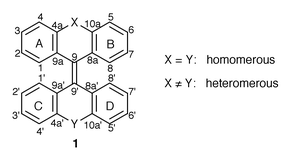
The stereochemistry of overcrowded homomerous bistricyclic aromatic enes with alkylidene bridges
Yitzhak
Tapuhi
,
Michal Rachel
Suissa
,
Shmuel
Cohen
,
P. Ulrich
Biedermann
,
Amalia
Levy
and
Israel
Agranat
*
Department of Organic Chemistry, The Hebrew University of Jerusalem, Jerusalem, 91904, Israel
First published on 14th January 2000
Abstract
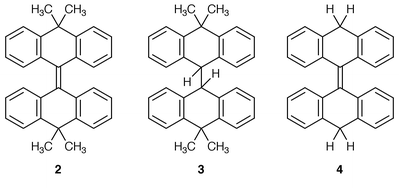
The objective of the research was to study the effects of alkylidene bridges on the conformations and the conformational behaviour of overcrowded homomerous bistricyclic aromatic ethenes (1). The isopropylidene-bridged bistricyclic ethene 2 and 3 were synthesized by a reductive “dimerization” of 7, using TiCl4–Zn–pyridine–THF. The methylene-bridged bistricyclic ethenes 4–6 were synthesized by LiAlH4–AlCl3–Et2O reductions of the corresponding bianthrones. The structures of 4–6 were established by 1H- and 13C-NMR spectroscopy and in the cases of 2 and 3, also by X-ray analysis. Compounds 2 and 3 adopted Ci-anti-folded conformations with 53.0° and 28.8° folding dihedrals between pairs of benzene rings of tricyclic moieties. The central C9![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) C9′ bond in 2 was essentially planar. A short C9
C9′ bond in 2 was essentially planar. A short C9![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) ⋯C10 distance of 2.81 Å in 2 indicated an intramolecular overcrowding effect in the highly folded bistricyclic ethene. Semiempirical PM3 and AM1 calculations of the anti-folded, syn-folded, twisted and orthogonally twisted conformations of 2 and 4 indicated that anti-folded 2 and 4 were the most stable conformations with folding dihedrals of 48.7° and 45.0°, respectively at AM1. A DNMR spectroscopic study of E, Z-isomerizations and conformational inversions gave ΔGc‡(E⇌Z) = 99.6 kJ mol−1 (CDBr3) and ΔGc# (inversion) = 97.9 kJ mol−1 (hexachlorobutadiene) in 5 and ΔGc‡ (inversion) > 108 kJ mol−1 (benzophenone) in 3. These high energy barriers were interpreted in terms of less overcrowded fjord regions in the anti-folded ground-state conformations.
⋯C10 distance of 2.81 Å in 2 indicated an intramolecular overcrowding effect in the highly folded bistricyclic ethene. Semiempirical PM3 and AM1 calculations of the anti-folded, syn-folded, twisted and orthogonally twisted conformations of 2 and 4 indicated that anti-folded 2 and 4 were the most stable conformations with folding dihedrals of 48.7° and 45.0°, respectively at AM1. A DNMR spectroscopic study of E, Z-isomerizations and conformational inversions gave ΔGc‡(E⇌Z) = 99.6 kJ mol−1 (CDBr3) and ΔGc# (inversion) = 97.9 kJ mol−1 (hexachlorobutadiene) in 5 and ΔGc‡ (inversion) > 108 kJ mol−1 (benzophenone) in 3. These high energy barriers were interpreted in terms of less overcrowded fjord regions in the anti-folded ground-state conformations.
Introduction
Bistricyclic aromatic enes (1) have served as attractive substrates for the study of the conformations and dynamic stereochemistry of overcrowded ethenes.1–3 Several of these systems, e.g., bianthrone (1, X = Y: CO) and dixanthylene (1, X = Y: O), display thermochromic, photochromic and piezochromic properties.3 The bistricyclic enes are nonplanar due to the intramolecular overcrowding in their fjord regions.3 The nonplanarity introduces the notion of chirality to the arena of these interesting compounds.4 Two principal modes of out-of-plane deviation were considered: twisting around the central carbon–carbon double bond and out-of-plane bending.5 In 1, the bending is realized by folding of the tricyclic moieties at both ends of the central ene about the C9⋯X and C9′⋯Y axes, resulting in boat conformations of the central rings. In addition, the atoms C9 and C9′ may be pyramidalized. Four pure conformations of 1 were considered: the twisted (t), the anti-folded (a), the syn-folded (s) and the orthogonally twisted (t⊥) conformations (Fig. 1).3 Homomerous bistricyclic enes (1, X = Y) with five-membered and six-membered central rings adopt twisted conformations (t) and anti-folded (a) conformations, respectively.3
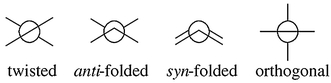 | ||
| Fig. 1 Schematic projection along C9C9′ of various types of conformations of bistricyclic enes (lines represent the peripheral benzene rings of the tricyclic moieties).
| ||
Bistricyclic enes may undergo the following fundamental dynamic processes:3
1. E,Z-isomerization (e.g., tE⇌tZ, aE⇌aZ),
2. Conformational inversion, e.g., inversion of the helicity in the twisted 1 (tP⇌tM) or inversion of the boat conformation in the central rings of folded 1,
3. syn, anti-isomerization (a⇌s), anti, twisted-isomerization (a⇌t) and syn, twisted-isomerization (s⇌t).
