (ΔG for eqn. (1) becomes more positive), we conclude that the change in −TΔS for the reaction of eqn. (1) is greater than the change in ΔH. In other words, the effect of the N-trityl group upon the base strength of glycine methyl ester is principally a sterically induced entropy of solvation effect.
| | | RR′NH2+ + H2O = RR′NH + H3O+; ΔG = ΔH − TΔS |
(1)
|
Literature pKa values for substituted anilinium cations are shown in Table 3;20 they range between 1 and 5.4, and are included as a Hammett correlation in Fig. 6. Use of the σ− parameter rather than σ for p-nitro leads to the better correlation indicating a resonance interaction between the nitro and amino groups in the unprotonated base, and ρ(σ–) = −2.9 for the amines acting as bases. The effect of substituents in the aniline ring upon the base strength of N-(dimethoxytrityl)aniline is exceptionally small (ρ = −0.07 for base strengths), and, as seen in Fig. 6, it makes little difference whether σ− or σ is used for the p-nitro substituent. Moreover, the values are all comparable with the base strengths of alkylamines, not arylamines (in contrast, N-methylaniline is only 0.25 pK units more basic than aniline).21 We conclude that, for steric reasons, the N-4,4′-dimethoxytrityl group requires a conformation of the bond between the amino nitrogen and the aromatic ring which effectively prevents any significant resonance interaction between them. If we assume that a difference of ca. 2–2.5 pK units upon tritylation of the nitrogen is again due to the solvation effect proposed above for glycine methyl ester, then the absence of a resonance interaction in the N-4,4′-dimethoxytritylanilines accounts for the other 2–6 pK units between the anilines and their N-4,4′-dimethoxytrityl analogues according to the extent of resonance in the unprotonated form of the aniline. An appreciably smaller effect is observed for N-(tert-butyl)aniline (pKBH+ = 7.0)22 reflecting the much smaller steric effect of tert-butyl compared with trityl. These compare with pKBH+ = 4.85 for N-methylaniline.21 Interestingly, N-tritylation of pyrrolidine and monoalkylamines is seen from Table 2 to have very little effect upon their base strengths (pKBH+ = 9.20 for pyrrolidine23 in aqueous ethanol and ca. 9–11 for simple alkylamines in water18).
Table 3 pKBH+ values of 4-substituted anilines (pKa values of 4-substituted anilinium ions)20
| X in 4-X-C6H4NH2 | pKBH+ | σX(σX−) |
|---|
| MeO | 5.34 | −0.28 |
| Me | 5.08 | −0.17 |
| H | 4.63 | 0 |
| F | 4.65 | 0.06 |
| Cl | 3.99 | 0.22 |
| CF3 | 2.60 | 0.54 |
| NO2 | 1.00 | 0.78 (1.27) |
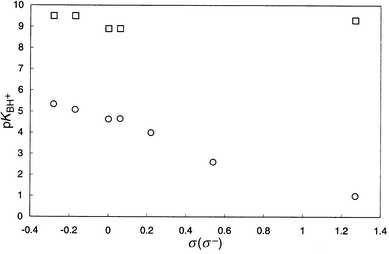 |
| | Fig. 6 Hammett plots for pKBH+ values of 4-X substituted anilines (○) and 4-X substituted N-(dimethoxytrityl)anilines (□).
| |
Experimental
Materials and methods
Solvents and reagents were standard laboratory grade; water was distilled using glass equipment. Aqueous sodium hydroxide (Convol, 0.100 mol dm−3) was used to standardize diluted aqueous perchloric acid (70%, Fisons) and hydrochloric acid. Separations by column chromatography were performed using silica gel (40–63 μm, Rhone-Poulenc) or activated basic alumina (Brockmann grade 1, 150 mesh). Fluka TLC-Card alumina (0.2 mm) and Merck silica gel 60 F254 plates were used for TLC analysis and visualized under UV light. Chemical shifts (δH and δC) are in ppm downfield from signals for TMS. Residual signals from deuteriated chloroform (7.25 ppm) and dimethyl sulfoxide (2.50 ppm) were used as 1H NMR references; for 13C NMR spectra, natural abundance signals from chloroform (77.0 ppm) and dimethyl sulfoxide (39.0 ppm) were used as references. Coupling constants (J) are given in Hz.Preparations
Tritylamine (1a).. Liquid ammonia was condensed into a cooled flask at −80 °C (acetone–CO2), poured (ca. 4 cm3, 20 mmol) into a measuring cylinder, then added to trityl bromide (200 mg, 0.60 mmol). The resulting yellow solution was stirred for several hours as the temperature rose and the excess of ammonia evaporated. Ether was added to the residual white solid, and the solution was stirred (3 h) then washed with water. The organic phase was separated, dried (MgSO4), then evaporated to dryness under reduced pressure. The title compound was obtained as a white solid (140 mg, 90%), mp 103–105 °C, lit.,24 103–105 °C; δH 3.41 (2H, s, NH2), 7.20–7.42 (15H, m, ArH); δC 66.35 (C1–NH2), 126.17 (C4′), 128.03 (C3′,C5′), 128.12 (C2′, C6′), 148.72 (C1′). Tritylammonium chloride (1b).. Tritylamine (200 mg, 0.77 mmol) was dissolved in acetone (5 cm3) and addition of aqueous HCl (3 mol dm−3, 2 cm3) caused precipitation of the title compound (160 mg, 70%), mp (recryst. methanol) 242–244 °C; vmax (KBr)/cm−1 3033, 2870, 1684, 1297, 1082; δH 7.10–7.33 (15H, m, ArH), 10.00 (3H, br s, –NH3+); δC 70.41 (C1–NH2), 128.15 (C4′), 