4. Enantiomerization and racemization may also be considered in these processes.
The dynamic stereochemistry of bistricyclic enes 1 with central six-membered rings have been studied by Agranat et al., and by Feringa et al., using dynamic NMR (DNMR) and equilibration techniques, when one of the conformational isomers is available in a pure form.3–9 The DNMR studies revealed low barriers for thermal E,Z-isomerization (ΔGc‡ = 75–115 kJ mol−1).3,6–10 The range of free energies of activation for thermal conformational inversion of bistricyclic enes with central six-membered rings was found to be similar. These remarkably low energy barriers were interpreted predominantly in terms of ground state destabilization due to steric strain and overcrowding.3 It has been shown that the barriers depend on the bridges X and Y, on the C–X and C–Y bond lengths, and on the C4a–C10a distances.6
The present article describes the syntheses, molecular and crystal structures, semiempirical calculations and DNMR study of homomerous bistricyclic enes with methylene and isopropylidene bridges (1, X = Y: CH2 and 1, X = Y: CMe2). In these systems, the Csp2–Csp3 bonds at the bridges (C4a–C10 and C10–C10a) are expected to be longer than the corresponding C4a–X and C4a–Y in dixanthylene, N,N′-dimethylbiacridan and bianthrone.3 The alkylidene-bridged 1 are thus expected to possess a higher degree of folding and to be less overcrowded in the fjord regions. Furthermore, the methylene and isopropylidene bridges contain axial and equatorial hydrogen atoms and methyl groups in the boat-shaped central six-membered rings, which in principle allow a determination of the inversion barriers of the parent bistricyclic enes. Heteromerous bistricyclic enes with one CMe2 bridge have previously been studied.11
Synthesis
The following compounds were synthesized: 10-[10,10-dimethyl-9(10H)-anthracenylidene]-9,10-dihydro-9,9-dimethylanthracene (2), 10-[10,10-dimethyl-9(10H)-anthracenyl]-9,10-dihydro-9,9-dimethylanthracene (3), 9-[9(10H)-anthracenylidene]-9,10-dihydroanthracene (4), 9-[2-methyl-9(10H)-anthracenylidene]-9,10-dihydro-2-methylanthracene (5) and 5-[3-methyl-5(12H)-naphthacenylidene]-5,12-dihydro-3-methylnaphthacene (6). The synthesis of 2 was accomplished by a low-valent titanium-induced reductive “dimerization” of 10,10-dimethylanthracen-9(10H)-one (7) using the Mukaiyama–Lenoir procedure12–14 of the McMurry reaction15 (TiCl4–Zn–pyridine–THF) (Scheme 1). The reaction afforded a 1∶3 mixture of 2 and 3 in 66% yield. The products were separated and purified by recrystallizations. Hydrocarbon 2 has been prepared by a reductive “dimerization” of 7, using zinc in AcOH–HCl.16 Hydrocarbon 3 has been prepared by hydrogenation of 2, using sodium in pentan-1-ol.16
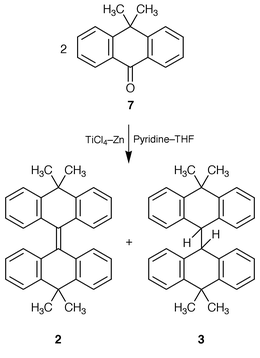 | ||
| Scheme 1 | ||
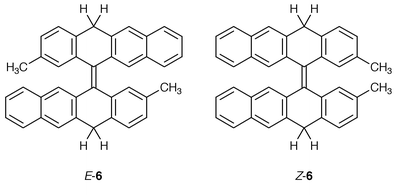
Compounds 4–6 were synthesized by LiAlH4–AlCl3 reduction of bianthrone, 2,2′-dimethylbianthrone and 12-[3-methyl-12-oxo-5(12H)-naphthacenylidene]-2-methylnaphthacen-5(12H)-one, respectively. The synthesis of 4 by a LiAlH4 reduction of bianthrone17 and by a reductive “dimerization” of anthrone using aluminium in CH3CN under the irradiation of ultrasonic wave18 has been reported. Compounds 5 and 6 were obtained as mixtures of E- and Z-diastereomers. The synthesis of (E)-5 and (Z)-5 is depicted in Scheme 2. The structures of 4–6 were established by NMR spectroscopy and elemental analysis. It is possible to distinguish qualitatively among the twisted, syn-folded and anti-folded conformations of homomerous bistricyclic enes (1, X = Y) in solution, using 1H-NMR spectroscopy. In the cases of 2 and 4, the fjord region protons appear at 7.010 and 6.984 ppm, respectively, indicating that these hydrocarbons adopt anti-folded conformations in solution.3,8 A similar picture appears in 5 and 6. In the case of 4, complete assignments were made through two-dimensional correlation spectroscopy (COSY, HSOC, HMBC, NOESY). In the 1H-NMR spectrum of 4, the methylene protons appear as an AB system at 4.240 and 3.865 ppm. The doublet at 4.240 ppm representing one of the protons of each methylene group was broad, while the second doublet at 3.865 ppm representing the other proton of each methylene group was sharp. In the aromatic region, the double doublet at 7.365 ppm representing H4, H5, H4′, H5′ was also broad. A COSY experiment indicated an allylic coupling between the 7.365 ppm doublet and the 4.240 ppm doublet. A NOESY experiment indicated through space interactions between the 7.365 ppm doublet and the 3.865 ppm doublet. On the basis of these experiments, it is concluded that the 3.865 ppm doublet is due to the equatorial H10eq while the 4.240 ppm doublet is due to the axial H10ax.
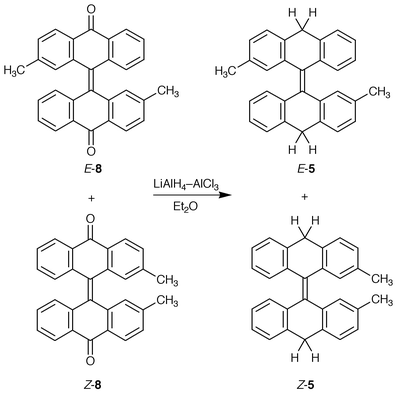 | ||
| Scheme 2 | ||
Molecular and crystal structures†
Compounds 2 and 3 crystallized in the space group P21/c.19Fig. 2 gives an ORTEP diagram of one molecule of 2 as determined by X-ray analysis. Table 1 gives the conformations and selected geometrical parameters of 2–4 derived from the crystal structures and from semiempirical calculations (vide infra).| Method | PG-Conf.a | ΔHf°/kJ mol−1 | ΔΔHf°/kJ mol−1 | Min/TSb |
A–B/deg | ω/deg | C9C9′/Å |
C8a–C9–C9a/deg | χ(Cg)/deg | C4a–C10/Å | C10a–C10/Å | C4a–C10–C10a/deg | C4a⋯C10a/Å
|
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Point group and conformation. b Minimum (Min), transition state (TS), or number of imaginary vibrational frequencies in case of a higher order saddle point. c H9′***H3C. | |||||||||||||