128.56 (C3′,C5′), 128.96 (C2′, C6′), 140.98 (C1′); found: C, 76.9; H, 6.1; N, 4.7. C19H18NCl requires C, 77.1; H, 6.1; N, 4.7%. N-Tritylglycine methyl ester (1c).. Triethylamine (2.20 g, 20.0 mmol) followed by trityl chloride (2.80 g, 10.0 mmol) were added to a stirred suspension of glycine methyl ester hydrochloride (1.25 g, 10.0 mmol) in dry chloroform (15 ml). The mixture was stirred for a further 6 h at room temperature then quenched with water. The organic phase was separated, washed with water, dried (Na2SO4), filtered, and rotary evaporated. Complete removal of the chloroform was ensured by the addition of a small volume of methanol and further evaporation under reduced pressure. The residue was recrystallised from methanol (2.70 g, 81%; mp 100–102 °C, lit.,8 106–107 °C); δH 2.30–2.40 (1H, br s, NH), 3.18 (2H, s, CH2), 3.62 (3H, s, CO2CH3), 7.16–7.30 (9H, m, C3′H, C4′H, C5′H), 7.42–7.48 (6H, d, J 8, C2′H, C6′H); δC 45.86 (CH2–NH), 51.82 (CH3–O), 70.75 (C1–NH), 126.5 (C4′), 128 (C3′, C5′), 128.7 (C2′, C6′), 145.3 (C1′), 172.8 (CO2); found: C, 79.9; H, 6.4; N, 3.9. C21H22NO2 requires C, 79.7; H, 6.4; N, 4.2%. N-Tritylglycine (1d).. N-Tritylglycine methyl ester (1.7 g, 10 mmol) was dissolved upon warming in ethanolic potassium hydroxide (0.5 mol dm−3, 20 cm3). After the solution had been stirred at room temperature (1 h), it was diluted to three times its volume with water, cooled, then acidified with acetic acid. The precipitated N-tritylglycine was filtered, washed several times with water, and recrystallised from ethanol. The title compound was obtained as white crystals (1.5 g, 47%) mp 172–174 °C, lit.,8 178–179 °C; δH(200 MHz; d6-DMSO) 2.60–2.62 (2H, m, NH2+), 3.00 (2H, s, CH2), 7.25–7.50 (15H, m, ArH); δC(50.32 MHz; d6-DMSO) 45.75 (CH2), 70.52 (C1–NH2+–), 126.78 (C4′), 128.26 (C3′, C5′), 128.56 (C2′, C6′), 145.76 (C1′), 173.18 (CO2−); found: C, 79.4; H, 6.0; N, 4.2. C21H19NO2 requires C, 79.5; H, 6.0; N, 4.4%. N-Tritylaniline (1g i).. A solution of trityl chloride (150 mg, 0.54 mmol) in dry pyridine (1 cm3) was added to a solution of aniline (0.048 cm3, 0.050 g, 0.54 mmol) in the same solvent (3 cm3) with cooling (ice–water). After standing for 24 h at room temperature, the reaction mixture was mixed with ice–water (5 cm3). A viscous product separated which became completely crystalline in the course of a few hours. This product was filtered, washed with water (2 cm3), then dissolved in chloroform (4 cm3); the solution was dried (Na2SO4), filtered, and evaporated at room temperature under reduced pressure to give the white product (80 mg, 44%), mp 150–151 °C, lit.,13 150–151 °C; δH 5.00 (1H, br s, NH), 6.38 (2H, d, J 8, C2′′H, C6′′H), 6.58 (1H, t, J 7, C4′′H), 6.90 (2H, t, J 7, C3′′H, C5′′H), 7.15–7.40 (15H, m, ArH); δC 71.45 (C-NH), 116.14 (C2′′, C6′′), 117.34 (C4′′), 126.83 (C4′), 127.96 (C3′, C5′), 128.23 (C3′′, C5′′), 129.26 (C2′, C6′), 145.43 (C1′), 146.00 (C1′′). N-Trityl-p-toluidine (1g ii).. Trityl bromide (71 mg, 0.22 mmol) was dissolved in chloroform (3 cm3) then freshly distilled p-toluidine (236 mg, 0.22 mmol) was added. The mixture was stirred overnight at room temperature then extracted between water (5 cm3) and more chloroform (5 cm3). The organic phase was dried (Na2SO4), filtered, and evaporated to give a brown crystalline compound which was purified on an alumina column (20% ethyl acetate∶petrol) to give the title compound (50 mg, 64%), mp 176–178 °C (lit.,13 180–181 °C, lit.,25 177 °C); δH 2.15 (3H, s, CH3), 4.90 (1H, br, NH), 6.25 (2H, d, J 8, C2′′H, C6′′H), 6.75 (2H, d, J 8, C3′′H, C5′′H), 7.20–7.45 (15H, m, ArH); δC 20.36 (CH3), 71.41 (C–NH), 116.10 (C2′′, C6′′), 126.76 (C4′), 127.90 (C3′, C5′), 127.97 (C2′, C6′), 128.79 (C4′′), 129.74 (C3′′, C5′′), 143.92 (C1′′), 145.61 (C1′). N-p-Nitrobenzyltritylamine (1f![[hair space]](https://www.rsc.org/images/entities/b_char_200a.gif) ).. Triethylamine (60 mg, 0.60 mmol) was added dropwise to a solution of p-nitrobenzylammonium chloride (50 mg, 0.30 mmol) and trityl bromide (100 mg, 0.30 mmol) in chloroform (2 cm3), and the reaction was stirred overnight at room temperature. TLC showed starting material so more triethylamine (8 drops) was added and the reaction was stirred for another 3 h. It was then extracted between ether (5 cm3) and water (5 cm3); the organic layer was separated, dried (MgSO4), filtered, and evaporated to give white crystals (78 mg, 66%); mp (recryst., chloroform) 174–176 °C; δH 1.85 (1H, br s, NH), 3.35 (2H, br s, CH2), 7.10–7.55 (17H, m, ArH), 8.10 (2H, d, J 9, C3"H, C5"H); δC (125.65 MHz; CDCl3) 47.49 (CH2), 71.07 (C–NH), 123.58 (C3", C5′′), 126.60 (C4′), 128.05 (C3′, C5′), 128.36 (C2", C6"), 128.47 (C2′, C6′), 145.53 (C1′), 146.94 (C1"), 148.67 (C4"); found: C, 79.2; H, 5.3; N, 6.9. C26H22N2O2 requires C, 79.2; H, 5.6; N, 7.1%.
).. Triethylamine (60 mg, 0.60 mmol) was added dropwise to a solution of p-nitrobenzylammonium chloride (50 mg, 0.30 mmol) and trityl bromide (100 mg, 0.30 mmol) in chloroform (2 cm3), and the reaction was stirred overnight at room temperature. TLC showed starting material so more triethylamine (8 drops) was added and the reaction was stirred for another 3 h. It was then extracted between ether (5 cm3) and water (5 cm3); the organic layer was separated, dried (MgSO4), filtered, and evaporated to give white crystals (78 mg, 66%); mp (recryst., chloroform) 174–176 °C; δH 1.85 (1H, br s, NH), 3.35 (2H, br s, CH2), 7.10–7.55 (17H, m, ArH), 8.10 (2H, d, J 9, C3"H, C5"H); δC (125.65 MHz; CDCl3) 47.49 (CH2), 71.07 (C–NH), 123.58 (C3", C5′′), 126.60 (C4′), 128.05 (C3′, C5′), 128.36 (C2", C6"), 128.47 (C2′, C6′), 145.53 (C1′), 146.94 (C1"), 148.67 (C4"); found: C, 79.2; H, 5.3; N, 6.9. C26H22N2O2 requires C, 79.2; H, 5.6; N, 7.1%. N-Tritylpyrrolidine (1e).. Triethylamine (100 mg, 1 mmol) was added in two portions to a solution of trityl bromide (164 mg, 0.5 mmol) in pyrrolidine (2 cm3), and the mixture was stirred for about 24 h at room temperature and monitored by TLC. It was then extracted between water (10 cm3) and ether (10 cm3) then the organic phase was dried (Na2SO4), filtered, and evaporated. The crude compound was chromatographed on silica gel (5% ethyl acetate∶petrol) to give white crystals (80 mg, 51%; mp 131–132 °C, lit.,11 126–127 °C; δH 1.60–1.70 (4H, m, C3′′H, C4′′H), 2.25–2.40 (4H, m, C2′′H, C5′′H), 7.10–7.35 (9H, m, ArH), 7.45–7.55 (6H, m, C2′H, C6′H); δC 22.38 (C3′′, C4′′), 46.30 (C2′′, C5′′), 74.38 (C–N), 125.91 (C4′), 127.27 (C3′, C5′),129.44 (C2′, C6′), 143.04 (C1′); found: C, 88.20; H, 7.36; N, 4.42. C23H23N requires C, 88.13; H, 7.39; N, 4.46%. 4-Methoxytrityl tetrafluoroborate.. Aqueous tetrafluoroboric acid (40%; 0.604 cm3) was added over 20 min to an ice-cold solution of 4-methoxytrityl alcohol (621 mg, 2.14 mmol) in acetic anhydride (0.820 cm3, 8.66 mmol). The solution was stirred (20 °C, 2 h) then addition of dry ether (10 cm3) to the resulting brown solution caused the product to precipitate out as a rust coloured solid (530 mg, 68%); mp (recryst., ether–acetone) 188–190 °C, lit.,26 206–208 °C; δH 4.21 (3H, s, OCH3), 7.25–7.95 (14H, m, ArH); δC 58.83 (OCH3), 119.20 (C3, C5), 129.81 (C3′, C5′), 133.45 (C1), 138.89 (C1′), 138.99 (C2′, C6′), 139.22 (C4′), 147.85 (C2, C6), 176.87 (C4), 198.47 (C+). 4-Methoxytritylamine (2a).. This was made from liquid ammonia and 4-methoxytrityl tetrafluoroborate (52 mg, 0.14 mmol) by the method described above for tritylamine. The resulting cream oil was chromatographed on silica gel (50% ethyl acetate∶petrol + 1% triethylamine) to give the title compound as a colourless oil (380 mg, 91%); δH 2.25 (2H, s, NH2), 3.78 (3H, s, OCH3), 6.80 (2H, d, J 9, C3H, C5H), 7.10 (2H, d, J 9, C2H, C6H), 7.18–7.22 (10H, m, ArH); δC 55.27 (OCH3), 65.93 (C–NH2), 113.24 (C3, C5), 126.61 (C4′), 127.94 (C3′, C5′), 128.15 (C2′, C6′), 129.36 (C2, C6), 142.20 (C1), 148.87 (C1′), 158.25 (C4). 4-Methoxytritylammonium chloride (2b).. Hydrogen chloride gas was bubbled through a solution of N-4-methoxytritylamine (200 mg, 0.691 mmol) in dry chloroform (2 cm3) for 10 min to give a white crystalline compound which was filtered off under reduced pressure (170 mg, 75.4%); δH 3.77 (3H, s, OCH3), 6.67 (2H, d, J 9, C3H, C5H), 7.10–7.28 (12H, m, ArH), 9.95 (3H, br s, –NH3+); δC 55.20 (OCH3), 70.09 (C–NH3+), 113.77 (C3, C5), 128.02 (C4′), 128.49 (C3′, C5′), 128.89 (C2′, C6′), 130.43 (C2, C6), 133.15 (C1), 141.31 (C1′), 159.17 (C4). Attempted recrystallisation was unsuccessful. N-4-Methoxytritylglycine (2d).. Trimethylsilyl chloride (0.13 cm3, 1.0 mmol) was added to a magnetically stirred suspension of glycine (75 mg, 1.0 mmol) in chloroform–acetonitrile (5∶1, 2 cm3) at room temperature. The reaction mixture was then heated under reflux for 3 h then allowed to cool to room temperature. Addition of triethylamine (0.28 cm3, 2.0 mmol) at a rate sufficient to maintain gentle reflux was followed by a solution of 4-methoxytrityl chloride (0.31 g, 1.0 mmol) in chloroform (2 cm3). The resulting mixture was stirred for 10 h at room temperature, and then methanol (0.9 cm3) was added. Evaporation under reduced pressure left a residue which was partitioned between ether (5 cm3) and a precooled aqueous solution of citric acid (5%, 5 cm3). The organic phase was separated and washed with aqueous sodium hydroxide (1 mol dm−3, 2 × 2 cm3) and water (2 × 1 cm3). The combined aqueous