| Compound 2 C(CH3)2 bridges | |||||||||||||
| X-Ray | C i-a | 53.0 | 0.0 | 1.347(3) | 110.2(1) | 0.0(3) | 1.533(2) | 1.530(2) | 106.7(1) | 2.458(2) | |||
| PM3 | C 2h-a | 416.594 | 0.000 | Min | 45.2 | 0.0 | 1.352 | 110.8 | 4.0 | 1.517 | 108.9 | 2.47 | |
| AM1 | C 2h-a | 479.428 | 0.000 | Min | 48.7 | 0.0 | 1.355 | 111.0 | 1.5 | 1.512 | 107.9 | 2.45 | |
| PM3 | C 2-s | 430.574 | 13.979 | Min | 46.8 | 8.6 | 1.356 | 108.9 | 10.6 | 1.518 | 1.516 | 109.0 | 2.47 |
| AM1 | C 2-s | 518.833 | 39.405 | Min | 50.7 | 24.7 | 1.357 | 109.6 | 9.5 | 1.516 | 1.511 | 107.9 | 2.45 |
| PM3 | C 2v-s | 431.221 | 14.627 | TS | 46.3 | 0.0 | 1.356 | 108.7 | 10.3 | 1.517 | 109.2 | 2.47 | |
| AM1 | C 2v-s | 534.682 | 55.254 | 2 | 41.3 | 0.0 | 1.359 | 109.4 | 12.1 | 1.513 | 110.7 | 2.49 | |
| PM3 | C 2-t | 491.539 | 74.945 | Min | 9.4 | 53.2 | 1.394 | 117.4 | −0.2 | 1.509 | 1.507 | 113.0 | 2.51 |
| PM3 | D 2-t | 498.399 | 81.805 | 2 | 3.6 | 53.3 | 1.395 | 117.4 | 0.0 | 1.509 | 113.4 | 2.52 | |
| AM1 | D 2-t | 542.010 | 62.582 | Min | 5.2 | 51.5 | 1.393 | 117.3 | 0.0 | 1.504 | 113.4 | 2.51 | |
| PM3 | D 2d-t⊥ | 519.938 | 103.343 | 3 | 0.0 | 90.0 | 1.467 | 120.4 | 0.0 | 1.512 | 113.7 | 2.53 | |
| AM1 | D 2d-t⊥ | 568.715 | 89.287 | TS | 0.0 | 90.0 | 1.460 | 120.1 | 0.0 | 1.506 | 113.7 | 2.52 | |
| Compound 3 C(CH3)2 bridges | |||||||||||||
| X-Ray | C i-a | 28.7 | 0.0 | 1.585(4) | 111.2(2) | 1.536(3) | 1.528(3) | 110.8(2) | 2.523(2) | ||||
| Compound 4 CH2 bridges | |||||||||||||
| PM3 | C 2h-a | 471.893 | 0.000 | Min | 47.9 | 0.0 | 1.352 | 110.9 | 3.5 | 1.496 | 109.7 | 2.45 | |
| AM1 | C 2h-a | 494.472 | 0.000 | Min | 45.0 | 0.0 | 1.356 | 111.7 | 2.7 | 1.491 | 110.4 | 2.45 | |
| PM3 | C 2v-s | 482.477 | 10.584 | Min | 52.2 | 0.0 | 1.350 | 109.5 | 13.0 | 1.497 | 109.0 | 2.44 | |
| AM1 | C 2v-s | 523.606 | 29.135 | Min | 50.1 | 0.0 | 1.353 | 109.9 | 14.9 | 1.494 | 109.9 | 2.44 | |
| PM3 | D 2-t | 545.962 | 74.069 | TS | 3.9 | 52.3 | 1.393 | 117.4 | 0.0 | 1.486 | 115.0 | 2.51 | |
| AM1 | D 2-t | 557.223 | 62.751 | TS | 4.3 | 50.9 | 1.392 | 117.4 | 0.0 | 1.483 | 115.0 | 2.50 | |
| PM3 | D 2d-t⊥ | 570.001 | 98.108 | TS | 0.0 | 90.0 | 1.466 | 120.3 | 0.0 | 1.488 | 115.2 | 2.51 | |
| AM1 | D 2d-t⊥ | 586.296 | 91.825 | TS | 0.0 | 90.0 | 1.459 | 120.1 | 0.0 | 1.485 | 115.3 | 2.51 | |
| Method | PG-Conf.a | C1⋯C1′/Å | C1⋯H1′/Å | C8⋯H8′/Å | H1⋯H1′/Å | C9⋯C10/Å | CH3⋯H3C/Å | H10⋯H10′/Å | C9⋯H3C/Å | C9⋯H10/Å | H4⋯H3C/Å | H5⋯H3C/Å |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound 2 C(CH3)2 bridges | ||||||||||||
| X-Ray | C i-a | 3.127(2) | 2.98 | 2.95 | 3.11 | 2.811(2) | 5.49 | 2.57 | 2.17 | 2.16 | ||
| PM3 | C 2h-a | 3.06 | 2.96 | 3.24 | 2.84 | 5.80 | 2.76 | 1.87 | ||||
| AM1 | C 2h-a | 2.99 | 2.83 | 3.07 | 2.82 | 5.44 | 2.57 | 2.03 | ||||
| PM3 | C 2-s | 3.00 | 2.62 | 2.41 | 1.72 | 2.86 | 1.67 | 2.92 | 1.84 | 1.84 | ||
| AM1 | C 2-s | 3.13 | 2.98 | 2.32 | 2.04 | 2.81 | 2.07 | 2.63 | 2.00 | 2.00 | ||
| PM3 | C 2v-s | 2.99 | 2.51 | 1.71 | 2.87 | 1.64 | 2.97 | 1.83 | ||||
| AM1 | C 2v-s | 2.97 | 2.59 | 1.85 | 2.89 | 1.81 | 3.13 | 1.93 | ||||
| PM3 | C 2-t | 2.93 | 2.51 | 2.88 | 2.53 | 2.98 | 7.76 | 3.54 | 1.83 | 1.84 | ||
| PM3 | D 2-t | 2.98 | 2.65 | 2.76 | 2.99 | 8.17 | 3.92 | 2.10 | ||||
| AM1 | D 2-t | 2.91 | 2.59 | 2.72 | 2.99 | 8.09 | 3.90 | 2.12 | ||||
| PM3 | D 2d-t⊥ | 3.78 | 3.49 | 3.53 | 2.95 | 8.47 | 3.88 | 2.17 | ||||
| AM1 | D 2d-t⊥ | 3.78 | 3.49 | 3.53 | 2.95 | 8.44 | 3.87 | 2.14 | ||||
| Compound 3 C(CH3)2 bridges | ||||||||||||
| X-Ray | C i-a | 3.070(4) | 3.15 | 3.08 | 3.43 | 2.973(3) | 6.24 | 2.26c |
2.04 | 2.06 | ||
| Compound 4 CH2 bridges | ||||||||||||
| PM3 | C 2h-a | 3.07 | 2.93 | 3.19 | 2.80 | 3.26 | 3.06 | 2.42 | ||||
| AM1 | C 2h-a | 2.94 | 2.77 | 3.02 | 2.82 | 3.24 | 3.11 | 2.41 | ||||
| PM3 | C 2v-s | 3.06 | 2.55 | 1.74 | 2.77 | 3.29 | 2.98 | 2.42 | ||||
| AM1 | C 2v-s | 3.10 | 2.69 | 1.94 | 2.76 | 3.28 | 2.99 | 2.42 | ||||
| PM3 | D 2-t | 2.98 | 2.64 | 2.74 | 2.96 | 2.60 | 3.74 | 2.60 | ||||
| AM1 | D 2-t | 2.91 | 2.58 | 2.69 | 2.96 | 2.61 | 3.75 | 2.61 | ||||
| PM3 | D 2d-t⊥ | 3.81 | 3.52 | 3.56 | 2.92 | 2.65 | 3.69 | 2.65 | ||||
| AM1 | D 2d-t⊥ | 3.81 | 3.52 | 3.56 | 2.92 | 2.66 | 3.71 | 2.66 | ||||
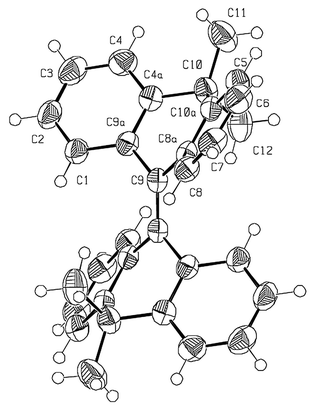 | ||
| Fig. 2 An ORTEP drawing of 2 derived from the X-ray crystal structure. | ||
The molecular and crystal structures of 2 show that the molecule adopts a Ci-anti-folded conformation (a-2). The folding dihedral angle (between the least-squares-planes of the two benzene rings of each tricyclic moiety) of 2 is 53.0°. For comparison, the degree of folding of a metalo-based (PdCl2 bridge) bistricyclic ene with the 9,9-dimethyl-9,10-dihydroanthracenylidene moiety is 48.8°.11 The degree of overcrowding in the fjord regions of 2, as reflected in the intramolecular distances C1⋯C1′, C1⋯H1′, and H1⋯H1′ is hardly significant: 3.13, 2.97 and 3.11 Å, respectively. For comparison, the van der Waals radii of carbon and hydrogen are 1.71 and 1.15 Å, resulting in van der Waals C⋯C, C⋯H and H⋯H contact distances of 3.42 Å, 2.86 and 2.30 Å, respectively.20 The above C1⋯C1′ distance in 2 reflects only an 8% penetration, while the above C1⋯H1′ and H1⋯H1′ distances do not indicate any penetration. On the other hand, the transannular C9⋯C10 distance, 2.81 Å is considerably shorter than the C⋯C, van der Waals contact distance, reflecting an 18% penetration. These short distances indicate an additional effect of intramolecular overcrowding in highly folded bistricyclic enes.