layers were washed with ether (20 cm3), cooled to 0 °C, then the pH was reduced to 7–8 with glacial acetic acid. The precipitated product was filtered and dried (50 mg, 15%), mp (recryst., methanol) 210–212 °C, lit.,9 76–77 °C; δH(200 MHz; d6-DMSO) 2.92 (2H, br s, –NH2+–), 3.40 (3H, s, OCH3), 6.57 (2H, d, J 7.4, C3H, C5H), 6.96–7.20 (12H, m, ArH); δC(50.32 MHz; D2O) 50.47 (CH2), 57.64 (OCH3), 72.99 (C–NH2+), 115.80 (C3, C5), 129.24 (C4′), 130.54 (C3′, C5′), 130.93 (C2′, C6′), 132.53 (C2, C6), 140.14 (C1), 147.97 (C1′), 159.91 (C4), 181.01 (CO2−). N-(p-Nitrobenzyl)-4-methoxytritylamine (2f).. Triethylamine (54 mg, 0.54 mmol) was added to a solution of p-nitrobenzylammonium chloride (100 mg, 0.27 mmol) in chloroform (3 cm3) followed by 4-methoxytrityl tetrafluoroborate (100 mg, 0.27 mmol). The reaction was stirred overnight at room temperature then extracted between ether (5 cm3) and water (5 cm3). The organic phase was separated, dried (Na2SO4), filtered, and evaporated to dryness. The crude product was chromatographed on silica gel (10% ethyl acetate∶petrol) to give white crystals (54 mg, 47%), mp (recryst., ether) 136–138 °C; δH 1.90 (1H, br s, NH), 3.42 (2H, s, CH2), 3.75 (3H, s, OCH3), 6.82 (2H, d, J 9, C3H, C5H), 7.05–7.30 (6H, m, ArH), 7.41 (2H, d, J 9, C2H, C6H), 7.51 (4H, d, J 7, C2′H, C6′H), 7.58 (2H, d, J 9, C2′′H, C6′′H), 8.17 (2H, d, J 9, C3′′H, C5′′H); δC 47.59 (CH2), 55.27 (OCH3), 70.64 (C–NH), 113.41 (C3, C5), 123.63 (C3′′, C5′′), 126.57 (C4′), 127.90 (C2′′, C6′′), 128.08 (C3′, C5′), 128.43 (C2′, C6′), 129.74 (C2, C6), 137.75 (C1), 145.91 (C1′), 147.01 (C1′′), 148.85 (C4′′), 158.15 (C4); found: C, 76.3; H, 5.6; N, 5.9. C27H24N2O3 requires C, 76.4; H, 5.7; N, 6.6%. N-4-Methoxytritylaniline (2g).. Aniline (0.045 g, 0.48 mmol, 0.05 cm3) in dry pyridine (0.16 cm3) was added to a solution of 4-methoxytrityl chloride (150 mg, 0.48 mmol) in the same solvent (0.23 cm3) and sufficient chloroform (less than 1 cm3) was then added to obtain a homogeneous solution. The solution was cooled with ice–water, then stirred at room temperature for 15 h. The chloroform was evaporated, water (1 cm3) was added and the mixture was stirred at room temperature for another 16 h before being extracted between water (3 cm3) and ether (3 cm3). The ether phase was separated, dried (Na2SO4), filtered, and evaporated. A colourless powder (95 mg, 54%) was obtained after chromatography on alumina (20% ethyl acetate in petrol), mp (recryst., ether) 137–138 °C, lit.,27 138–139 °C; δH 3.80 (3H, s, OCH3), 5.00 (1H, b, NH), 6.38 (2H, d, J 9, C2′′H, C6′′H), 6.58 (1H, t, J 7, C4′′H), 6.82 (2H, d, J 9, C3H, C5H), 6.90 (2H, t, J 7, C3′′H, C5′′H), 7.15–7.40 (12H, m, ArH); δC 59.61 (OCH3), 71.41 (C–NH), 113.17 (C2′′, C6′′), 113.27 (C3, C5) 117.35 (C4′′), 126.82 (C4′), 128.23 (C3′, C5′), 128.80 (C2′, C6′), 129.19 (C2, C6), 129.27 (C3′′, C5′′), 143.29 (C1), 143.92 (C1′′), 145.61 (C1′), 158.77 (C4). N-4-Methoxytritylpyrrolidine (2e).. 4-Methoxytrityl tetrafluoroborate (246 mg, 0.80 mmol) was dissolved in pyrrolidine (2 cm3) and triethylamine (120 mg, 1.20 mmol) was added. The reaction was stirred overnight at room temperature then extracted between water (5 cm3) and ether (5 cm3). The organic phase was dried (MgSO4), filtered, and evaporated. The crude product was chromatographed on silica gel (20% ethyl acetate : petrol) to give a colourless oil (222 mg, 81%). Crystals (mp 88–90 °C) were deposited from a solution of this oil in ether; δH 1.55–1.70 (4H, m, C3′′H, C4′′H), 2.25–2.35 (4H, m, C2′′H, C5′′H), 3.75 (3H, s, OCH3), 6.78 (2H, d, J 9, C3H, C5H) 7.05–7.30 (6H, m, ArH), 7.39 (2H, d, J 9, C2H, C6H), 7.50 (4H, d, J 7, C2′H, C6′H); δC 22.37 (C3′′, C4′′), 46.24 (C2′′, C5′′), 55.13 (OCH3), 73.80 (C–N), 112.48 (C3, C5), 125.77 (C4′), 127.20 (C3′, C5′), 129.23 (C2′, C6′), 130.62 (C2, C6), 134.92 (C1), 143.48 (C1′), 157.48 (C4); found: C, 83.6; H, 7.3; N, 3.9. C24H25NO requires C, 83.9; H, 7.3; N, 4.1%. N-4-Methoxytritylglycine methyl ester (2c).. Triethylamine (2.2 g, 0.02 mol) was added to a stirred suspension of glycine methyl ester hydrochloride (1.25 g, 0.01 mol) in dry chloroform (15 cm3) followed by 4-methoxytrityl chloride (3.08 g, 0.01 mol). The mixture was allowed to react for 6 h at room temperature then was washed twice with water, dried (Na2SO4), and filtered. The solvent was evaporated, complete removal of the chloroform being ensured by the addition of a small volume of methanol and reconcentration under reduced pressure. The product was chromatographed on silica gel (10% ethyl acetate : petrol) to give a colourless oil (2.88 g, 80%); δH 2.42–2.45 (1H, br s, NH), 3.18 (2H, s, CH2), 3.70 (3H, s, CO2CH3), 3.80 (3H, s, OCH3), 6.78 (2H, d, J 9, C3H, C5H), 7.10–7.35 (6H, m, ArH), 7.41 (2H, d, J 9, C2H, C6H), 7.51 (4H, d, J 8, C2′H, C6′H); δC 45.94 (CH2), 51.82 (CO2CH3), 55.28 (OCH3), 70.37 (C–NH), 113.38 (C3, C5), 126.55 (C4′), 128.04 (C3′, C5′), 128.64 (C2′, C6′), 129.96 (C2, C6), 137.60 (C1), 145.78 (C1′), 158.18 (C4), 172.89 (CO2Me). 