In the previously reported structures of anti-folded homomerous bistricyclic enes, pyramidal C9C9′ carbon atoms have been noted. However, the central carbon–carbon double bond in 2 is essentially planar, with negligible values of the pyramidalization angles χ9 and χ9′3 and a pure twist of zero.3 The C9C9′ bond length is 1.347 Å. The relatively long C4a–C10 bonds at the bridges, 1.534/1.531 Å, probably enhance the degree of folding. Overcrowding is also evident in the short contact distances between a methyl hydrogen and the corresponding peri-hydrogens (H4, H5): 2.16 Å.
Fig. 3 gives an ORTEP diagram of 3 as determined by X-ray analysis. The molecular and crystal structure of 3 show that the molecule adopts a Ci-anti-folded conformation. The folding dihedral angle (A–B, C–D) is 28.8°. The length of the central C9–C9′ is 1.587 Å. There is a certain degree of overcrowding: the C1⋯C1′ and C9⋯C10 distances are 3.07 and 2.97 Å, respectively. The short H4⋯H3C distance, 2.04 Å should also be noted.
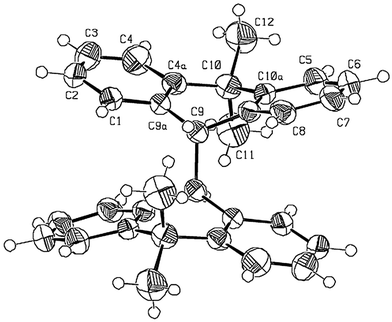 | ||
| Fig. 3 An ORTEP drawing of 3 derived from the X-ray crystal structure. | ||
Semiempirical calculations
Recently, a systematic theoretical survey of overcrowded homomerous and heteromerous bistricyclic enes 1 has been carried out, using MOPAC 6.00 and the semiempirical method PM3.3 The methylene- and isopropylidene-bridged bistricyclic enes 4 and 2 have been included, inter alia, in the modelling.3 The present article reports the results of the PM321 and AM122 calculations of 4 and 2, using MOPAC 93.23 Comparisons of AM1 with PM3 have been commented on.24,25 The following conformations of 2 and 4 have been considered: C2h-anti-folded-2 (a-2), C2v-syn-folded-2 (s-2), C2-syn-folded-2 (C2-s-2), D2-twisted-2 (t-2), C2-twisted-2 (C2-t-2), D2d-orthogonally twisted-2 (t⊥-2), and the corresponding conformations of 4, a-4, s-4, t-4 and t⊥-4. The various conformations have been fully optimized (Keywords: EF SYMMETRY PRECISE GNORM = 0.1 LET DDMIN = 0.0; for orthogonal conformations BIRADICAL was added) and their vibrational frequencies calculated (Keywords: FORCE PRECISE SYMMETRY; for orthogonal conformations BIRADICAL was added). The following conformations were found to be bona fide minima (positive vibrational frequencies): C2h-a-2, C2-s-2, C2-t-2, C2h-a-4, C2v-s-4 at PM3, C2h-a-2, C2-s-2, D2-t-2, C2h-a-4, C2v-s-4 at AM1. The following conformations were found to be transition states (one imaginary frequency): C2v-s-2, D2-t-4 and D2d-t⊥-4 at PM3, D2d-t⊥-2, D2-t-4 and D2d-t⊥-4 at AM1. On the other hand, D2-t-2 at PM3 and C2v-s-2 at AM1 were found to be second order saddle points. Surprisingly, D2d-t⊥-2 at PM3 was found to be a third order saddle point.
The semiempirical enthalpies of formation (ΔHf°) of the conformations of 2 and 4, the conformational energies (ΔΔHf°) of 2 and 4 (relative to the respective anti-folded global minimum conformation) and selected geometrical parameters of the conformations of 2 and 4 derived from the PM3 and AM1 calculations are included in Table 1, together with the corresponding geometrical parameters derived from the crystal structures of 2 and 3. The 3D structures of the AM1 optimized s-2 and a-4 are given in Fig. 4 and Fig. 5. The most stable conformations of 2 and 4 are the anti-folded conformations a-2 and a-4. Their folding dihedrals are 48.7° and 45.0° at AM1, as compared with 53.0° in the crystal structures of 2. In a-2, the C4a–C10 bond at the bridge is elongated, 1.517 Å (PM3) and 1.512 (AM1) as compared with a-4, 1.496 Å (PM3) and 1.491 Å (AM1).