4,4′-Dimethoxytritylamine (3a).. This compound was prepared as described above for 4-methoxytritylamine. The resulting cream oil was chromatographed on silica gel (50% ethyl acetate∶petrol + 1% triethylamine) to give the title product as a colourless oil (70 mg, 85%); δH 2.31 (2H, br s, NH2), 3.78 (6H, s, 2 × OCH3), 6.79 (4H, d, J 9, C3H, C5H), 7.15 (4H, d, J 9, C2H, C6H), 7.22–7.27 (5H, m, ArH); δC 55.28 (OCH3), 65.38 (C–NH2), 113.19 (C3, C5), 126.51 (C4′), 127.90 (C3′, C5′), 128.07 (C2′, C6′), 129.26 (C2, C6), 141.17 (C1), 149.22 (C1′), 158.18 (C4). N-4,4′-Dimethoxytritylpropylamine (3f ii).. Triethylamine (0.09 cm3) was added to a solution of 4,4′-dimethoxytrityl chloride (405 mg, 1.2 mmol) in n-propylamine (0.2 cm3, 2.4 mmol) and the solution was stirred under nitrogen at room temperature for 24 h. After the usual work-up, the crude product was chromatographed on alumina (20% ethyl acetate∶ petrol) to give a colourless oil (310 mg, 71.5%); δH 0.91 (3H, t, J 7, C3′′H), 1.49 (2H, sextet, J 7, C2′′H), 2.11 (2H, t, J 7, C1′′H), 3.78 (6H, s, 2 × OCH3), 6.82 (4H, d, J 9, C3H, C5H), 7.10–7.50 (5H, m, ArH), 7.39 (4H, d, J 9, C2H, C6H); δC 12.05 (C3′′), 24.07 (C2′′), 45.57 (C1′′), 55.23 (OCH3), 69.86 (C–NH), 113.08 (C3,C5), 126.06 (C4′), 127.76 (C3′, C5′), 128.56 (C2′, C6′), 129.80 (C2, C6), 138.96 (C1), 147.04 (C1′′), 157.81 (C4). N-(4,4′-Dimethoxytrityl)-p-nitroaniline (3g v).. A solution of 4,4′-dimethoxytrityl chloride (405 mg, 1.2 mmol) in pyridine (1 cm3) was added to a solution of p-nitroaniline (166 mg, 1.2 mmol) in dry pyridine (0.88 cm3). The mixture became warm so was cooled to room temperature and stirred under nitrogen for 26 h. After addition of water–ice (9 cm3), an oil separated and the mixture was extracted between water (5 cm3) and ether (5 cm3); the ether phase was separated, dried (Na2SO4), filtered, and evaporated. The residual crude product was chromatographed on alumina (20% ethyl acetate∶petrol) to give dark yellow crystals (226 mg, 51%), mp (recryst., ether) 102–104 °C; vmax (KBr)/cm−1 3473, 3057, 3000, 2930, 2905, 1084; δH 3.80 (6H, s, 2 × OCH3), 5.70 (1H, br s, NH), 6.30 (2H, d, J 9, C2′′H, C6′′H), 6.80 (4H, d, J 9, C2H, C6H), 7.17 (4H, d, J 9, C3H, C5H), 7.20–7.25 (5H, m, ArH), 7.80 (2H, d, J 9, C3′′H, C5′′H); δC 55.28 (OCH3), 71.06 (C–NH), 113.56 (C3, C5), 114.61 (C2′′, C6′′), 125.28 (C3′′, C5′′), 127.35 (C4′), 128.30 (C3′, C5′), 128.80 (C2′, C6′), 130.14 (C2, C6), 136.36 (C1), 138.30 (C4′′), 144.77 (C1′), 152.16 (C1′′), 158.66 (C4); found: C, 73.5; H, 5.4; N, 6.1; C27H24N2O4 requires C, 73.6; H, 5.5; N, 6.3%. N-(4,4′-Dimethoxytrityl)-p-fluoroaniline (3g ii).. As described above, a solution of p-fluoroaniline (270 mg, 2.4 mmol, 0.23 cm3), 4,4′-dimethoxytrityl chloride (810 mg, 2.4 mmol), and pyridine (0.66 cm3) was stirred for 24 h to give, upon work-up, a white powder (630 mg, 64%), mp (recryst., ether) 132–134 °C; vmax (KBr)/cm−1 3409, 3057, 3032, 3000, 2932, 2905, 1083; δH 3.75 (6H, s, 2 × OCH3), 4.80 (1H, br s, NH), 6.20–6.25 (2H, m, C2′′H, C6′′H), 6.55–6.65 (2H, t, J 8.5, C3′′H, C5′′H), 6.70–6.80 (4H, d, J 8, C3H, C5H), 7.10–7.40 (9H, m, C2H, C6H, C2′H, C3′H, C4′H, C5′H, C6′H); δC 55.24 (OCH3), 70.74 (C–NH), 113.21 (C3, C5), 114.49 (C3′′, C5′′), 116.93 (C2′′, C6′′), 126.73 (C4′), 127.93 (C3′, C5′), 129.19 (C2′, C6′), 130.31 (C2, C6), 137.67 (C1), 142.69 (C1′′), 145.94 (C1′), 153.29 (C4′′), 158.25 (C4); m/z (+EI) 412 ((M − 1)+, 21%), 360 (M+ − 2 × OMe, 20), 303 (M+ − NHC6H4F, 100), 111 (H2NC6H4F, 17), 77 (Ph, 9); found: C, 78.6; H, 5.8; N, 3.3. C27H24NO2F requires C, 78.4; H, 5.8; N, 3.4%. N-(4,4′-Dimethoxytrityl)-p-methoxyaniline (3g iv).. As described above, a solution of p-anisidine (147 mg, 1.2 mmol) and 4,4′-dimethoxytrityl chloride (400 mg, 1.2 mmol) in pyridine (0.88 cm3) was stirred for 48 h to give, upon work-up, pale yellow crystals (332 mg, 68%), mp (recryst., ether) 68–70 °C; δH 3.62 (3H, s, OCH3), 3.75 (6H, s, 2 × OCH3), 4.67 (1H, br s, NH), 6.27 (2H, d, J 9, C2′′H, C6′′H), 6.49 (2H, d, J 9, C3′′H, C5′′H), 6.76 (4H, d, J 9, C3H, C5H), 7.15–7.40 (9H, m, ArH); δC 55.21 (OCH3), 55.50 (OCH3), 70.66 (C–NH), 113.10 (C3, C5), 113.83 (C3′′, C5′′), 117.20 (C2′′, C6′′), 126.57 (C4′), 127.82 (C3′, C5′), 129.07 (C2′, C6′), 130.33 (C2, C6), 138.00 (C1), 140.43 (C1′′), 146.28 (C1′), 151.70 (C4′′), 158.11 (C4); found: C, 79.3; H, 6.6; N, 3.2. C28H27NO3 requires C, 79.0; H, 6.4; N, 3.3%. N-(4,4′-Dimethoxytrityl)-p-methylaniline (3g iii).. 