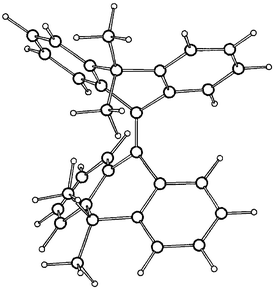 | ||
| Fig. 4 3D structure of C2-s-2 calculated by AM1. | ||
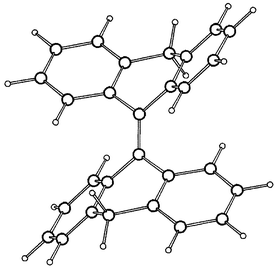 | ||
| Fig. 5 3D structure of C2h-a-4 calculated by AM1. | ||
In a-2, severe methyl, peri-H interactions are noted: the CH3⋯H4 contact distance is 1.87 Å (PM3) and 2.03 Å (AM1). The short CH3⋯C9 contact distance in a-2 should also be noted: 2.76 Å (PM3) and 2.57 Å (AM1). These values are only slightly shorter than the van der Waals hydrogen⋯ carbon contact distance (2.86 Å).20 In a-4, the C9⋯C10 distance, 2.84 Å (PM3) and 2.82 Å (AM1) reflects considerable penetration of the van der Waals carbon⋯carbon contact distance, 3.42 Å,20 17% (PM3) and 18% (AM1). The anti-folded conformations are only slightly pyramidalized. However, both syn-folded conformations are markedly pyramidalized at C9: 10.6° (PM3) and 9.5° (AM1) in C2v-s-2 and 13.0° (PM3) and 14.9° (AM1) in s-4. The conformational energies of the syn-folded conformation are 14.0 (PM3) and 39.4 kJ mol−1 (AM1) in C2-s-2 and 10.6 (PM3) and 29.1 kJ mol−1 (AM1) in s-4. DFT ab initio calculations at B3LYP/6-31G*//HF/6-31G* on the related 10,10′-dimethylene derivative of 4 indicated that the syn-folded conformation is less stable than the anti-folded conformation by 39.1 kJ mol−1.26 The syn-folded conformations show also short H1⋯H1′ contact distances at the fjord regions (1.72 Å (PM3) and 2.04 Å (AM1) in C2-s-2 and 1.74 Å (PM3) and 1.94 Å (AM1) in s-4). In C2-s-2, very short CH3⋯H3C distances between the bridges are noted: 1.67 Å (PM3) and 2.07 Å (AM1). The conformational energies of the twisted conformations t-2 and t-4 are practically identical, 63 kJ mol−1 (AM1). The a-4→s-4 photoisomerization and a CFF-π electron-CI calculation of s-4 have been reported.27,28
DNMR Spectroscopy
The synthesis of 5 and 6 allowed a DNMR spectroscopic study of the conformational inversions and E,Z-isomerizations of these methylene-bridged bistricyclic enes. The 1H-NMR spectrum of 5 included two methyl singlets, indicating the presence of the E- and Z-diastereomers. In an anti-folded conformation of 5, the symmetries of the E- and Z-diastereomers are Ci and C2 respectively; causing both methyl substituents in each diastereomer to be isochronous. The thermal E,Z-isomerization was studied by monitoring the coalescence of the two methyl signals representing the E- and the Z-diastereomers, respectively. The conformational inversion of 5 and 6 was studied by monitoring the 1H-NMR coalescence of the AB system representing the diastereotopic axial and equatorial hydrogens of the CH2 bridges. It should be noted that in 5 and 6, there was not any difference in the chemical shifts of the hydrogens of the CH2 bridges between the E- and the Z-diastereomers. The results of the DNMR study are summarized in Table 2. In the case of 5, ΔGc‡ (E⇌Z) = 99.6 kJ mol−1 (in CDBr3) and ΔGc‡ (inversion) = 97.9 kJ mol−1 (in hexachlorobutadiene). In 6, the corresponding ΔGc‡ values were 100.4 and 96.7 kJ mol−1 (in 1-bromonaphthalene). In the isopropylidene-bridged 2, the 1H-NMR and 13C-NMR spectra led in each case to two separated singlets representing the axial and equatorial methyl groups. Upon a conformational inversion of 2, a Meax⇌Meeq interconversion would be expected. A DNMR experiment in naphthalene (Δν = 35 Hz) up to 207 °C did not show any significant broadening of the two 1H NMR methyl singlets, indicating that ΔGc‡ > 92 kJ mol−1. Likewise, in benzophenone (Δν = 3 Hz) at 190 °C, the two methyl 1H-NMR singlets could still be resolved indicating that ΔGc‡ > 108 kJ mol−1. Feringa et al. have reported on the conformational inversion of a few cases of heteromerous bistricyclic enes with isopropylidene bridges.6 As in the case of 2, a DNMR study of metalo-bridged bistricyclic enes with CMe2 and ZnCl2 bridges and with CMe2 and PdCl2 bridges did not result in a coalescence of the methyl signals even at 200 °C (in nitrobenzene-d5).11 The authors suggested that in these cases the results indicated considerable steric hindrance for the isomerization process inverting the folded conformations, and that metal binding results in folded structures completely inert towards interconversions.11 In the case of 9-[10,10-dimethyl-9(10H)-anthracenylidene]-2-methyl-9H-thioxanthene in which the bridges are CMe2 and S, the racemization barrier ΔGc‡ = 105 ± 1 kJ mol−1.6
Table 3 gives the free energies of activation for thermal E,Z-isomerizations and for thermal conformational inversions of various homomerous bistricyclic enes. The most remarkable result of the present DNMR studies is the relatively high and essentially identical values of the free energies of activation for the E,Z-isomerization and for the conformational inversion. These results are consistent with the mechanisms of the conformational processes of anti-folded bistricyclic enes depicted in Fig. 6 and 7.3 These mechanisms involve a common “edge passage” highest transition state. The higher energy barriers observed in the cases of the methylene-bridged and isopropylidene-bridged bistricyclic enes (e.g., 5 and 2) as compared with dixanthylenes8 and bianthrones9 are primarily due to the less overcrowded fjord regions in the anti-folded ground states of these systems. The longer C–X bonds in 2, 5 and 6 and the longer C4a⋯C10a bridge distance allow a higher degree of folding and thus a reduced overcrowding in the fjord regions.6 Such an effect is more pronounced in the case of the isopropylidene-bridged 2 as compared with the methylene-bridged 4. The significantly higher barrier for the conformational inversion of 2versus 5 (ΔΔGc‡ > 8 kJ mol−1e) may also be ascribed to unfavorable steric interactions between one of the methyl groups of 2 and the fjord regions at the opposing ring (e.g., CH3⋯H1′) in the transition state, during the “edge passage”.
3
| System1, X = Y | ΔG‡ (E⇌Z)/kJ mol−1 |
Substituent | ΔG‡ (inversion)/kJ mol−1 |
Substituent |
|---|---|---|---|---|
| a This work. | ||||
| — | 104.4 | 2,2′-di-CH3 | 43.9 | 2-CH(CH3)2 |
| O | 73.2 | 2,2′-di-CH(CH3)2 | 74.1 | 2,2′-di-CH(CH3)2 |
| CO |
83.7 | 2,2′-di-CH3 | 87.9 | 2,2′-di-OCH(CH3)2 |
| NCH3 | 87.0 | 2,2′-di-CH3 | — | — |
| S | 114.6 | 2-CH3 | 114.6 | 2-CH3 |
| CH2a |
99.6 | 2,2′-di-CH3 | 97.9 | 2,2′-di-CH3 |
| C(CH3)2a |
— | — | 108 | — |
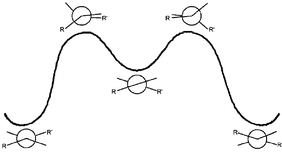 | ||
| Fig. 6 Mechanism of the thermal conformational inversion of anti-folded bistricyclic enes. | ||
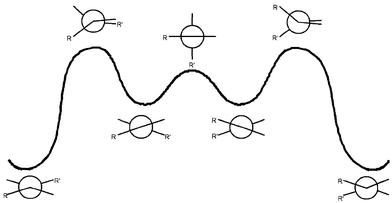 | ||
| Fig. 7 Mechanism of the thermal E,Z-isomerization of anti-folded bistricyclic enes. | ||
Experimental
Melting points are uncorrected. All NMR spectra were recorded with a Bruker DRX 400 spectrometer, unless otherwise stated. 1H NMR spectra were recorded at 400.1 MHz using CDCl3 as solvent and as internal standard (δ(CHCl3) = 7.26 ppm). 13C NMR spectra were recorded at 100.6 MHz using CDCl3 as solvent and as internal standard (δ(CHCl3) = 77.01 ppm). The barriers were determined by DNMR spectroscopy, substituting the coalescence temperature Tc and the extrapolated chemical shift difference Δνc at Tc in the following equation for the Eyring Equation ΔGc‡ = 4.47 Tc [9.97 − log (Tc/Δνc)].10-[10,10-Dimethyl-9(10H![[hair space]](https://www.rsc.org/images/entities/h3_char_200a.gif) )-anthracenylidene]-9,10-dihydro-9,9-dimethylanthracene (2) and 10-[10,10-dimethyl-9(10H)-anthracenyl]-9,10-dihydro-9,9-dimethylanthracene (3)29
)-anthracenylidene]-9,10-dihydro-9,9-dimethylanthracene (2) and 10-[10,10-dimethyl-9(10H)-anthracenyl]-9,10-dihydro-9,9-dimethylanthracene (3)29
The reaction was carried out under an argon atmosphere in a 500 mL three-necked round-bottomed flask equipped with a reflux condenser (protected from moisture), a dropping funnel, and a magnetic stirrer. Freshly distilled dry THF (200 mL distilled over sodium diphenyl ketyl) was added to the flask and cooled to −5 °C. Dropwise slow injection of TiCl4 (1.85 mL, 16.9 mmol) using a plastic syringe with continuous stirring gave a yellow complex, which was treated with Zn dust (2.26 g, 35.5 mmol). The temperature was gradually raised to rt and kept for another 1 h to give a greenish suspension. The mixture was then refluxed for 5 h to complete the reduction, giving a green–black suspension. After cooling to 0 °C, the mixture was treated with pyridine (2 mL), followed by dropwise addition over 3.5 h of a solution of ketone 7 (3.50 g, 15.5 mmol)22 in dry THF (100 mL). The resulting mixture was refluxed for 48 h. The disappearance of the starting material was monitored by TLC. After being cooled to room temperature, the mixture was added to dichloromethane (300 mL), stirred vigorously and treated with aqueous HCl (0.1 M) to give two layers. The organic layer was separated, and the aqueous layer was extracted with dichloromethane. The organic layers were dried (MgSO4), and the solvents were removed in vacuum. The crude products (3.0 g) were dissolved in dichloromethane and chromatographed on a silica column, using a hexane–diethyl ether gradient (up to 20% diethyl ether) as eluent. The chromatography afforded 2.1 g (65.6% yield) of the fluorescent yellow products. The 1H-NMR spectrum (vide infra) indicated a 1∶3 mixture of 2 and 3. Recrystallization from benzene gave single crystals of 2 (1∶1 complex with benzene), mp 325 °C, suitable for X-ray analysis (lit.,16 mp 312–313 °C).
Anal. Calcd for C38H34 (C32H28·C6H6): C, 93.02; H, 6.98. Found: C, 93.26; H, 6.87%. 1H-NMR (300 MHz, CDCl3δ = 7.26 ppm) δ: 7.526 (br d, 3J = 7.7 Hz, 4H, H4, H5, H4′, H5′), 7.153 (dt, 3J = 7.5 Hz, 4J = 1.5 Hz, 4H, H3, H6, H3′, H6′), 7.010 (dd, 3J = 7.7 Hz, 4J = 1.5 Hz, 4H, H1, H8, H1′, H8′), 6.885 (dt, 3J = 7.4 Hz, 4J = 1.0 Hz, 4H, H2, H7, H2′, H7′), 1.974 (s, 6H, CH3), 1.851 (s, 6H, CH3). 13C-NMR (50.29 MHz, CDCl3δ = 77.008 ppm) δ 147.338, 137.885, 130.516, 128.851, 126.479, 124.572, 122.970, 40.438, 30.831, 24.257. MS (140 °C, % P) m/z 412 (C32H28+·, 97), 411 (C32H27+·, 100), 382 (C30H22+·, 98), 367 (C29H19+·, 17), 352 (C28H16+·, 25), 206 (C16H14+·, 15), 191 (C30H282+, 66), 176 (C28H162+, 89), 175 (C28H142+, 90).
21/c (no.14), Z = 2, μ(Mo-Kα) = 0.32 cm−1, 2306 unique reflections of which 1823 were observed (I > 2σI), R = 0.033, RW = 0.045.
Compound 3 was isolated from the filtrate of the recrystallization of 2. The solvent was removed in vacuum and the remaining solid was recrystallized three times from dichloromethane. Sublimation at 170 °C/0.1 mmHg gave single crystals of 3, mp 258 °C, suitable for X-ray analysis (lit.,16 mp 260 °C).
Anal. Calcd for C32H30: C, 92.70; H, 7.30. Found: C, 92.42; H, 6.99%. 1H-NMR (400 MHz, 363 K, Cl2CDCDCl2δ = 5.15) δ 7.396 (d, 3J = 8.0 Hz, 4H, H4, H5, H4′, H5′), 7.179 (t, 3J = 7.1 Hz, 4H, H3, H6, H3′, H6′), 6.915 (t, 3J = 7.5 Hz, 4H, H2, H7, H2′, H7′), 6.419 (br d, 3J = 7.4 Hz, 4H, H1, H8, H1′, H8′), 4.440 (s, 2H, H9, H9′), 1.562 (s, 6H, CH3), 0.971 (s, 6H, CH3). 13C-NMR (100 MHz, 363 K, Cl2CDCDCl2, δ = 77.007 ppm) δ: 145.71 (C4a, C10a, C4a′, C10a′), 135.19 (C8a, C9a, C8a′, C9a′), 129.75 (C1, C8, C1′, C8′), 126.92 (C3, C6, C3′, C6′), 126.35 (C4, C5, C4′, C5′), 125.27 (C2, C7, C2′, C7′), 55.78 (C9, C9′), 38.62 (C10, C10′), 33.85 (CH3), 33.64 (CH3). MS (150 °C, % P). m/z 208 (C16H16+·, 70), 207 (C16H15+·, 100), 193 (C15H13+·, 71), 192 (C15H12+·, 97), 191 (C15H11+·, 74), 190 (C15H10+·, 26), 189 (C15H9+·, 69), 178 (C14H10+·, 33), 165 (C13H9+·, 49).