4,4′-Di-methoxytrityl chloride (405 mg, 1.2 mmol) and p-toluidine (128 mg, 1.2 mmol) in pyridine (1.40 cm3) were stirred for two days, and white crystals of the title compound (365 mg, 75%, mp 101–102 °C) were obtained following the usual work-up and purification on an alumina column (20% ethyl acetate∶petrol); vmax (KBr)/cm−1 3405, 3052, 2997, 2930, 2909, 1083; δH 2.15 (3H, s, CH3), 3.78 (6H, s, 2 × OCH3), 4.85 (1H, br s, NH), 6.25 (2H, d, J 8, C2′′H, C6′′H), 6.72 (2H, d, J 8, C3′′H, C5′′H), 6.78 (4H, d, J 9, C3H, C5H), 7.20–7.45 (9H, m, ArH); δC 20.36 (CH3), 55.21 (OCH3), 70.41 (C–NH), 113.13 (C3, C5), 116.06 (C2′′, C6′′), 126.13 (C4′), 126.61 (C4′′), 127.86 (C3′, C5′), 128.82 (C2′, C6′), 129.10 (C3′′, C5′′), 130.35 (C2, C6), 138.00 (C1), 144.07 (C1′′), 146.23 (C1′), 158.14 (C4); found: C, 81.96; H, 6.46; N, 3.17. C28H27NO2 requires C, 82.12; H, 6.54; N, 3.42%. N-4,4′-Dimethoxytritylaniline (3g i).. By the procedure described above, 4,4′-dimethoxytrityl chloride (405 mg, 1.2 mmol) and aniline (0.11 cm3) in pyridine (0.88 cm3) gave crystals of the title compound (316 mg, 67%); vmax (KBr)/cm−1 3402, 3052, 3001, 2932, 2907, 1089; δH 3.75 (6H, s, 2 × OCH3), 4.93 (1H, br s, NH), 6.33 (2H, d, J 7, C2′′H, C6′′H), 6.54 (1H, t, J 7, C4′′H), 6.77 (4H, d, J 9, C2H, C6H), 6.90 (2H, t, J 7, C3′′H, C5′′H), 7.24 (4H, d, J 9, C3H, C5H), 7.18–7.30 (5H, m, ArH); δC 55.21 (OCH3), 70.46 (C–NH), 113.17 (C3, C5), 116.09 (C2′′, C6′′), 117.13 (C4′′) 126.68 (C4′), 127.89 (C3′, C5′), 128.23 (C2′, C6′), 129.08 (C3′′, C5′′), 130.33 (C2, C6), 137.83 (C1), 146.05 (C1′), 146.44 (C1′′), 158.22 (C4); found: C, 81.9; H, 6.3. C27H25NO2 requires C, 82.2; H, 6.4%. N-(p-Nitrobenzyl)-4,4′-dimethoxytritylamine (3f i).. Triethylamine (77 mg, 0.72 mmol) was added to a stirred suspension of p-nitrobenzylammonium chloride (72 mg, 0.38 mmol) in dry chloroform (3 cm3) followed by 4,4′-dimethoxytrityl tetrafluoroborate (150 mg, 0.38 mmol), and the mixture was allowed to react for 12 h at room temperature. It was then quenched with water and the organic phase was dried (Na2SO4), filtered, and evaporated to dryness. The resulting oil was chromatographed on alumina (20% ethyl acetate∶petrol) to give a colourless crystalline compound (70 mg, 41%), mp (recryst., ethyl acetate–petrol) 127–129 °C; vmax (KBr)/cm−1 3446, 2905, 2835, 1606, 1581, 1508, 1109, 1033, 829; δH 1.90 (1H, br s, NH), 3.50 (2H, s, CH2), 3.80 (6H, s, 2 × OCH3), 6.85 (4H, d, J 9, C3H, C5H), 7.05–7.38 (3H, m, ArH), 7.40 (4H, d, J 9, C2H, C6H), 7.41 (2H, d, J 9, C2′H, C6′H), 7.58 (2H, d, J 9, C2′′H, C6′′H), 8.15 (2H, d, J 9, C3′′H, C5′′H); δC 47.59 (CH2), 55.27 (OCH3), 70.14 (C–NH), 113.38 (C3, C5), 123.62 (C3′′, C5′′), 126.49 (C4′), 128.08 (C3′, C5′), 128.32 (C2′′, C6′′), 128.41 (C2′, C6′), 129.64 (C2, C6), 138.04 (C1), 146.20 (C1′), 146.97 (C1′′), 148.94 (C4′′), 158.08 (C4); m/z (+EI) 454 (M+, 11%), 423 (M+ − OMe, 11), 377 (M+ − Ph, 13), 347 (M+ − C6H4OMe, 12), 333 ((M − 1)+ − C6H4NO2, 72), 303 (M+ − NHCH2C6H4NO2, 100), 107 (C6H4OMe, 29), 77 (Ph, 51); found: C, 74.0; H, 5.8; N, 6.2. C28H26N2O4 requires C, 74.20; H, 5.7; N, 6.0%. 4,4′,4′′-Trimethoxytritylamine (4a).. This compound was prepared as described above for 4-methoxytritylamine. The resulting oil was chromatographed on silica gel (50% ethyl acetate∶petrol + 1% triethylamine) to give the title product as pale yellow crystals (96 mg, 75%), mp (recryst., ether–petrol) 103–105 °C, lit.,28 105–106 °C; δH 2.10 (2H, br s, NH2), 3.78 (9H, s, 3 × OMe), 6.79 (6H, d, J 9, C3H, C5H), 7.15 (6H, d, J 9, C2H, C6H); δC 55.27 (OCH3), 64.86 (C–NH2), 113.16 (C3, C5), 129.18 (C2, C6), 141.46 (C1), 158.11 (C4). N-(p-Nitrobenzyl)-4,4′,4′′-trimethoxytritylamine (4f i).. Triethylamine (72 mg, 0.70 mmol) was added to a stirred suspension of p-nitrobenzylammonium chloride (67 mg, 0.35 mmol) in dry chloroform (3 cm3) followed by 4,4′,4′′-trimethoxytrityl tetrafluoroborate (150 mg, 0.35 mmol); the mixture was allowed to react for 12 h at room temperature. It was then quenched with water and the organic phase was dried (Na2SO4), filtered, and evaporated. The resulting oil was chromatographed on alumina (20% ethyl acetate∶petrol) to give pale yellow crystals (73 mg, 43%); mp (recryst., ether) 138–139 °C; vmax (KBr)/cm−1 3311, 2906, 2835, 1605, 1579, 1507, 1111, 1033, 827; δH 1.90 (1H, br s, NH), 3.45 (2H, s, CH2), 3.80 (9H, s, 3 × OCH3), 6.85 (6H, d, J 9, C3H, C5H), 7.40 (6H, d, J 9, C2H, C6H), 7.60 (2H, d, J 9, C2′′H, C6′′H), 8.15 (2H, d, J 9, C3′′H, C5′′H); δC 47.52 (CH2), 55.27 (OCH3), 69.65 (C–NH), 113.37 (C3, C5), 123.61 (C3′′, C5′′), 128.42 (C2′′, C6′′), 129.53 (C2, C6), 138.34 (C1), 146.98 (C1′′), 149.03 (C4′′), 158.04 (C4); m/z (+EI) 484 (M+, 30%), 333 (TMT+, 100), 303 (MH+ − OCH3, 22), 227 (MH+ − Ph, 30); found: C, 72.0; H, 5.8; N, 5.8. C29H28N2O5 requires C, 71.9; H, 5.8; N, 5.8%. X-Ray crystallography![[hair space]](https://www.rsc.org/images/entities/h3_char_200a.gif) †
†
Crystal data and some structure refinement details are presented in Table 4. Measurements for 4a were made on a Stoe-Siemens four-circle diffractometer equipped with Cu-Kα radiation (λ = 1.54184 Å). Those for 1b·Me2CO, 1c and 1d were made on a Bruker SMART CCD area detector diffractometer equipped with Mo-Kα radiation (λ = 0.71073 Å). All data sets were collected at low temperature (160 K) and suffered from no measurable crystal decay. Data were corrected for Lorentz and polarisation effects and for absorption in the cases of 1b and 4a. All non-H atoms were refined anisotropically on F2 values. H atoms were constrained using a riding model except for: H(1A), H(1B) and H(1C) in 1b·Me2CO; H(1) in 1c; and H(1A), H(1B), H(2A) and H(2B) in 4a; for which the coordinates were freely refined. H-atom Uiso values were set to be 1.2 times that of the carrier atom (1.5 times for methyl-H). The absolute structure of 1d could not be determined reliably. In 1d, two-fold disorder in the group N(1) C(2) C(3) O(1) O(2) was modelled successfully with restraints on geometry, the two components having occupancies of 54.6(6) and 45.3(6)%. Software: Bruker SMART and SAINT for data collection and frame integration; Stoe DIF4 for data collection; Bruker SHELXTL for structure solution, refinement and molecular graphics; and local programs.
Table 4 Summary of crystal data and structure determination for compounds 1b·Me2CO, 1c, 1d and 4a
| Compound | 1b·Me2CO | 1c | 1d | 4a |
|---|
| Cu-Kα radiation. Mo-Kα radiation. Conventional R = Σ||Fo| − |Fc||/Σ|Fo| for “observed” reflections having Fo2 > 2σ(Fo2). Rw = [Σw(Fo2 − Fc2)2/Σw(Fo2)2]1/2 for all data. |
|---|
| Mol. form. | C22H24ClNO | C22H21NO2 | C21H19NO2 | C22H23NO3 |
| M | 353.87 | 331.40 | 317.37 | 349.41 |
| Cryst. syst. | Triclinic | Monoclinic | Orthorhombic | Monoclinic |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) | C2/c | P212121 | P21/c |
| a/Å | 8.7123(12) | 16.574(2) | 9.2290(18) | 10.497(2) |
| b/Å | 9.3000(13) | 12.5221(18) | 9.6725(18) | 18.498(2) |
| c/Å | 13.4414(19) | 17.769(3) | 18.700(3) | 19.597(2) |
| α/deg | 88.445(4) | | | |
| β/deg | 75.459(3) | 105.757(3) | | 102.780(17) |
| γ/deg | 64.682(3) | | | |
| V/Å3 | 948.9(2) | 3549.3(9) | 1669.3(5) | 3710.9(10) |
| Z | 2 | 8 | 4 | 8 |
| μ/mm−1 | 0.210b | 0.079b | 0.081b | 0.664a |
| No. of reflns. measd. | 6335 | 10748 | 8809 | 11167 |
| No. of unique reflns. | 4164 | 4053 | 2939 | 6506 |
| No. of reflns. with F2 > 2σ(F2) | 3090 | 2711 | 1964 | 5007 |
| Rint (on F2) | 0.0261 | 0.0331 | 0.0472 | 0.0320 |
| Rc | 0.0466 | 0.0511 | 0.0598 | 0.0435 |
| Rwd | 0.1139 | 0.1579 | 0.1575 | 0.1105 |
Determination of pKa values
Two routine techniques were used. One involved a combined glass and reference electrode, and directly reading the pH from a pH meter in the titration of the base in aqueous sodium hydroxide against standard aqueous perchloric or hydrochloric acid. The other method involved automatic recording of the pH using an automatic titrator connected to a microcomputer, a Russell pH glass electrode, and calomel reference electrode.The isoelectric point of N-tritylglycine (1d)
A solution of N-tritylglycine in acetonitrile (1.45 × 10−3 mol dm−3), aqueous sodium hydroxide (1.98 mol dm−3), aqueous sodium perchlorate (3.00 mol dm−3), and water was titrated against aqueous perchloric acid (0.25 cm3 aliquots, 0.031 mol dm−3) to give pKa1 = 9.55 and pKa2 = 3.23 at 21 °C and ionic strength = 0.1 mol dm−3. This led to pI = 6.4 which compares with pI = 6.1 for unprotected glycine in water.29References
- B. Helferich, Adv. Carbohydr. Chem., 1948, 3, 79 Search PubMed; J. F. W. McOmie
, Protective Groups in Organic Chemistry, Plenum Press, London and New York, 1973;
Search PubMed; T. W. Greene
and P. G. M. Wuts
, Protective Groups in Organic Synthesis, Wiley-Interscience, 3rd edn., New York, 1999.