21/c (no.14), Z = 2, μ(Mo-Kα) = 0.33 cm−1, 2269 unique reflections of which 1553 were observed (I > 2σI), R = 0.055, RW = 0.068.
9-[2-Methyl-9(10H)-anthracenylidene]-9,10-dihydro-2-methylanthracene (5)
The reaction was carried out in an argon atmosphere in a round-bottomed flask equipped with a reflux condenser (protected from moisture) and a magnetic stirrer. Dry diethyl ether (30 ml) was added to the flask followed by anhydrous AlCl3 (3.6 g, 27 mmol) in small portions and then by LiAlH4 (0.6 g, 15.8 mmol) in small portions. The mixture was stirred for 30 min at rt, dry benzene (50 ml) was added followed by 2,2′-dimethylbianthrone (8)9 (0.25 g, 0.6 mmol) in small portions. The mixture was then refluxed at 80 °C for 3 h. The excess LiAlH4 was decomposed by adding dropwise ethyl acetate and the mixture treated by water, followed by dilute aqueous HCl. The organic layer was separated and the aqueous layer extracted with dichloromethane. The combined organic layers were dried (MgSO4), and the solvents were removed in vacuum. The crude product was purified by preparative chromatography on a PLC silica plate, using benzene as eluent (Rf = 0.9). Recrystallization from benzene–petrol ether (40–60 °C) gave 5 as a colorless powder, mp 265–272 °C, in 65% yield. The E∶Z ratio was 55∶45 (or vice versa).
Anal. Calcd for C30H24: C, 93.70; H, 6.30. Found: C, 93.44; H, 6.08%. 1H-NMR (400 MHz, CDCl3, δ = 7.26 ppm) 7.349 (d, 3J = 7.5 Hz, 2H), 7.247 (d, 3J = 7.5 Hz, 2H), 7.076–7.122 (t, 3J = 7.4 Hz, 4J = 1.5 Hz, 2H), 6.912–6.975 (m, 4H), 6.877 (t, 3J = 7.4 Hz, 2H), 6.787 (br s, 0.45 × 2H), 6.759 (br s, 0.55 × 2H), 4.214 (br d, J = 16.2 Hz, 2H), 3.827 (sd, J = 16.2 Hz, 2H), 2.052 (s, 0.45 × 6H CH3), 2.037 (s, 0.55 × 6H, CH3). δ (CH3) νE − νZ (270 MHz, CDBr3, CH3 signals) 3.75 Hz. Δδ (CH2, AB system) 100 MHz, hexachlorobutadiene). νA − νB = 41.2 Hz, 2J = 16.3 Hz. DNMR (270 MHz), CDBr3, Tc = 4.32 ± 5 K, Δνc = 3.75 ± 0.05 Hz, (CH3), ΔGc‡ (E⇌Z) = 99.6 kJ mol−1. DNMR (100 MHz), hexachlorobutadiene, Tc = 471 ± 8 K, Δνc = 41.2 ± 0.5 (CH2), ΔGc‡ (inversion) = 97.9 kJ mol−1. MS (% P) m/z: 385 (13C12C29H24+·, 32), 384 (12C30H24+·, 100), 383 (12C30H23, 26), 382 (12C30H22+·, 45), 192 (12C30H24+2, 73). UV (CH2Cl2) λmax nm (log ε): 253 (4.24), 323 (4.13). 13C-NMR (100 MHz, CDCl3) δ: 139.24, 139.12, 137.90, 137.77, 137.56, 137.39, 135.99, 135.87, 134.14, 133.05, 131.84, 129.63, 129.57, 129.02, 128.93, 127.04, 126.80, 126.65, 126.63, 126.28, 126.26, 124.76, 124.55, 37.15 (C10′, C10′), 37.14 (C10′, C10′), 20.95 (CH3), 20.85 (CH3).
9-[9(10H)-Anthracenylidene]-9,10-dihydroanthracene (4)
Hydrocarbon 4 was prepared by a LiAlH4–AlCl3 reduction of bianthrone (1, X = Y: CO) analogously to 5 in 50% yield and was obtained after recrystallization from toluene as colorless powder, mp 288–291 °C (decomp.). (Lit.,17 mp 328 °C).
Anal. Calcd for C28H20: C, 94.34; H, 5.66. Found: C, 94.75; H, 5.76%. 1H-NMR (400 MHz, CDCl3) δ: 7.364 (dd, 3J = 7.5 Hz, 4H, H4, H5, H4′, H5′), 7.117 (dt, 3J = 7.4 Hz, 4J = 1.4 Hz, 4H, H3, H6, H3′, H6′), 6.984 (dd, 3J = 7.8, 4J = 1.0 Hz, 4H, H1, H8, H1′, H8′), 6.894 (dt, 3J = 7.5 Hz, 4J = 1.1 Hz, 4H, H2, H7, H2′, H7′), 4.240 (br d, 2J = 16.2 Hz, 2H, CH2), 3.865 (d, J = 15.8 Hz, 2H, CH2). 13C-NMR (100 MHz, CDCl3) δ 37.60 (CH2, C10, C10′), 124.83 (C2, C7, C2′, C7′), 126.39 (C3, C6, C3′, C6′), 128.96 (C1, C8, C1′, C8′), 131.97 (C9, C9′), 137.70 (C8a, C9a, C8a′, C9a′), 139.06 (C4a, C10a, C4a′, C10a′). MS (% P) m/z 357 (13C12C27H20+·, 32), 356 (12C28H20+·, 100), 355 (12C28H19+·, 20), 354 (12C28H18, 12), 179 (36), 178 (12C30H20+2, 96). UV (CH2Cl2) λmax nm (log ε): 253 (4.34), 320 (4.20).
5-[3-Methyl-5(12H)-naphthacenylidene]-5,12-dihydro-3-methylnaphthacene (6)
Hydrocarbon 6 was prepared by a LiAlH4–AlCl3 reduction of 2-methyl-12-[3-methyl-12-oxo-5(12H)-naphthacenylidene]naphthacen-5(12H)-one,9 analogously to 4. The product was purified on a silica PLC plate, using dichloromethane–petrol ether (40–60 °C) 1∶1 as eluent (Rf = 0.7). Recrystallization from benzene–petrol ether (40–60 °C) gave 6 as a pale yellowish powder, mp >300 °C (slow decomposition during heating to give a brown–red color). The yield was 72%. The E∶Z ratio was 52∶48 (or vice versa).
Anal. Calcd for C38H28: C, 94.18; H, 5.82. Found: C, 94.33; H, 5.99%. 1H-NMR (400 MHz, CDCl3) δ: 1.834 (s, 0.52 × 6H, (CH3)2), 2.059 (s, 0.48 × 6H, (CH3)2), 4.061 (d, 2J = 15.9, 2H, H10, H10′), 4.451* (br d, 2J = 16.0, 2H, H10, H10′), 6.784 (br s, 0.52 × 2H), 6.809 (br s, 0.48 × 2H), 6.914 (d, 3J = 7.6, 4J = 1.0, 0.52 × 2H), 6.960 (d, 3J = 7.5, 4J = 0.8, 0.48 × 2H), 7.136 (t, 3J = 7.9, 4J = 1.1, 0.52 × 2H), 7.209 (d, 3J = 8.1, 0.52 × 2H), 7.278–7.346 (m, 5.8H), 7.386 (t, 3J = 8.0, 3J = 6.8, 4J = 1.3, 0.52 × 2H), 7.432 (d, J = 8.1 Hz, 0.52 × 2H), 7.489 (s, 2H), 7.751 (t, 2H), 7.838 (d, J = 7.9, 2H). DNMR (100 MHz), 1-bromonaphthalene, Tc = 4.67 ± 8 K, Δνc = 25 ± 1 Hz, (CH3), ΔGc‡ (E⇌Z) = 100.4 kJ mol−1; Tc = 465 ± 10 K, Δνc = 49 ± 1 (CH2), ΔGc‡ (inversion) = 96.7 kJ mol−1. 13C-NMR (100 MHz, CDCl3) δ: 20.85 (CH3), 20.89 (CH3), 37.45 (C10, C10′), 37.48 (C10, C10′), 124.60, 124.86, 124.89, 124.98, 125.69, 125.73, 126.62, 126.77, 126.86, 126.87, 127.33, 127.34, 127.48, 127.78, 127.84, 127.92, 129.32, 129.59, 131.44, 131.50, 132.10, 132.10, 132.18, 132.71, 132.75, 134.24, 134.51, 135.89, 135.91, 135.92, 135.94, 137.29, 137.32, 137.43, 137.51. MS (% P) m/z: 485 (13C12C37H28+·, 42), 484 (12C38H28+·, 100), 483 (10), 482 (9), 469 (12C37H25+·, 5), 242 (12C38H28+2, 30). UV (CH2Cl2) λmax nm (log ε) 237 (4.19), 275 (3.81), 348 (3.38).
References
- G. Shoham , S. Cohen , R. M. Suissa and I. Agranat , in Molecular Structure: Chemical Reactivity and Biological Activity, eds. J. J. Stezowski, J.-L. Huang and M.-C. Shao, Oxford University Press, Oxford, 1988, p. 290. Search PubMed.
- J. Sandström , in The Chemistry of Double-Bonded Functional Groups, Supplement A3, ed. S. Patai, Wiley, New York, 1997, p. 1253. Search PubMed.
- P. U. Biedermann , J. J. Stezowski and I. Agranat , in Advances in Theoretically Interesting Molecules, Vol. 4, ed. R. P. Thummel, JAI Press, Stamford, CT, 1998, p. 245. Search PubMed.
- P. U. Biedermann, A. Levy, J. J. Stezowski and I. Agranat, Chirality, 1995, 7, 199 CrossRef CAS.
- A. Z.-Q. Khan and J. Sandström, J. Am. Chem. Soc., 1988, 110, 4843 CrossRef CAS.
- B. L. Feringa, W. F. Jager and B. de Lange, Tetrahedron Lett., 1992, 33, 2887 CrossRef CAS.
- I. Agranat and Y. Tapuhi, J. Am. Chem. Soc., 1978, 100, 5604 CrossRef CAS.
- I. Agranat and Y. Tapuhi, J. Am. Chem. Soc., 1979, 101, 665 CrossRef CAS.
- I. Agranat and Y. Tapuhi, J. Org. Chem., 1979, 44, 1941 CrossRef CAS.
- I. O. Sutherland, Annu. Rep. N.M.R. Spectrosc., 1971, 4, 71 Search PubMed.
- (a) W. I. Smid, A. M. Schoevaars, W. Kruizinga, N. Veldman, W. J. J. Smeets, A. L. Spek and B. L. Feringa, Chem. Commun., 1996, 2265 RSC; (b) 3D Search and Research Using the Cambridge Structural Database, F. H. Allen and O. Kennard , Chem. Design Autom. News, 1993, 8(1), 31. Search PubMed.
- T. Mukaiyama, T. Sato and J. Hanna, Chem. Lett., 1973, 1041 CAS.
- D. Lenoir and P. Lemmen, Chem. Ber., 1980, 113, 3112 Search PubMed.
- I. Agranat, S. Cohen, R. Isaksson, J. Sandström and M. R. Suissa, J. Org. Chem., 1990, 55, 4943 CrossRef CAS.
- J. March , Advanced Organic Chemistry: Reactions, Mechanisms and Structure, 4th edn., Wiley, New York, 1992, p. 1227. Search PubMed.
- C. Herberg, H.-D. Beckhaus and C. Rüchardt, Chem. Ber., 1994, 127, 2065 CAS.
- A. F. A. Ismail and Z. M. El-Chafei, J. Chem. Soc., 1957, 796 RSC.
- R. Sato, T. Nagaoka and M. Saito, Tetrahedron Lett., 1990, 31, 4165 CrossRef CAS.
- Preliminary results of the crystal structure of 2 have been previously reported, see references 1 and 3..
- Y. V. Zefirov, Crystallogr. Rep., 1997, 42, 111.
- J. J. P. Stewart, J. Comput. Chem., 1989, 10, 209 CrossRef CAS.
- M. J. S. Dewar, E. G. Zoebisch, E. F. Healy and J. J. P. Stewart, J. Am. Chem. Soc., 1985, 107, 3902 CrossRef.
- J. J. P. Stewart , MOPAC 93; Fujitsu Limited: Tokyo, Japan; Search PubMedall rights reserved..
- M. J. S. Dewar, E. F. Healy, A. J. Holder and Y.-C. Yuan, J. Comput. Chem., 1990, 11, 541 CrossRef CAS.
- J. J. P. Stewart, J. Comput. Chem., 1990, 11, 543 CrossRef CAS.
- S. Kammermeier and R. Herges, Angew. Chem., Int. Ed. Engl., 1996, 35, 417 CrossRef CAS.
- R. Korenstein, K. A. Muszkat and G. Seger, J. Chem. Soc., Perkin, Trans. 2, 1976, 1536 RSC.
- R. Korenstein, K. A. Muszkat and E. Fischer, J. Chem. Soc., Perkin, Trans. 2, 1977, 564 RSC.
- M. R. Suissa , PhD Thesis, The Hebrew University of Jerusalem, Jerusalem, 1988. Search PubMed.
Footnote |
| † CCDC reference number 188/192. See http://www.rsc.org/suppdata/p2/a9/a906696i for crystallographic files in .cif format. |
| This journal is © The Royal Society of Chemistry 2000 |