Search PubMed.
- M. Canle L., J. Crugeiras M., I. Demirtas, H. Maskill and E. Stix, Org. React., 1997, 31, 71 Search PubMed.
- C. Bleasdale, B. T. Golding, W. H. Lee, H. Maskill, J. Riseborough and E. Smits, J. Chem. Soc., Chem. Commun., 1994, 93 RSC.
- J. Crugeiras and H. Maskill, J. Chem. Soc., Perkin Trans. 2, 1998, 1901 RSC.
- R. W. Hanson and H. D. Law, J. Chem. Soc., 1965, 7285 RSC.
- H.-B. Bürgi and J. D. Dunitz, J. Am. Chem. Soc., 1987, 109, 2924 CrossRef; P. G. Jones and A. J. Kirby, J. Am. Chem. Soc., 1984, 106, 6207 CrossRef CAS; H.-B. Bürgi and J. D. Dunitz, Acc. Chem. Res., 1983, 16, 153 CrossRef; F. H. Allen, O. Kennard and R. Taylor, Acc. Chem. Res., 1983, 16, 146 CrossRef CAS.
- C. Bleasdale, S. B. Ellwood and B. T. Golding, J. Chem. Soc., Perkin Trans. 1, 1990, 803 RSC; A. P. Henderson, J. Riseborough, C. Bleasdale, W. Clegg, M. R. J. Elsegood and B. T. Golding, J. Chem. Soc., Perkin Trans. 1, 1997, 3407 RSC.
- L. Zervas and D. M. Theodoropoulos, J. Am. Chem. Soc., 1956, 78, 1359 CrossRef CAS.
- Y. Lapidot, N. De Groot, M. Weiss, R. Peled and Y. Wolman, Biochim. Biophys. Acta, 1967, 138, 241 CAS.
- K. Barlos, D. Papaioannou and D. Theodoropoulos, J. Org. Chem., 1982, 47, 1324 CrossRef CAS.
- K. C. Schreiber and V. P. Fernandez, J. Org. Chem., 1961, 26, 1744 CrossRef CAS.
- M. G. Siskos, S. K. Garas, A. K. Zarkadis and E. P. Bokaris, Chem. Ber., 1992, 125, 2477 CAS.
- P. E. Verkade, H. Nijon, F. D. Tollenaar, J. H. Van Rij and M. Van Leeuven, Recl. Trav. Chim. Pays-Bas, 1952, 71, 1007 Search PubMed.
- C. Glidewell and G. Ferguson, Acta Crystallogr., Sect. C, 1994, 50, 924 CrossRef.
- F. H. Allen and O. Kennard, Chem. Des. Autom. News, 1993, 8, 31 Search PubMed.
- A. Carpy, J.-M. Leger and J.-C. Colleter, Acta Crystallogr., Sect. B, 1980, 36, 2837 CrossRef.
- P. Huszthy, J. S. Bradshaw, K. E. Krakowiak, T. Wang and N. K. Dalley, J. Heterocycl. Chem., 1993, 30, 1197 Search PubMed.
- H. Maskill
, The Physical Basis of Organic Chemistry, Oxford University Press, 1985.
Search PubMed.
- P. Moinjoint and M. F. Ruasse, Tetrahedron Lett., 1984, 25, 3183 CrossRef; J. T. Edsall and M. H. Blanchard, J. Am. Chem. Soc., 1933, 55, 2337 CrossRef CAS.
- G. N. Okafo, R. Brown and P. Camilleri, J. Chem. Soc., Chem. Commun., 1991, 864 RSC; A. I. Biggs and R. A. Robinson, J. Chem. Soc., 1961, 388 RSC.
- A. Castro, P. Sánchez and J. A. Santaballa, Bull. Soc. Chim. Fr., 1988, 364 Search PubMed.
- G. Girault-Vexlearschi, Bull. Soc. Chim. Fr., 1956, 589 Search PubMed.
- J. Oszczapowicz, W. Krawczyk and P. Łyźwiński, J. Chem. Soc., Perkin Trans. 2, 1990, 311 RSC.
- D. Martin and A. Weise, Liebigs. Ann., 1967, 702, 86 Search PubMed.
- K. Elbs, , 1884, 17, 701 Search PubMed; M. Gomberg, Ber., 1902, 35, 1822 Search PubMed.
- J. Riseborough
, PhD Thesis, University of Newcastle, 1993.
Search PubMed.
- A. Baeyer and V. Villiger, Chem. Ber., 1904, 37, 597 Search PubMed.
- C. A. Bunton and S. K. Huang, J. Am. Chem. Soc., 1974, 96, 515 CrossRef CAS.
- E. J. King, J. Am. Chem. Soc., 1945, 67, 2178 CrossRef CAS; E. J. King, J. Am. Chem. Soc., 1951, 73, 155 CrossRef CAS.
- R. A. Robinson and A. K. Kiang, Trans. Faraday Soc., 1956, 52, 327 RSC; H. K. Hall, J. Am. Chem. Soc., 1957, 79, 5439 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2000 |
Click here to see how this site uses Cookies. View our privacy policy here.